

Abstract
In eukaryote genomes, the polyadenylation site marks termination of mature RNA transcripts by a poly-adenine tail. The polyadenylation site is recognized by a dynamic protein complex, among which the poly-adenine-binding protein nuclear1 plays a key role. Reduced poly-adenine-binding protein nuclear1 levels are found in aged muscles and are even lower in oculopharyngeal muscular dystrophy patients. Oculopharyngeal muscular dystrophy is a rare, late onset autosomal dominant myopathy, and is caused by an alanine expansion mutation in poly-adenine-binding protein nuclear1. Mutant poly-adenine-binding protein nuclear1 forms insoluble nuclear aggregates leading to depletion of functional poly-adenine-binding protein nuclear1 levels. In oculopharyngeal muscular dystrophy models, increased utilization of proximal polyadenylation sites has been observed in tandem 3′-untranslated regions, and most often cause gene upregulation. However, global alterations in expression profiles canonly partly be explained by polyadenylation site switches within the most distal 3′-untranslated region. Most poly-adenine signals are found at the distal 3′-untranslated region, but a significant part is also found in internal gene regions, like introns, exons, and internal 3′-untranslated regions. Here, we investigated poly-adenine-binding protein nuclear1’s role in polyadenylation site utilization in internal gene regions. In the quadriceps muscle of oculopharyngeal muscular dystrophy mice expressing expPABPN1 we found significant polyadenylation site switches between gene regions in 17% of genes with polyadenylation site in multiple regions (N = 574; 5% False Discovery Rate). Polyadenylation site switches between gene regions were associated with differences in transcript expression levels and alterations in open reading frames. Transcripts ending at internal polyadenylation site were confirmed in tibialis anterior muscles from the same mice and in mouse muscle cell cultures overexpressing expPABPN1. The polyadenylation site switches were associated with nuclear accumulation of full-length transcripts. Our results provide further insights into the diverse roles of poly-adenine-binding protein nuclear1 in the post-transcriptional control of muscle gene expression and its relevance for oculopharyngeal muscular dystrophy pathology and muscle aging.
Muscle degeneration: RNA structures as modulator of muscle function
Transcriptional and post-transcriptional regulation of gene expression has a fundamental role in shaping the protein landscape. In skeletal muscles, the RNA landscape plays an important role in muscle function in both health and disease conditions. PABPN1 is a key regulator of mRNA processing in a late-onset muscular dystrophy and in aging. Here, the authors demonstrate that a decrease in PABPN1 function results in genome-wide alterations in mRNA structures. Reduced PABPN1 function alters both mRNA levels and RNA structures leading to altered protein sequences. Importantly, among the affected genes are muscle function regulators. Together, this study calls for a better understanding of alterations in RNA structures in disease and aging.
Introduction
The maturation of a messenger RNA (mRNA) is a multistep process influencing the RNA landscape of a cell. Polyadenylation is an essential step in mRNA maturation, affecting mRNA stability, nuclear export, and translation efficiency.1 Polyadenylation includes endonucleolytic cleavage of mRNA at the polyadenylation site (PAS) and is completed by addition of an adenine tail. The majorityof genes have multiple PASs, generating distinct transcripts.2 The majority of PASs are located in the distal 3′-untranslated region (3′-UTR) and alternative PAS in the distal 3′-UTR most often result in transcripts with a shorter 3′-UTR, suggesting to affect mRNA stability.3 Although PASs are most abundant in the distal 3′-UTR, polyadenylation within internal gene regions (introns, exons, and internal 3′-UTRs) were also found.2, 4 Genes with PASs in internal gene regions could generate a wide range of alternative transcripts, among those with alternative coding sequence (CDS).5
Polyadenylation is regulated by a dynamic protein complex and the poly-adenine (poly(A))-binding protein nuclear 1 (PABPN1) plays a central role.6 PABPN1 is ubiquitously expressed and is primarily recognized for regulation of the poly(A) tail length.7 PABPN1 regulates PAS utilization in the distal 3′-UTR by suppressing proximal PASs.8, 9 Reduced levels of functional PABPN1 are found in aged muscles and in muscles from oculopharyngeal muscular dystrophy (OPMD) patients.10 OPMD is caused by an alanine expansion at the N-terminus of PABPN1 (expPABPN1).11 ExpPABPN1 forms insoluble aggregates in myonuclei,12 which reduces levels of soluble PABPN1 (ref. 13). Reduced levels of functional PABPN1 cause switches in PAS utilizationin the distal 3′-UTR, with most prominent switches from distal to proximal PAS 3′-UTR.8, 9 Shorter transcripts lead to a loss of microRNA (miRNA)-binding sites, which could generally explain upregulation of shorter transcripts from proximal PAS.8 More recently, Li et al.,5 suggested a role for PABPN1 in internal PAS utilization. Yet, this was not investigatedin the context of disease.
In this study, we investigated a role for expPABPN1 in PAS utilization in internal gene regions in muscles. We employed our previously generated genome-wide single molecule polyA-Seq data9 where dealt only with PAS switches within the same 3′-UTR region. Here, we extended PAS analysis across the genome and found internal PASs in a quarter of muscle genes. We show that changes in PAS utilization between gene regions occur in both upstream and downstream directions (Fig. 1). The expPABPN1-induced PAS switches are associated with few non-canonical PAS motifs. We show that PAS switches in genes involved in muscle pathologies are affected by expPABPN1 expression. Our study suggests that the role of PABPN1 in regulation of mRNA processing is broad and can contribute to muscle weakness in OPMD via different mechanisms.
Fig. 1.

Schematic representation of PAS switches. Switches in PAS utilization are shown with arrows: upstream switches are depicted in blue and downstream switches in red. An upstream switch is most often from distal 3′-UTR to any internal gene regions (intron, exon, and internal 3′-UTR), and a downstream switch is most often from any internal gene regions to distal 3′-UTR. Switches between tandem PASs in the distal 3′-UTR are also shown. The most prominent switches are depicted with continues line. PASs are depicted with vertical bars, the gene regions, exon (E), intron (I), and 3′-UTR are indicated. Illustrated are the most abundant switch possibilities
Results
Polyadenylation within gene regions is affected by expPABPN1 in muscles
To investigate the effect of expPABPN1 on PAS usage in internal gene regions we employed our previously generated single molecule polyA sequencing data from the mouse quadriceps muscles of A17.1 mice expressing expPABPN1 and their matched wild-type FVB controls.9 Only PASs with coverage of at least two reads per PASs in at least 6 out of 12 samples were included in the analysis, leaving 35,351 PASs located in 13,685 genes. Confirming previous studies,8, 9 the majority of PAS (69%) mapped to the most distal 3′-UTR region, and the rest (31%; Fig. 2a) mapped into internal gene regions, including introns, exons, and internal 3′-UTRs. Three-thousand two-hundred and ninety six genes (24%) contained PASs in multiple regions (Fig. 2b), and those were used to investigate a role for expPABPN1 in PAS utilization in internal regions.
Fig. 2.

Distribution of PAS locations. a Pie chart showing the percentage of PASs distribution in different regions of the gene (24,248; 6273; 3106, and 1724 PASs located at distal 3′-UTRs, introns, exons, and internal 3′-UTR, respectively). b Pie chart showing the percentage of genes containing PAS in single or multiple regions (3296 genes have multiple PAS regions and 10,355 genes have single PAS region)
In our previous study, we reported the effect of expPABPN1 on PAS utilization in the distal 3′-UTR region (PASs in distal 3′-UTR). To investigate expPABPN1 effect on internal PAS utilization, PAS reads within a gene region were summed and the relative usage of the different PAS regions were assessed with a generalized linear mixed model on the binomial distribution of the counts for each region in a gene. Per gene, a PAS region that was more used in FVB control compared to A17.1 is dubbed as a default region, and a PAS region that was more used in A17.1 compared to FVB is referred to as a switch region.
The possible switches in PAS utilization could be from a default distal PAS to an upstream PAS (named here an upstream switch) or from a default PAS in internal region to a downstream PAS (named here a downstream switch) (Fig. 1). Examples of upstream and downstream switches are shown in Fig. 3. In 2300009A05Rik (shortened here to RikA05), the default distal PAS was located in the distal 3′-UTR and a secondary PAS was found within the intron (Fig. 3a; FVB). The PAS within the intronic region was relatively more utilized in A17.1 compared to FVB (Fig. 3a), indicating an upstream switch. In Arih1, additionally to the previously described PAS switch within the 3′-UTR,9 a PAS was found within an intron, which was relatively more utilized in FVB. In A17.1 the PAS in the distal 3′-UTR was relatively more utilized, indicating a downstream switch (Fig. 3b).
Fig. 3.

Upstream and downstream switches in PAS regions. a–b USCS Genome Browser screenshots displaying PASs in RikA05 (upstream switch) and Arih1 (downstream switch) genes. The y-axis represents the coverage of the peaks, corresponding to the number of reads mapped at each PAS. One of the FVB control and A17.1 mice are depicted in independent traces. PASs are depicted by arrows; distal or internal PASs are indicated. The gene structure based on Ensembl annotation is presented in red. The amplified regions that were used for validation are indicated below the gene structure; c Pie chart on left side shows the percentage of genes with changes in PAS utilization region (567 genes) and the right pie chart shows the percentage of genes with downstream or upstream switches in PAS utilization (193 and 374 genes, respectively)
From the 3296 genes with two or more PAS regions, 17% showed a significant switch in PAS utilization between regions (N = 574; false discovery rate (FDR) < 0.05; Fig. 3c). An upstream switch was found in 66% of those genes (Fig. 3c; list in Supplementary Table S2).
PASs within gene regions affect gene expression levels
Within the same 3′-UTR, switches from distal to proximal PAS predominantly affect mRNA stability leading to transcript upregulation.8, 9 Here, we investigated whether PAS utilization between gene regions also affects mRNA expression levels. For each gene fold changes were determined (Supplementary Table S2), and we found that gene dysregulation was significantly higher in genes with a PAS switch between gene regions compared to genes without a switch (50% and 28%, respectively; Fisher’s exact test p-value = 8.2E10-12) (Fig. 4a). The majority of genes (66%) with PAS switch were found to be upregulated (Fig. 4a). We then examined whether PAS switch direction specifically affect gene dysregulation direction, and found in genes with a downstream switch a slightly higher proportion of dysregulated genes compared with an upstream switch (56.5% and 46%, respectively).
Fig. 4.
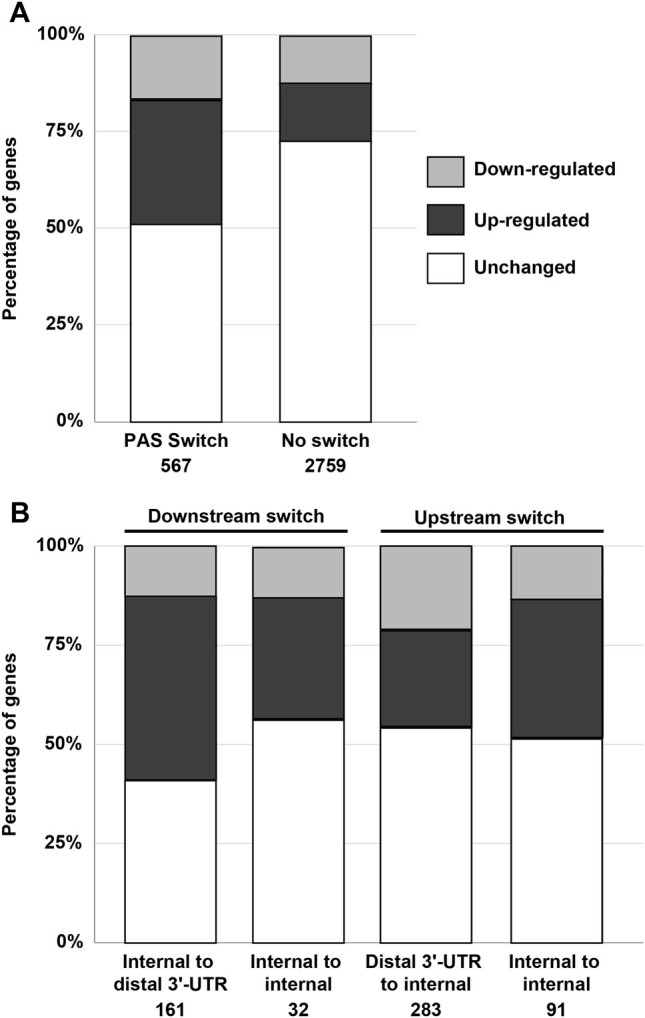
Differential expression analysis in upstream and downstream switch groups. Bar chart shows differential expression analysis in genes with or without a significant switch in PAS utilization (a) and in genes with upstream or downstream switches (b). The number of genes for each category is indicated
We further investigated whether switches to distinct gene regions may affect dysregulation direction. As expected, PAS switches to/from the distal 3′-UTR were predominant (78%) (Fig. 4b). In internal gene regions PAS switches from/to introns was the most abundant (Supplementary Table S3). The majority PAS switches were upstream (66%) compared with a downstream switch (34%). In both switch direction, the proportion of upregulated genes was higher compared with downregulation, but in the downstream switch relatively more genes had upregulation compared with the upstream switch (77% and 59%, respectively). Notably, an upstream switch from distal 3′-UTR to internal gene regions was not associated with a specific dysregulation direction (Fig. 4b). Thus, in contrast to upstream PAS switches within the 3′-UTR, which predominantly lead to increase in transcript levels, an upstream switch between 3′-UTR and introns or other gene regions doesnot specifically affect dysregulation direction.
To validate PAS switches between gene regions that were found in quadriceps muscles, reverse transcription quantitative PCR (RT-qPCR) was performed on RNA from tibialis anterior muscles, which are severely affected in A17.1 mouse.14 PAS switches between gene regions were validated in six genes containing one distinct PAS per gene region. Arih1 represents a downstream switch, whereas RikA05, Vezf1, Lmna, Figf, and Ttn genes represent an upstream switch (Fig. 5a). Usp19 was selected as control as it possesses internal and distal PASs without switch in utilization in A17.1 (Fig. 5a). We first confirmed the expression of transcripts from internal PAS using a primer set specific to the internal 3′-UTR or intronic region (Supplementary Fig. S3). PAS usage in intronic regions resulted in alternative transcripts for RikA05, Vezf1, Figf, and Arih1 transcripts (Supplementary Fig. S3). Alternative transcripts from internal 3′-UTR region were found for Lmna and Usp19 (Supplementary Fig. S3). The intronic region near PAS in Ttn was not suitable for qRT-PCR, and the proximal primer set was, therefore, designed on the nearby exon (Supplementary Fig. S2). PAS switch was assessed by the ratio of PCR products from the internal region and the distal 3′-UTR. In accordance with the polyA-Seq data, we found a downstream switch between internal PAS and distal PAS for Arih1 and an upstream switch for RikA05, Lmna, Figf, and Ttn (Fig. 5b). For Vezf1 only a trend suggesting an upstream switch was found (Fig. 5b). As expected, no switch in PAS utilization was found for Usp19 (Fig. 5b). For those seven genes we then also assessed whether PAS switch is accompanied by a change in expression levels. Overall, fold change direction was found to be consistent between RNA-seq and qRT-PCR for 6 out of the 7 genes (Arih1, RikA05, LmnA, Figf, Ttn, and Usp19; Fig. 5c, d). Only for Vezf1 fold change was not validated using qRT-PCR (Fig. 5c, d). Among the five genes with a downstream switch in three genes upregulation and in two genes downregulation was found in A17.1.
Fig. 5.
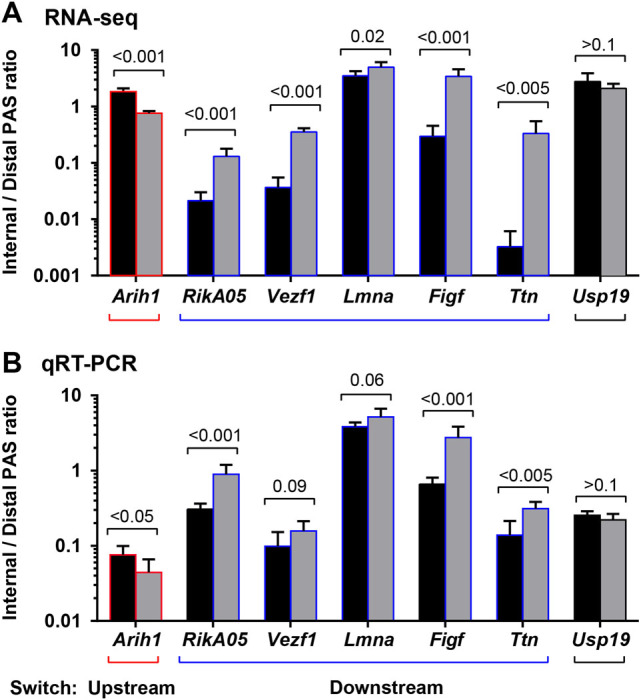
PAS switch between regions in quadriceps and tibialis anterior muscles. a Bar chart shows the ratio between distal to internal PAS obtained from read counts from polyA-sequencing in quadriceps muscle. Expression values were obtained after TMM normalization. Averages and SD are from six mice. b Bar chart shows the ratio between distal to internal PAS obtained from qRT-PCR analysis in tibialis anterior muscle. Averages and SD are from six mice. p-values for the differences between A17.1 and WT tibialis anterior muscles were assessed with the Student’s t-test. Red outlines depict a downstream switch, and blue outlines an upstream switch
To confirma role of expPABPN1 in intronic PAS utilization, we performed qRT-PCR in the IM2 muscle cell culture model for OPMD. In this model, wild-type PABPN1 (WT) or expPABPN1 were stably overexpressed at similar levels.13 Significant switches between gene regions were found for 4 out of the 5 genes (Fig. 6a). Switch direction was similar between the muscle cell culture and muscles for RikA05 and Vezf1,and Arih1, but the fold change was smaller in the cell culture model (Fig. 6a). In Lmna a downstream switch, instead of an upstream switch, was found (Fig. 6a). No PAS switch was found in Figf, and PAS switch also remained unchanged in Usp19 (Fig. 6a). Ttn was excluded from this study, since the intronic region near PAS could not be amplified. Together those results further support a role for expPABPN1 in PAS switch between regions.
Fig. 6.
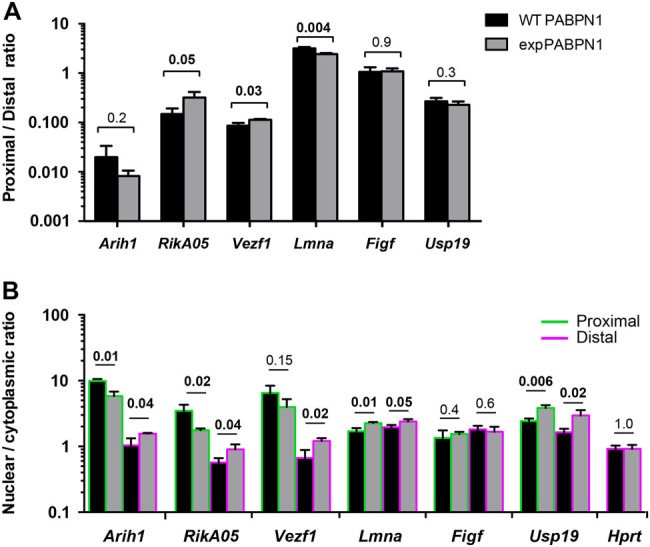
The effect of expPABPN1 on PAS utilization in muscle cell culture. a Bar chart shows the ratio between distal and internal PAS from qRT-PCR analysis. Red outlines are depicted for a downstream switch, and blue outlines for an upstream switch and unchanged by a black line. b Bar chart shows changes in transcript abundance between nuclear and cytoplasmic fractions. Primer set amplifying intronic regions are depicted by green outlines and from primer set in the distal 3′-UTR by purple outlines. Averages and SD are from three biological replicates. Differences between WT-PABPN1 and expPABPN1 cultures were assessed with the Student’s t-test. Significant p-values are in bold
Nuclear export of transcripts is affected by expPABPN1
PABPN1 shuttles between the nuclear and cytoplasm, and expPABPN1 has a higher nuclear accumulation compared with its wild-type counterpart.15 A higher nuclear accumulation of expPABPN1 compared with WT PABPN1 was also found in the stable IM2 cell model.13 To assess whether subcellular distribution of transcripts from internal or distal 3′-UTRPAS is affected by nuclear retention of expPABPN1, the ratio between nuclear and cytosolic abundance of transcripts from distal or proximal PAS was investigated using qRT-PCR. RNA levels were normalized to Hprt whose levels were similar in both fractions and expression levels were unaffected by expPABPN1 expression (Fig. 6c). In 5 out of the 6 genes (Arih1, RikA05, Vezf1, Lmna, Usp19) the nuclear to cytoplasmic ratio was affected by expPABPN1 expression (Fig. 6c). Most consistently, the transcript from distal 3′-UTR showed a higher nuclear accumulation in expPABPN1 expressing cells compared with WT-PABPN1 cell cultures (Fig. 6c). Nuclear accumulation of transcripts from proximal PAS was reduced in expPABPN1 cell culture for Arih1 and RikA05 but was increased in Lmna and Usp19 (Fig. 6c). High nuclear accumulation could suggest aberrant nuclear export of transcripts or higher mRNA decay in the cytoplasm. Our results suggest that nuclear accumulation of transcripts from distal PAS is associated with nuclear retention of expPABPN1. Nuclear accumulation of transcripts from distal PAS was also confirmed in A17.1 tibialis anterior muscles (Supplementary Fig. S5).
Polyadenylation signal motifs utilization is associated with expPABPN1
Previously, we showed that in the distal 3′-UTR the non-canonical polyadenylation signals are more frequently utilized when expPABPN1 is expressed. The non-canonical polyadenylation signals are found near proximal PASs in the distal 3′-UTR, whereas the canonical polyadenylation signals (AATAAA and ATTAAA) are mostly present near distal PASs in the distal 3′-UTR.9 Here, we investigated whether any of the 2 canonical and 11 non-canonical motifs were associated with expPABPN1-mediated PAS switches. We counted the 13 polyadenylation signal motifs < 50 upstream of the identified PAS, and assessed their frequencies in gene regions demonstrating a PAS switch. The frequency of the motifs was investigated separately in upstream or downstream regions and default or switch regions and was compared with frequencies in random selections from the identified PAS (Fig. 7a and Supplementary Table S4). Only the motif frequencies in PAS in downstream switch regions were found to be significantly different from the randomly selected PAS (p-value = 0.003; chi-square test; Fig. 7a).
Fig. 7.

Utilization of polyadenylation signal motifs in switch regions. a Density histograms of p-values of PAS motif distribution in switch or default PAS regions. Analysis was stratified for downstream and upstream switches. In each group motif frequency was compared to the frequency of all PASs, and statistical significance was assessed with Pearson’s chi-square test. p-values obtained by comparing between motifs distribution in 1000 random subsets of PASs and motifs distribution of all PASs. The red line shows the calculated p-value for each group. b The ratio between switch (A17.1) and default (FVB) PAS utilization regions for 13 polyadenylation signal motifs. Numerical values are shown in Supplementary Table S4. Downstream or upstream switches are depicted by open circles or closed squares, respectively. Hashed line marks ratio = 1. Canonical motifs are underlined. Arrows mark motifs with consistent switches in A17.1 mice: up arrows represents more utilized in A17.1 and down arrow less utilized in A17.1. c Bar chart shows the proportion of genes with up/down or unchanged expression in each PAS motif
To identify the specific polyadenylation signal motifs associated with expPABPN1 expression, we analysed the differences between the frequencies in the switch and default regions for each motif separately. In five non-canonical motifs, we found a consistent effect in both up- and downstream switches: AATATA, AATACA, GATAAA, and AATAGA motifs were more utilized in A17.1, whereas AAGAAA was less utilized in A17.1 (Fig. 7b). In six polyadenylation signal motifs switch to default PAS utilization was affected in A17.1, but this was not consistent between downstream and upstream switches (Fig. 7b). The CATAAA motif utilization was markedly decreased by downstream. PAS utilization in two motifs, including the major canonical motif AATAAA, was not affected in A17.1 (Fig. 7b). Together this suggests that few PAS motifs are associated with switch direction, and others are not.
We then also investigated whether motifs are associated with changes in expression. Gene dysregulation was found to be significantly higher across all motifs (p = 0.007; chi-square test). Higher proportion of upregulated genes was found for AATAAA, ATTAAA, AATGAA, AATACA, and GATAAA motifs, whereas a higher proportion of downregulated genes was found dysregulation was found for AAGAAA and ACTAAA motifs (Fig. 7c). Overall, comparing between switch directions and dysregulation directions per motif a clear consensus was not found. This suggests that gene dysregulation is associated with PAS motifs but not with PAS switch direction.
Discussion
Levels of functional PABPN1 are reduced in aged muscles and to a greater extent in OPMD muscles.10 The expanded PABPN1 is prone to aggregation leading to depletion of the soluble protein.13 Previous studies, including ours, demonstrated a regulatory role for PABPN1 in alternative PAS in the distal 3′-UTR.8, 9, 16 Although PASs are most abundant in the 3′-UTR, here we show that in skeletal muscles PAS reads in internal gene regions were found in one quarter of the genes. Most of the PASs in internal gene regions are mapped to introns. We demonstrate that expPABPN1 also alters PAS utilization between internal gene regions. This indicates that expPABPN1 effect on mRNA processing is not limited to the distal 3′-UTR, and implies that the spectrum of alternative transcripts in OPMD is broader than shortening of transcripts at the distal 3′-UTR. Our study demonstrates that expPABPN1-mediated switches in PAS utilization between gene regions affect not only expression levels in general but also the levels of transcripts variants with different open reading frames (ORFs). We suggest that PAS switches between gene regions potentially contribute to altered muscle homeostasis as a consequence of expPABPN1 expression.
Upstream PAS switches within the distal 3′-UTR result in shorter transcripts and generally upregulation of gene expression.8, 9 Transcripts from proximal PAS in the 3′-UTR could lose regulatory elements, like miRNA-binding sites, and lead to increase in transcript levels.8, 17, 18 Here, we show that downstream switches between gene regions are predominantly associated with gene upregulation. However, gene upregulation was mostly found in the downstream switches, whereas upstream switches were not associated with dysregulation direction. This suggests that PAS switches between gene regions may alter the nature of the coding sequence rather than expression levels. Since most PAS switches were found from/to introns, it is possible that those alternative transcripts could result in altered ORFs and/or non-sense transcripts.
Among the genes with PAS switch between gene regions we identified muscle genes that are associated with muscular disorders like Dmd, Lmna, Ttn, Ano5, and Sgcb (Supplementary Table S3). It is possible that altered transcript levels of those genes may contribute to muscle weakness in OPMD. Since PABPN1 levels are also reduced in muscles of the elderly,19 it is possible that those altered transcripts also contribute to sarcopenia. In agreement, complete knockdown of PABPN1 in cell culture also leads to internal PAS utilization.5 In addition to alterations in transcript levels, usage of PASs within internal gene regions could generate alternative transcripts with alternative coding sequences or be associated with lower levels of intact proteins and/or production of truncated proteins. This may also contribute to lower muscle strength in OPMD.
Increased internal PAS usage may be explained by changes in the rate of splicing, alternative or incomplete splicing (intron retention).20 Alternative splicing is regulated by the spliceosome ribonucleoprotein complex.21 Although PABPN1 is not considered as a splicing factor, a regulatory role for PABPN1 in splicing was first suggested in non-muscle cells.22 More recently, it was demonstrated that splicing of intron 15 of Troponin T3 (TNNT3) is affected in OPMD muscle cells and can be regulated by PABPN1 expression levels.23 In our study, we found an upstream switch in Tnnt3 (Supplementary Fig. S4). This supports alternative splicing in Tnnt3 in A17.1 quadriceps muscles. However, our PolyA-Seq data reveals internal PAS in several exons and introns in Tnnt3. A PAS was found in intron 15 in the A17.1, but the most significant switch was found in intron 14 (Supplementary Fig. S4). Additionally, we found intronic PAS in A17.1 of Clcn1 gene (Supplementary Fig. 4). CLCN1 mis-splicing causes delayed muscle relaxation (myotonia) in Myotonic dystrophy 1 (ref. 24). This suggests that a switch is PAS utilization possibly cause altered polypeptides in genes regulating muscle contraction, and this can contribute to muscle weakness in OPMD.
Here, we show that transcripts from distal 3′-UTR are retained in the nuclei in expPABPN1 expressing cells, while transcripts from internal PAS have a higher cytoplasmic to nuclear ratio. Our observations are in agreement with a recent study showing nuclear retention of longer transcript isoforms, while the shorter transcript isoforms tend to be overrepresented in the cytoplasm.25 Since expPABPN1 has a higher nuclear accumulation,26 it is possible that the nuclear residence of expPABPN1 and expPABPN1 aggregation in the nucleus lead to nuclear retention of transcripts. Lower levels of full-length cytoplasmic transcripts could contribute to alterations in protein levels in the OPMD muscle.
In our previous study, we found that the two canonical polyadenylation signals are enriched in the distal PAS in the distal 3′-UTR, and those are less efficiently utilized in expPABPN1-expressing muscles. Instead the non-canonical motifs in tandem PAS sites in the distal3′-UTR are utilized in OPMD conditions.9 Here, we extend this observation and show that a shift in PAS utilization is associated with increased utilization of non-canonical motifs, although the majority of transcripts from internal PAS are associated with canonical polyadenylation signal motifs, indicating that they represent bona fide PASs. The non-canonical AATATA, GATAAA, AATACA, and AATAGA motifs were more used in A17.1 mouse, whereas the non-canonical AAGAAA motif was found to be repressed in A17.1 switch regions. Those motifs were not specifically associated with a switch direction (upstream or downstream). This suggests that binding of polyadenylation factors to those motifs is increased in presence of low levels of PABPN1.
We show alterations in internal PAS utilization in OPMD mouse and cell models, and the results in muscles were partly replicated in the muscle cell culture. In muscles expPABPN1 is highly overexpressed (30-folds),14 whereas in the muscle cell culture the expPABPN1 overexpression is only 1.3-fold over endogenous PABPN1 (ref. 13). Additional differences, like different types of stress to which the cells and the muscles are exposed, could contribute to the observed differences between the two models. These may signify a role for PABPN1 in coordinating the splicing and polyadenylation machineries, and these may add to the altered mRNA landscape in OPMD and muscle aging. In conditions with mutant PABPN1, internal gene PAS utilization affects transcript identity and its cytoplasmic abundance. The change in the transcriptome and nuclear export of transcripts could lead to widespread changes in the proteome. Alternative transcripts of contractile proteins, like Tnnt3, were indeed found in our switch analysis. Alteration in those proteins could contribute to muscle pathology.
Material and methods
PAS single molecule sequencing method and sequencing
Helicos single-molecule PAS sequencing data were generated from quadriceps mouse muscles overexpressing the expPABPN1 (A17.1) (ref. 27) and control mice (FBV) aged 6 and 26 weeks (N = 3 per group) as previously described.9 No significant differences in PAS region usage were found between young (6 weeks) and adult (26 weeks) mice, therefore, the two age groups were combined (N = 6). PASs with low counts (<2 reads in<6 samples) were excluded. The genomic coordinates of mouse PASs (previously assigned and annotated as described in de Klerk, et al.9) were converted from genome assembly mm9 to mm10 using the LiftOver utility in UCSC (http://genome.ucsc.edu). Only 54% of the PASs were found in genes annotated in Ensembl version 84 (located in 3′-UTRs, exons, or introns).
Differential expression analysis of genes
Differential expression analysis between A17.1 and FVB control mice was carried out in R programming language (version 3.2.5). The analysis was performed on the sum of all reads mapping between the start and stop coordinates of a gene (on the same strand) using the R Bioconductor package edge R28 (version 3.14.0). A negative binomial model was fitted and a common and tag-wise dispersion was estimated for all tags prior testing procedures. p-values were computed using the exact test and adjusted for multiple testing using Benjamini and Hochberg’s false discovery rate.29 In our polyA-sequencing procedure, in contrast to regular RNA-sequencing, alterations in transcript length do not affect the number of counts. Gene dysregulation was determined by normalized counts per gene. Therefore, the definition of gene dysregulation (expression levels) is independent of transcript length.
Statistical model to identify genes with PAS utilization changes between gene regions
PAS counts within each gene region (intron, exon, 3′-UTR) were summed, and a logistic generalized linear mixed model was fitted to identify genes with a switch in PAS usage between different regions within a gene using the R Bioconductor package lme4 (ref. 30). The model for statistical evaluation of differential PAS ustilization was described in.9 In brief, for each gene with multiple PAS regions, the counts n ij for region j of mouse i were modeled as binomial using parameters N i and p ij. Here, , k is the number of regions for a particular gene, and N i is the total number of reads for all regions of a given gene in mouse i. The log odds of the parameter p ij was modeled using fixed effects for PAS region and OPMD status, with their interaction, combined with a random intercept and PAS region effect within a mouse. We tested for the presence of an OPMD effect with a chi-squared likelihood ratio test, using as the null hypothesis the same model, but with the OPMD effect and the OPMD-PAS interaction set to zero. Significant p-values (FDR < 0.05) indicate genes with a relative change in PAS usage between different regions in a gene.
To find the direction of the PAS switch, default and switch region were identified. After adding up the count for all PASs within a region, default and switch region were defined as regions with maximum Σ PASFVB—Σ PASA17.1 and minimum Σ PASFVB—Σ PASA17.1, respectively. With downstream switch we refer to genes where the default region is located 5′ from the switch region and with upstream switch we refer to genes where the default region is located 3′ from the switch region.
Cell culture, RNA isolation, and qRT-PCR
Stable cell cultures of immortalized mouse myoblasts (clone IM2) over-expressing wild-type (WT) PABPN1 or expPABPN1 are described in ref. 13. PABPN1 transgene expression is driven by the Desmin promoter, and was induced by switching proliferating cultures to fusion conditions using 4% Horse serum in Dulbecco's modified Eagle medium. Cells were seeded to 80% confluence and 2 days after incubation with 4% horse serum medium, cells were collected for RNA isolation.
Total RNA isolation from A17.1 and FBV tibialis anterior mouse muscles at 18 weeks was carried out as described in de Klerk, et al.9 cDNA was generated using oligo(dT) and random primer mix as described in de Klerk, et al.9 qRT-PCR was carried out with the LightCycler 480 (Roche) using 2× SensiMix reagent (Bioline). Primers for PAS switches validation in qRT-PCR were designed in the sequences upstream to the detected PAS (in both default and switch regions) of six candidate genes. Primers for qRT-PCR were designed using the Primer3Plus program (version: 2.4.0) (ref. 31). Primer sequences are listed in Supplementary Table S1. Relative expression levels of candidate genes were calculated after normalizing for Hprt housekeeping gene. Ratios between PCR products were calculated to verify a switch in PAS utilization. Fold change was calculated after normalization to Hprt FVB control.
Subcellular fractionation in cell culture was carried out as described in ref. 32 but RNasout® Ribonuclease Inhibitor (Promega) was added for all extraction and washing buffers. RNA was isolated from each fraction as described, using the phenol-chloroform method. Fractionation between the nuclear and cytoplasmic fractions was verified using western blotting, with Emerin (nuclear, 1:5000; anti-Rabbit EPR11071, AbCam) and Gapdh (cytosolic, 1:10,000; anti-mouse G8795, Sigma, Miss. USA). Quality of RNA from both fractions was assessed using the Bioanalyzer RNA 6000 Nano kit (Agilent Genomics). Hprt, whose expression remained unchanged in A17.1, was used for normalization.
Polyadenylation signal motifs analysis
Polyadenylation signal motif analysis was carried out for genes with a significant switch in PAS utilization. Every read in our data set marks a specific PAS, and the total number of reads associated with a specific PAS were used as input in the R Bioconductor package edgeR to identify the ones that were differentially utilized between A17.1 and WT mice (as previously described in gene expression analysis section). Low utilized PASs were defined as those PASs which were significantly less used in A17.1 compared with WT mice and high utilized PASs were those which were used significantly more in A17.1 than WT mice. Only PASs that were low utilized in default regions and high utilized in switch regions were considered in genes with upstream and downstream switches. Thirteen polyadenylation signal motifs were included in the analysis: 2 canonical and 11 non-canonical, and those motifs were counted within ~50 nt upstream of all PASs.2 For 24,153 PASs (68% of all PASs) at least one of the 13 known motifs was found (39,389 motifs in total) and those were included in an analysis assessing an association with PAS switch. Frequency distribution of motifs in each group was compared with motif frequencies of all PASs using Pearson’s chi-squared test. To assess whether the observed significant differences differ from random, 1000 random subsets of PASs for each group were generated and the frequency distribution of motif in each subset was compared with motif frequencies of all PASs.
Electronic supplementary material
Acknowledgements
This study is supported by The Prinses Beatrix Spierfonds (grant nr.: W OR13-13).
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Supplementary Information accompanies the paper on the npj Aging and Mechanisms of Disease website (doi:10.1038/s41514-017-0007-x).
References
- 1.Lee JY, Ji Z, Tian B. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucleic Acids Res. 2008;36:5581–5590. doi: 10.1093/nar/gkn540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandberg R, et al. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25:1770–1782. doi: 10.1101/gad.17268411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, et al. Systematic profiling of poly(A)+transcripts modulated by core 3’ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11:e1005166. doi: 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 7.Kühn U, et al. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J. Biol. Chem. 2009;284:22803–22814. doi: 10.1074/jbc.M109.018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenal M, et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–553. doi: 10.1016/j.cell.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 9.de Klerk E, et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012;40:9089–9101. doi: 10.1093/nar/gks655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anvar SY, et al. A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging (Albany NY) 2013;5:412–426. doi: 10.18632/aging.100567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brais B, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 12.Calado A, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum. Mol. Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- 13.Raz V, et al. Modeling oculopharyngeal muscular dystrophy in myotube cultures reveals reduced accumulation of soluble mutant PABPN1 protein. Am. J. Pathol. 2011;179:1988–2000. doi: 10.1016/j.ajpath.2011.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trollet C, et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum. Mol. Genet. 2010;19:2191–2207. doi: 10.1093/hmg/ddq098. [DOI] [PubMed] [Google Scholar]
- 15.Abu-Baker A, et al. Cytoplasmic targeting of mutant poly(A)-Binding protein nuclear 1 suppresses protein aggregation and toxicity in oculopharyngeal muscular dystrophy. Traffic. 2005;6:766–779. doi: 10.1111/j.1600-0854.2005.00315.x. [DOI] [PubMed] [Google Scholar]
- 16.Li W, et al. Systematic profiling of Poly(A)+Transcripts modulated by core 3’ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11:e1005166. doi: 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura P, et al. Brain-derived neurotrophic factor expression is repressed during myogenic differentiation by miR-206. J. Neurochem. 2012;120:230–238. doi: 10.1111/j.1471-4159.2011.07583.x. [DOI] [PubMed] [Google Scholar]
- 18.Boutet SC, et al. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem. Cell. 2012;10:327–336. doi: 10.1016/j.stem.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raz Y, Raz V. Oculopharyngeal muscular dystrophy as a paradigm for muscle aging. Front. Aging Neurosci. 2014;6:317. doi: 10.3389/fnagi.2014.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braunschweig U, et al. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–1269. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Muniz L, Davidson L, West S. Poly(A) polymerase and the nuclear poly(A) binding protein, PABPN1, coordinate the splicing and degradation of a subset of human Pre-mRNAs. Mol. Cell. Biol. 2015;35:2218–2230. doi: 10.1128/MCB.00123-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein P, et al. Nuclear poly(A)-binding protein aggregates misplace a pre-mRNA outside of SC35 speckle causing its abnormal splicing. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oana K, et al. Manumycin A corrects aberrant splicing of Clcn1 in myotonic dystrophy type 1 (DM1) mice. Sci. Rep. 2013;3:2142. doi: 10.1038/srep02142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neve J, et al. Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation. Genome Res. 2016;26:24–35. doi: 10.1101/gr.193995.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uyama E, et al. Nuclear accumulation of expanded PABP2 gene product in oculopharyngeal muscular dystrophy. Muscle Nerve. 2000;23:1549–1554. doi: 10.1002/1097-4598(200010)23:10<1549::AID-MUS11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Davies JE, et al. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat. Med. 2005;11:672–677. doi: 10.1038/nm1242. [DOI] [PubMed] [Google Scholar]
- 28.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. 1995;57:289–300. [Google Scholar]
- 30.Douglas Bates MM, Bolker Ben, Walker Steve. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015;67:1–48. [Google Scholar]
- 31.Untergasser A, et al. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimauro I, Pearson T, Caporossi D, Jackson MJ. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res. Notes. 2012;5:513. doi: 10.1186/1756-0500-5-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.