

Abstract
Propofol is an anesthetic drug commonly used in the clinical practice. The aim of this study is to explore the effect of propofol on the aggressive behaviors of rheumatoid arthritis fibroblast-like synoviocytes (RA-FLSs). Propofol treatment for 48 or 72 h significantly inhibited the viability of RA-FLSs, but a 24-h treatment did not produce cytotoxic effects. Propofol exposure for 48 h led to reduction of proliferation and induction of apoptosis in RA-FLSs, which was coupled with increased Bax and decreased Bcl-2 and survivin levels. Additionally, treatment with propofol for 24 h significantly suppressed the migration and invasion of RA-FLSs. Mechanistically, propofol inhibited nuclear factor-κB (NF-κB) activity. Overexpression of constitutively active NF-κB p65 reversed the inhibitory effects of propofol on RA-FLSs. Taken together, propofol exerts anti-proliferative and anti-invasive effects on RA-FLSs via the NF-κB pathway and may have therapeutic potential in treatment of RA.
Keywords: Apoptosis, growth, invasion, NF-κB signaling, propofol, rheumatoid arthritis fibroblast-like synoviocytes
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that is characterized by hyperplasia of the synovium and destruction of cartilage and bone [1]. Fibroblast-like synoviocytes (FLSs) play an important role in the pathogenesis of RA by releasing a large amount of proinflammatory cytokines and matrix metalloproteinases [2]. Moreover, RA-FLSs show aggressive phenotypes including hyperproliferation, apoptosis defect, and invasiveness [3,4]. Therefore, many efforts have been made to modulate the biological behaviors of RA-FLSs in the treatment of RA [5,6].
Activation of the nuclear factor-κB (NF-κB) pathway is responsible for the aggressive properties of RA-FLSs [6]. Pharmacological inhibition of NF-κB has been documented to suppress inflammation response and proliferation in human RA-FLSs [7]. NF-κB is predominantly a heterodimeric transcription factor composed of p50 and p65, which can regulate a lot of genes involved in inflammation, cell proliferation, survival, and invasion [8]. At baseline, NF-κB is sequestered in the cytoplasm by inhibitory IκB proteins. Upon activation, IκB proteins are degraded, thereby allowing NF-κB to undergo nuclear translocation and activate target gene expression [8]. Phosphorylation of p65 at Ser-536 can increase the NF-κB transcriptional activity [9].
Propofol, an anesthetic drug commonly used in the clinical practice, has shown multiple biological activities including anti-inflammatory [10] and anti-tumor [11] properties. It has been reported that propofol reduced the production of proinflammatory cytokines in a rat model of lipopolysaccharide (LPS)-induced acute lung injury [12]. In a rat model of ischemic brain damage, administration of propofol was found to attenuate inflammatory response in the brain [13]. In addition, propofol was reported to exert growth-suppressive effects against esophageal squamous cell carcinoma [14] and prostate cancer [15] cells. However, the effects of propofol on the aggressive phenotypes of RA-FLSs remain unclear.
Therefore, in this study, we explored the effects of propofol on RA-FLS proliferation, apoptosis, and invasion, and examined associated molecular pathways.
Materials and methods
Cell culture and propofol treatment
RA-FLSs were obtained from Cell Applications (San Diego, CA, USA) and cultured in RPMI 1640 medium (Solarbio, Beijing, China) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA). Propofol was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in RPMI 1640 medium. For propofol treatment, RA-FLSs were seeded in triplicate at a density of 3 × 103 cells/well in 96-well plates or 2 × 104 cells/well in 24-well plates and allowed to attach overnight. The cells were then exposed to propofol at a final concentration ranging from 10 to 80 μM for 24, 48, or 72 h. Afterwards, the cells were collected and tested for viability, proliferation, apoptosis, and gene expression.
MTT assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to determine cell viability. In brief, cells were incubated with MTT solution (0.5 mg/mL; Sigma-Aldrich) at 37°C for 4 h and then added with dimethyl sulfoxide to dissolve formazan crystals. The number of viable cells was determined by measuring absorbance at 570 nm.
BrdU incorporation assay
Cell proliferation was measured using the 5-bromo-2’-deoxy-uridine (BrdU) Cell Proliferation Assay Kit (BioVision, Milpitas, CA, USA) following the manufacturer’s protocol. BrdU incorporation was determined spectrophotometrically by measuring absorbance at 450 nm.
Apoptosis analysis by flow cytometry
For apoptosis detection, cells were incubated with propidium iodide (PI) and Annexin-V (Sigma-Aldrich) for 30 min in the dark. Stained cells were analyzed on a flow cytometer (Becton Dickinson Biosciences, San Jose, CA, USA).
Caspase-3 activity assay
Caspase-3 activity was measured using the Caspase 3 Colorimetric Assay Kit (Sigma-Aldrich), following the manufacturer’s protocols. Absorbance was recorded at 405 nm.
Western blot analysis
Cells were lysed in a lysis buffer containing the complete protease inhibitor mixture (Roche Diagnostics, Indianapolis, IN, USA). Protein concentrations were determined using the BCA protein assay reagent kit (Pierce, Rockford, IL, USA). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred electrophoretically to polyvinylidene difluoride membranes. Membranes were incubated with anti-Bax, anti-Bcl-2, anti-survivin, anti-phospho-NF-κB p65 (Ser536), anti-NF-κB p65, anti-IκBα, and anti-β-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h at room temperature, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Pierce). Proteins were visualized by chemifluorescence using the ECL Plus Western Blotting Detection Reagents (Amersham Biosciences/GE Healthcare, Piscataway, NJ, USA). Quantitation of the protein bands was done with Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA).
In vitro cell migration and invasion assays
Cell migration was determined using wound healing assays as previously reported [17]. In brief, RA-FLSs were seeded onto 6-well plates and grew to confluence. Alinear wound was generated on the cell monolayer using a 200-μl pipette tip. The cell cultures were added with indicated concentrations of propofol and incubated for 24 h. Cells were photographed by a microscope. Cell migration was determined by calculating a percent recovery of wound area.
Cell invasion assays were performed using Transwell inserts (8 μm in pore size) that were pre-coated with Matrigel (Becton Dickinson Biosciences). The inserts were placed in 24-well plates. The upper chamber was added with RA-FLSs in serum-free medium (5 × 104 cells/well). The lower chamber was filled with RPMI 1640 medium containing 10% FBS and indicated concentrations of propofol. After incubation for 24 h, the cells on the top membrane surface were removed. The invaded cells were stained with 0.5% crystal violet and counted under a microscope.
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was extracted from RA-FLSs using TRIzol reagent (Invitrogen) following the manufacturer’s protocol. RNA was reverse-transcribed to cDNA using the SuperScript III reverse transcription kit and random hexamers (Invitrogen). Real-time PCR was performed using the SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA). PCR primers are as follows: MMP3 forward, 5’-AGCAAGGACCTCGTTTTCATT-3’ and MMP3 reverse, 5’-GTCAATCCCTGGAAAGTCTTCA-3’ [18]; MMP9 forward, 5’-TGACAGCGACAAGAAGTG-3’ and MMP9 reverse, 5’-CAGTGAAGCGGTACATAGG-3’ [19]; β-actin forward, 5’-ATCCACGAAACTACCTTCAACTC-3’ and β-actin reverse, 5’-GAGGAGCAATGATCTTGATCTTC-3’. Gene expression results were normalized to that of β-actin mRNA.
Luciferase reporter assay
RA-FLSs (2 × 104 cells/well) seeded on 24-well plates were co-transfected with the NF-κB-dependent reporter construct pNF-κB-Luc (0.2 μg; Agilent Technologies, Santa Clara, CA, USA) together with the Renilla luciferase reporter pRL-TK (0.02 μg; Promega, Madison, WI, USA) using FuGENE 6 (Roche Diagnostics, Indianapolis, IN, USA). At incubation for 12 h, transfected cells were exposed to 20 or 80 μM of propofol for additional 48 h. Cells were lysed and luciferase activities were measured using the dual-luciferase assay kit (Promega). The NF-κB-driven reporter activity was normalized to Renilla luciferase activity.
Overexpression of constitutively active NF-κB p65
A constitutively active p65 mutant (S536D) was generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), as described previously [20]. For rescue experiments, RA-FLSs (2 × 106) were seeded on 60-mm dishes and transfected with 5 μg of the p65 (S536D) construct using FuGENE 6. Twenty-four hours after transfection, the transfected cells were treated with 80 μM of propofol for additional 24 or 48 h and subjected to BrdU incorporation and Transwell invasion assays.
Statistical analysis
Values are presented as means ± standard deviation. Statistical differences were analyzed using one-way analysis of variance (ANOVA) with Tukey’s post-hoc test. P<0.05 was considered statistically significant.
Results
Propofol inhibits cell viability and proliferation in RA-FLSs
MTT assays showed that treatment with propofol for 24 h did not impact the viability of RA-FLSs after 24-h treatment (P>0.05 vs. control; Figure 1A). However, treatment with propofol for 48 or 72 h led to a significant inhibition of the viability of RA-FLSs, compared to control cells (P<0.05; Figure 1B and 1C). The IC50 values of propofol at 48 and 72 h were 62.3 ± 1.8 and 29.6 ± 1.2 μM, respectively. However, BrdU incorporation assays confirmed concentration-dependent inhibitory effects of propofol on RA-FLS proliferation after 48-h treatment (P<0.05 vs. control; Figure 1D).
Figure 1.
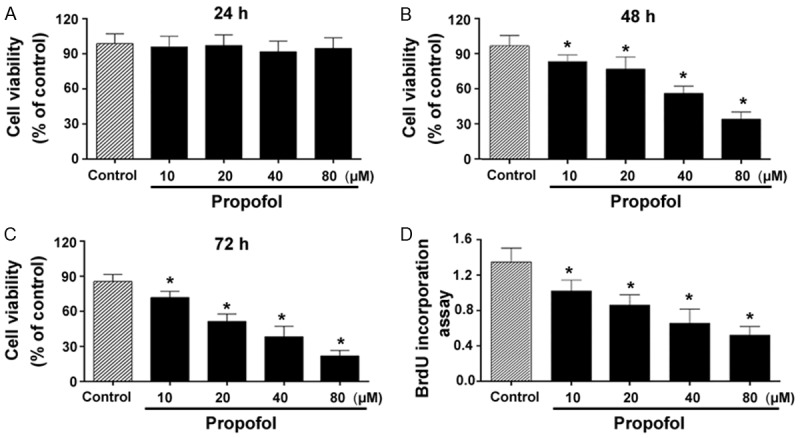
Propofol inhibits cell viability and proliferation in RA-FLSs. A-C. RA-FLSs were treated with indicated concentrations of propofol for 24, 48, or 72 h, and cell viability was measured using the MTT assay. D. BrdU incorporation assays were conducted to assess cell proliferation in RA-FLSs after 48-h treatment with propofol. *P<0.05 vs. vehicle-treated control.
Propofol promotes apoptosis in RA-FLSs
Propofol at 20 and 80 μM caused a 2.4- and 5.2-fold, respectively, in the percentage of apoptosis after 48-h treatment, as determined by flow cytometry with annexin-V/PI staining (P<0.05 vs. control; Figure 2A and 2B). Moreover, the caspase-3 activity was significantly higher in propofol-treated RA-FLSs than in control cells (P<0.05; Figure 2C). In addition, Western blot analysis of key apoptosis-related proteins revealed that propofol treatment significantly raised the level of Bax and reduced the levels of Bcl-2 and survivin (Figure 2D).
Figure 2.
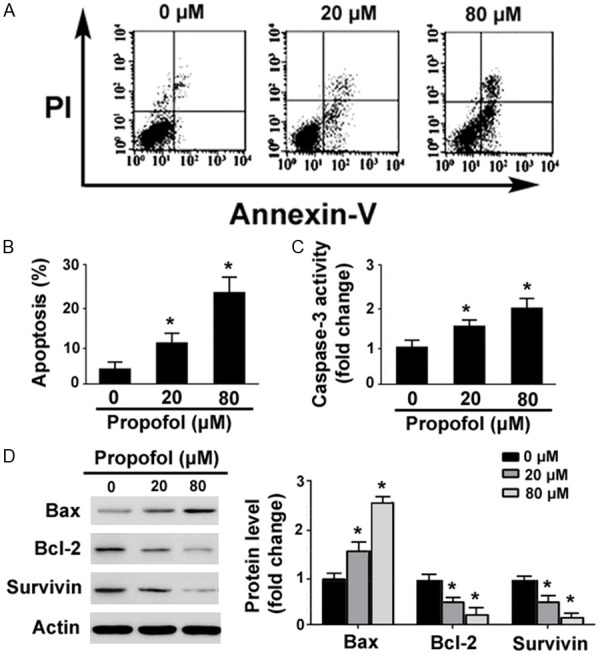
Propofol promotes apoptosis in RA-FLSs. A. Flow cytometric analysis of apoptosis in RA-FLSs treated with 20 or 80 μM of propofol for 48 h after annexin-V/PI staining. B. Quantification of apoptosis from three independent experiments. C. Measurement of caspase-3 activity using Colorimetric Caspase Assays. D. Western blot analysis of indicated proteins. Bar graphs show densitometric analysis of protein levels after normalization to β-actin levels. *P<0.05 vs. vehicle-treated cells.
Propofol suppresses the migration and invasion of RA-FLSs
In vitro wound-healing assays demonstrated that the addition of propofol significantly impaired the migration capacity of RA-FLSs (Figure 3A). After 24-h incubation, the percentage of wound closure was significantly lower in propofol (80 μM)-treated cells than in control cells (22 ± 4% vs. 63 ± 7%, P<0.05). In Transwell invasion assays (Figure 3B), RA-FLSs showed a high capacity to invade through the Matrigel membrane in response to serum stimulation. Propofol at the low concentration of 20 μM inhibited RA-FLS invasion by 25%, whereas a higher concentration of propofol (80 μM) led to a 70% reduction in cell invasion. It should be mentioned that after 24-h treatment, propofol up to 80 μM significantly blocked cell migration and invasion (Figure 3), without causing noticeable cytotoxic effects to RA-FLSs (Figure 1B).
Figure 3.
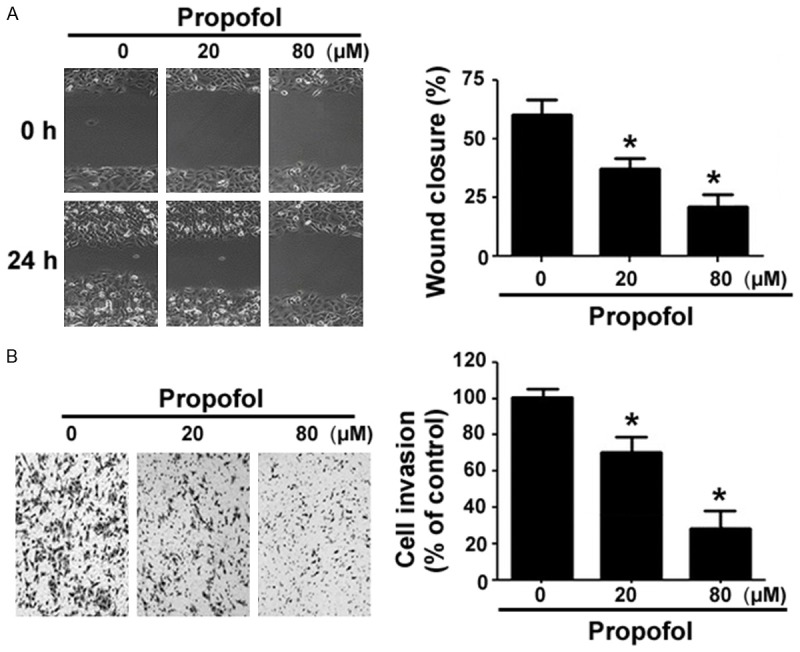
Propofol suppresses the migration and invasion of RA-FLSs. A. In vitro wound-healing assays. Wound closure was quantified after 24-h culturing in the presence or absence of 80 μM of propofol. B. Transwell invasion assays were done to determine the invasion of RA-FLSs after treatment with 80 μM of propofol for 24 h. *P<0.05 vs. vehicle-treated cells.
Propofol inhibits NF-κB activity in RA-FLSs
As detected by Western blot analysis, propofol treatment led to 35-67% decrease in the phosphorylation of NF-κB p65 on Ser-536 and 1.8-3-fold increase in IκBα protein levels (Figure 4A). To assess the effect of propofol on the transcriptional activity of NF-κB, RA-FLSs were pre-transfected with the NF-κB p65-dependent reporter construct 12 h before propofol treatment. We found that the addition of propofol significantly decreased NF-κB-dependent transcription from reporter constructs (Figure 4B). The expression of NF-κB target genes MMP3 and MMP9 was significantly downregulated by propofol (Figure 4C).
Figure 4.
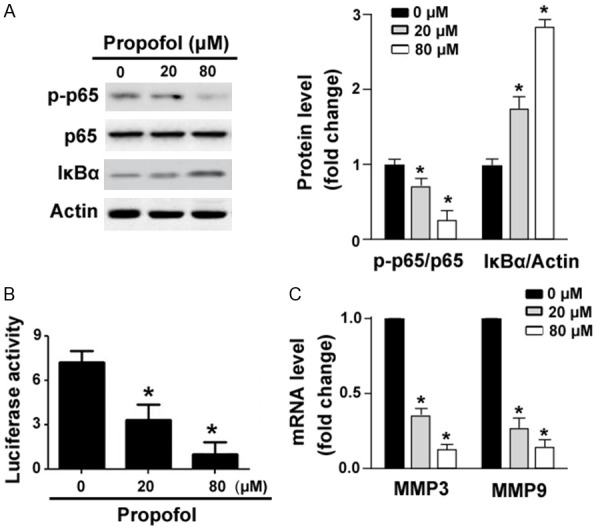
Propofol inhibits NF-κB activity in RA-FLSs. (A) Western blot analysis of indicated proteins in RA-FLSs treated with 20 or 80 μM of propofol for 48 h. (B)NF-κB p65-dependent luciferase reporter assays. RA-FLSs were pre-transfected with the NF-κB p65-dependent reporter construct 12 h before propofol treatment and luciferase activities were then measured. (C) Quantitative real-time PCR analysis of MMP3 and MMP9 mRNA levels in RA-FLSs treated as in (A). *P<0.05 vs. vehicle-treated cells.
Overexpression of constitutively active NF-κB p65 reverses the inhibitory effects of propofol on RA-FLSs
To determine whether inhibition of NF-κB activity was required for the biological effects of propofol on RA-FLSs, we overexpressed a constitutively active mutant of NF-κB p65 in RA-FLSs before propofol exposure. Western blot analysis verified that RA-FLSs transfected with the p65 (S536D) construct had significantly greater levels of phosphorylated p65 in the presence of propofol (Figure 5A). Notably, overexpression of p65 (S536D) almost completely abolished the inhibitory effects of propofol (80 μM) on RA-FLS proliferation (Figure 5B) and invasion (Figure 5C).
Figure 5.
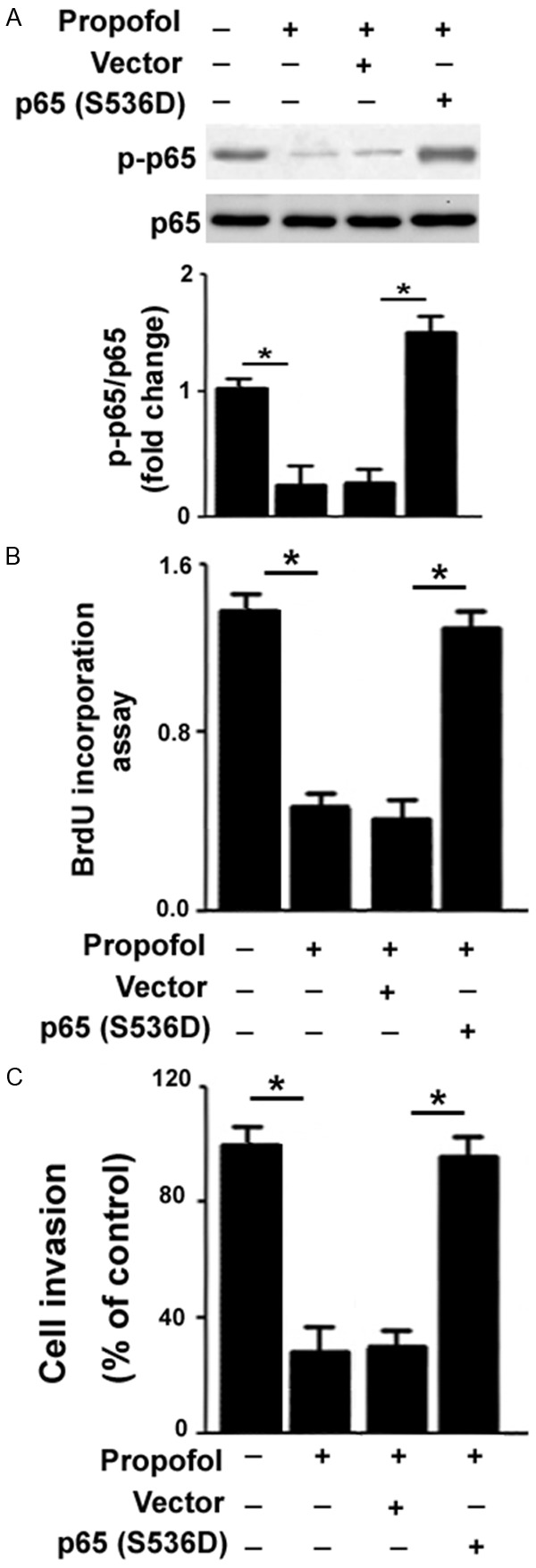
Overexpression of constitutively active NF-κB p65 reverses the inhibitory effects of propofol on RA-FLSs. (A) Western blot analysis of NF-κBp65 phosphorylation in propofol-treated RA-FLSs with or without pre-transfection with the p65 (S536D) construct. (B) BrdU incorporation assays were performed to measure cell proliferation in RA-FLSs treated as in (A). (C) Cell invasion was determined by Transwell invasion assays. Bar graphs show quantitative data from three independent experiments. *P<0.05.
Discussion
Propofol has shown anti-proliferative activity in hippocampal neurons [21] and cardiac fibroblasts [22], as well as several types of malignant cells [14,15]. In this study, we provided first evidence for the growth-suppressive activity of propofol in RA-FLSs. We found that exposure to propofol for longer than 24 h caused a significant inhibition of viability and proliferation in RA-FLSs. Moreover, propofol treatment for 48 h significantly triggered apoptotic death and increased caspase-3 activity in RA-FLSs. At the molecular level, multiple apoptosis-related proteins including Bax, Bcl-2 and survivin were deregulated by propofol. These results suggest that propofol-mediated apoptosis of RA-FLSs may be causally linked to increased expression of the pro-apoptotic protein Bax and decreased expression of the anti-apoptotic proteins Bcl-2 and survivin. In line with our findings, propofol has been reported to induce apoptosis by downregulating Bcl-2 in neurons [21]. However, it should be mentioned that in some cellular contexts, propofol can upregulate the expression of Bcl-2 and suppress the expression of Bax [23,24], consequently protecting from apoptosis.
Apart from suppression of cell proliferation, propofol showed the ability to hamper the migration and invasion of RA-FLSs. MMPs are important mediators of cell invasiveness [25]. Inhibition of MMP3 and MMP9 has been linked to reduced RA-FLS migration and invasion [26]. Notably, we found that both MMP3 and MMP9 were downregulated in propofol-treated RA-FLSs, which provided an explanation for its anti-invasive property. Propofol-mediated downregulation of MMPs has also been described in esophageal squamous cell carcinoma cells [14].
NF-κB signaling plays a critical role in the pathogenesis of RA and contributes to the aggressive properties of RA-FLSs [6]. NF-κB can transcriptionally activate a lot of target genes including Bcl-2, survivin, MMP3 and MMP9 [27]. Consistent with downregulation of Bcl-2, survivin, MMP3, and MMP-9, there was a significant decrease in the phosphorylation of NF-κB p65 in propofol-treated RA-FLSs. Luciferase reporter assays further demonstrated that NF-κB-dependent transcriptional activity was significantly inhibited in the presence of propofol. These observations suggest that propofol may exert its biological activities in RA-FLSs by interfering with the activation of NF-κB signaling. In support of this hypothesis, rescue experiments using a constitutively active construct of NF-κB p65 demonstrated that overexpression of p65 (S536D) almost completely reversed the inhibitory effects of propofol on RA-FLS proliferation and invasion. Propofol can also modulate signaling pathways other than NF-κB signaling, such as mTOR [11] and ERK [14]. Therefore, it is possible that several other signaling pathways may be involved in the action of propofol in RA-FLSs.
In conclusion, our results show that propofol has inhibitory effects on the aggressive behaviors of RA-FLSs, which is, at least partially, ascribed to inactivation of NF-κB signaling. These findings suggest that propofol may have therapeutic benefits in the treatment of RA.
Disclosure of conflict of interest
None.
References
- 1.van Vollenhoven RF. Rheumatoid arthritis in 2012: Progress in RA genetics, pathology and therapy. Nat Rev Rheumatol. 2013;9:70–72. doi: 10.1038/nrrheum.2012.232. [DOI] [PubMed] [Google Scholar]
- 2.Xiao Y, Liang L, Huang M, Qiu Q, Zeng S, Shi M, Zou Y, Ye Y, Yang X, Xu H. Bromodomain and extra-terminal domain bromodomain inhibition prevents synovial inflammation via blocking IκB kinase-dependent NF-κB activation in rheumatoid fibroblast-like synoviocytes. Rheumatology (Oxford) 2016;55:173–184. doi: 10.1093/rheumatology/kev312. [DOI] [PubMed] [Google Scholar]
- 3.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim Y, Yi H, Jung H, Rim YA, Park N, Kim J, Jung SM, Park SH, Park YW, Ju JH. A dual target-directed agent against interleukin-6 receptor and tumor necrosis factor α ameliorates experimental arthritis. Sci Rep. 2016;6:20150. doi: 10.1038/srep20150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mu N, Gu J, Huang T, Zhang C, Shu Z, Li M, Hao Q, Li W, Zhang W, Zhao J, Zhang Y, Huang L, Wang S, Jin X, Xue X, Zhang W, Zhang Y. A novel NF-κB/YY1/microRNA-10a regulatory circuit in fibroblast-like synoviocytes regulates inflammation in rheumatoid arthritis. Sci Rep. 2016;6:20059. doi: 10.1038/srep20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi L, Zhang X, Wang X. Heparin inhibits the inflammation and proliferation of human rheumatoid arthritis fibroblast-like synoviocytes through the NF-κB pathway. Mol Med Rep. 2016;14:3743–3748. doi: 10.3892/mmr.2016.5719. [DOI] [PubMed] [Google Scholar]
- 8.Park MH, Hong JT. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells. 2016:5. doi: 10.3390/cells5020015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279:55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 10.Celik MG, Saracoglu A, Saracoglu T, Kursad H, Dostbil A, Aksoy M, Ahiskalioglu A, Ince I. Effects of propofol and midazolam on the inflammation of lungs after intravenous endotoxin administration in rats. Eurasian J Med. 2015;47:109–114. doi: 10.5152/eajm.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang D, Zhou XH, Zhang J, Zhou YX, Ying J, Wu GQ, Qian JH. Propofol promotes cell apoptosis via inhibiting HOTAIR mediated mTOR pathway in cervical cancer. Biochem Biophys Res Commun. 2015;468:561–567. doi: 10.1016/j.bbrc.2015.10.129. [DOI] [PubMed] [Google Scholar]
- 12.Gokcinar D, Ergin V, Cumaoglu A, Menevse A, Aricioglu A. Effects of ketamine, propofol, and ketofol on proinflammatory cytokines and markers of oxidative stress in a rat model of endotoxemia-induced acute lung injury. Acta Biochim Pol. 2013;60:451–456. [PubMed] [Google Scholar]
- 13.Shi SS, Yang WZ, Chen Y, Chen JP, Tu XK. Propofol reduces inflammatory reaction and ischemic brain damage in cerebral ischemia in rats. Neurochem Res. 2014;39:793–799. doi: 10.1007/s11064-014-1272-8. [DOI] [PubMed] [Google Scholar]
- 14.Xu YB, Du QH, Zhang MY, Yun P, He CY. Propofol suppresses proliferation, invasion and angiogenesis by down-regulating ERK-VEGF/MMP-9 signaling in Eca-109 esophageal squamous cell carcinoma cells. Eur Rev Med Pharmacol Sci. 2013;17:2486–2494. [PubMed] [Google Scholar]
- 15.Huang H, Benzonana LL, Zhao H, Watts HR, Perry NJ, Bevan C, Brown R, Ma D. Prostate cancer cell malignancy via modulation of HIF-1α pathway with isoflurane and propofol alone and in combination. Br J Cancer. 2014;111:1338–1349. doi: 10.1038/bjc.2014.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu M, Chen J, Jiang H, Miao C. Propofol protects against high glucose-induced endothelial adhesion molecules expression in human umbilical vein endothelial cells. Cardiovasc Diabetol. 2013;12:13. doi: 10.1186/1475-2840-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernaudo S, Salem M, Qi X, Zhou W, Zhang C, Yang W, Rosman D, Deng Z, Ye G, Yang B, Vanderhyden B, Wu Z, Peng C. Cyclin G2 inhibits epithelial-to-mesenchymal transition by disrupting Wnt/β-catenin signaling. Oncogene. 2016;35:4816–4827. doi: 10.1038/onc.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien M, O’Shaughnessy D, Ahamide E, Morrison JJ, Smith TJ. Differential expression of the metalloproteinase MMP3 and the alpha5 integrin subunit in human myometrium at labour. Mol Hum Reprod. 2007;13:655–661. doi: 10.1093/molehr/gam047. [DOI] [PubMed] [Google Scholar]
- 19.Chou YT, Wang H, Chen Y, Danielpour D, Yang YC. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–5560. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- 20.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 21.Zhong Y, Liang Y, Chen J, Li L, Qin Y, Guan E, He D, Wei Y, Xie Y, Xiao Q. Propofol inhibits proliferation and induces neuroapoptosis of hippocampal neurons in vitro via downregulation of NF-κB p65 and Bcl-2 and upregulation of caspase-3. Cell Biochem Funct. 2014;32:720–729. doi: 10.1002/cbf.3077. [DOI] [PubMed] [Google Scholar]
- 22.Cheng TH, Leung YM, Cheung CW, Chen CH, Chen YL, Wong KL. Propofol depresses angiotensin II-induced cell proliferation in rat cardiac fibroblasts. Anesthesiology. 2010;112:108–118. doi: 10.1097/01.anes.0000365960.74268.21. [DOI] [PubMed] [Google Scholar]
- 23.Lin C, Sui H, Gu J, Yang X, Deng L, Li W, Ding W, Li D, Yang Y. Effect and mechanism of propofol on myocardial ischemia reperfusion injury in type 2 diabetic rats. Microvasc Res. 2013;90:162–168. doi: 10.1016/j.mvr.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Xie CL, Pan YB, Hu LQ, Qian YN. Propofol attenuates hydrogenperoxide-induced apoptosis in human umbilical vein endothelial cells via multiple signaling pathways. Korean J Anesthesiol. 2015;68:488–495. doi: 10.4097/kjae.2015.68.5.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou JJ, Ma JD, Mo YQ, Zheng DH, Chen LF, Wei XN, Dai L. Down-regulating peroxisome proliferator-activated receptor-gamma coactivator-1 beta alleviates the proinflammatory effect of rheumatoid arthritis fibroblast-like synoviocytes through inhibiting extracellular signal-regulated kinase, p38 and nuclear factor-kappaB activation. Arthritis Res Ther. 2014;16:472. doi: 10.1186/s13075-014-0472-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing R, Jin Y, Sun L, Yang L, Li C, Li Z, Liu X, Zhao J. Interleukin-21 induces migration and invasion of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Clin Exp Immunol. 2016;184:147–158. doi: 10.1111/cei.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]