

Abstract
Prostaglandin E1 (PGE1) is used as a pretreatment for ischemia reperfusion injury in many biological systems. However, its value as a pretreatment for coronary microembolization (CME) is unknown. The goal of this study was to determine whether PGE1 would protect against CME. In a CME rat model, we observed microthrombi and early myocardial ischemia, with endothelium appearing exfoliated and mitochondria having irregular morphology and decreased internal complexity. The level of fibrinogen-like protein 2 prothrombinase was increased and superoxide dismutase and catalase levels were decreased. Moreover, mitochondria copy number and mitochondrial permeability transition pore (mPTP) opening were increased. Pretreatment with PGE1 (1 or 2 μg/kg) significantly improved these cardiological deficits, acting via the glycogen synthase kinase 3β (GSK-3β)-mPTP pathway. Unexpectedly, the phosphorylation of Akt at Ser473 decreased in the PGE1 at high dose. Overall, our findings suggested an important role for PGE1 in pretreatment of coronary microvascular dysfunction.
Keywords: Coronary microembolization, prostaglandin E1, sodium laurate, mitochondrial permeability transition pore, coronary microvascular dysfunction, GSK-3β, Akt
Introduction
Coronary microembolization (CME) induces microvascular dysfunction by occluding coronary arterioles (vessels <200 µm in diameter) [1-3]. CME is often detected following coronary microcirculation assessments, such as coronary flow reserve and index of microvascular resistance, but its pathophysiology is unclear. Therefore, an animal model of CME is needed to explore its pathological and physiological processes. In recent years, investigators [4,5] developed a CME model by injecting sodium laurate into the left ventricle, causing myocardial ischemia, inflammatory cell infiltration and microthrombosis. Sodium laurate also caused coronary microvascular endothelial exfoliation. Moreover, we referenced the dose of 1 mg/kg for sodium laurate [4,5] to establish our model. In our preliminary experiments, the results showed that 1 mg/kg sodium laurate injected into the left ventricle caused endothelial injuries and coronary microvascular thrombosis (affecting vessels <100 µm in diameter) with no thrombosis detected in coronary vessels (>150 µm). Therefore, this model is suitable for simulating the pathological process of CME.
Prostaglandin E1 (PGE1) has many physiological and pharmacological activities contributing to microvascular dysfunction [6-8], but its ability to protect coronary microcirculation from CME injury and the associated mechanisms remain unclear. PGE1 has anti-thrombotic effects [9] that have not yet been characterized at the molecular level. Recent studies indicated that fibrinogen-like protein 2 prothrombinase (fgl2) was highly expressed during coronary microvascular dysfunction [10,11]. This protein was recently identified as a coagulation factor, belonging to the fibrinogen family and expressed in microvascular endothelial cells. Therefore, in our study, we investigated whether cardioprotective mechanisms of PGE1 involve inhibition of fgl2 expression. PGE1 also has protective effects against oxidative stress injury and inflammation [12,13].
Mitochondria are important organelles in the heart and, under normal physiological conditions, produce ATP to provide energy for the heart and also balance production of Ca2+ and ATP [14,15]. Putative signaling pathways for protecting coronary microvascular function include opening of sarcolemmal and/or mitochondrial ATP-dependent potassium channels and activation of pro-survival kinases (Akt and extracellular regulated protein kinases 1/2 (ERK 1/2)), protein kinases C and G and endothelial nitric oxide synthase. These effects ultimately lead to blockade of the mitochondrial permeability transition pore (mPTP) [16-20]. Therefore, we also investigated whether cardioprotective effects of PGE1 in the rat heart were mediated by the Akt-GSK3-β-mPTP pathway, known to play an important role in the beneficial effects of ischemic preconditioning during myocardial ischemia-reperfusion injury [21-23].
The goal of our study was to determine whether PGE1 is a potential pharmacological target for protecting the myocardium from CME injury, as well as to identify the mechanisms involved in this protection, including the role of the Akt-GSK3-β-mPTP signaling pathway.
Materials and methods
Reagents
Sodium laurate was from Shanghai Jiang Lai Biological Technology Co., Ltd. (Shanghai, China). PGE1 was from the Beijing Tide Pharmaceutical Co Ltd. (Beijing, China). Atractyloside dipotassium salt (Atr) was from ApexBIO (Houston, TX, USA). Enhanced Bicinchoninic acid (BCA) Protein Assay Kit, Total Superoxide Dismutase (SOD) Assay Kit with WST-8, SDS-PAGE loading buffer (5×), RIPAlysis buffer and protease inhibitors were from Beyotime. (Jiangsu, China). Catalase (CAT) Assay Kit (Visible Light) was from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). An ELISA kit for fibrinogen-like protein 2 prothrombinase (fgl2) was from Shanghai Xin Fan Biological Technology Co., Ltd. (Shanghai, China). Mitochondria Isolation Kit for Tissue and phosphatase inhibitor were from Thermo Fisher Scientific (Waltham, MA, USA). Chemiluminescent HRP Substrate was from Merck Millipore (Billerica, MA, USA). Antibodies against t-Akt, p-Akt Ser473, p-Akt Thr308, t-GSK-3β and p-GSK-3β Ser9 were from Cell Signaling Technology (Boston, MA, USA). GoTaq® quantitative real-time polymerase chain reaction (qPCR) Master Mix was from Promega Corporation (Madison, WI, USA).
Animals
Adult male Sprague-Dawley (SD) rats (250-300 g) were from the Laboratory Animal Center of Zhejiang Chinese Medical University Laboratory Animal Research Center (certificate number SYXK(Zhe) 2013-0184). All experimental procedures were approved by the Animal Care and Use Committee of ZheJiang Chinese Medical University and conformed to the Guide for the Care and Use of Laboratory Animals (updated version, National Institutes of Health (NIH), Bethesda, MD, USA). The animals were acclimated for one week under standard housing conditions and given free access to food and water.
Induction of CME and PGE1 administration
Previous reports [24-26] described a CME model injecting microparticles into the left ventricle, with the microparticles forming a physical embolism in distal vasculature. Prior to building a CME model, we investigated related literature [4], ultimately choosing a protocol involving injection of sodium laurate into the left ventricle. We found that, compared with other models reported in the literature, this procedure better simulated the pathological process of CME observed in the clinic. This is because this method first induces vascular endothelial injury and fibrin formation, leading to CME. Sixty rats were randomly assigned to four groups: sham, CME model, high dose PGE1 (PGE1(H), 2 µg/kg) and low dose PGE1 (PGE1(L), 1 µg/kg). PGE1 (1 or 2 µg/kg) was administered by tail vein injection 20 min before induction of CME. The sham and CME model groups received equal doses of physiological saline instead of PGE1. Rats were anesthetized with pentobarbital sodium (45 mg/kg, intraperitoneally) and ventilated with a small animal ventilator (Hallowell, Rossville, IN, USA), with respiratory parameters adjusted to an airway pressure of 12-15 cmH2O and a respiratory frequency of 60 times/min. Body temperature was maintained at 37±1°C with a heating pad. A thoracotomy was performed at the midline of the chest, followed by a sternotomy between the second and fourth intercostal spaces. The pericardium was opened and the ascending aorta fully exposed. Sodium laurate (1 mg/kg) was injected into the left ventricle using a 30-gauge needle during a 20 sec occlusion of the ascending aorta in the CME groups, with the sham receiving equal doses of physiological saline. All rats were killed after 24 h and blood samples collected for biochemical analysis, qPCR and ELISA. Hearts from five rats per group were prepared for histopathology and others were prepared for protein analyses and mitochondrial assays.
Transmission electron microscopic analyses
Transmission electron microscopy was performed as described [27]. Briefly, selected areas of heart tissue, located near the apex, were collected. In preliminary experiments, with surgery performed by the same investigator and consistent injection sites in the left ventricle, pathological changes were conspicuous in this region. The tissue was cut into blocks and fixed in glutaraldehyde, post-fixed in osmium tetroxide and embedded in epoxy resin. Changes in microvascular ultrastructure and mitochondria were observed with a transmission electron microscope (Hitachi, Tokyo, Japan).
Light microscopic analyses
The remaining heart tissue was dissected from near the apex, in parallel with the coronary ditch, cut into 4 µm sections, fixed in 10% buffered formalin solution and embedded in paraffin. Various aspects of the disease were scored according to results of hematoxylin eosin (HE), Heidenhain’s hematoxylin and fgl2 immunohistochemical staining. Some samples were stained with HE to evaluate microthrombosis. The number of microthrombosis/50 coronary arterioles (vessels <100 µm in diameter) was calculated for each group. The scores were: 0, no microthrombosis; 1+, microthrombosis area <25% of vascular area; 2+, microthrombosis area 25-50% of vascular area; 3+, microthrombosis area 50-75% of vascular area; 4+, microthrombosis area >75% of vascular area [4]. Fgl2 was detected by streptavidin-peroxidase immunohistochemistry. Early myocardial ischemia was identified with Heidenhain’s hematoxylin staining. The scores were: 0, no myocardial ischemia; 1+, ischemia involving <5% of the myocardium; 2+, ischemia involving 5-10% of the myocardium; 3+, ischemia involving 10-15% of the myocardium; 4+, ischemia involving >15-20% of the myocardium. 5+, ischemia involving >20% of the myocardium.
Measurement of circulating Fgl2 levels by sandwich ELISA
Blood samples were obtained from the abdominal aorta of all rats prior to sacrifice and centrifuged at 5000×g for 10 min. Supernatant was removed from each sample and stored at -20°C. Serum fgl2 levels were determined by antigen-based sandwich ELISA, according to the manufacturer’s instructions. Briefly, 96-well plates were coated with 50 µl anti-fgl2 monoclonal antibody per well and plates washed three times. After a 30 min preincubation with washing buffer at 37°C, 50 µl of a fgl2 standard solution, control solution or rat supernatant was added to each well and plates were incubated for 30 min at 37°C. A goat polyclonal anti-murine fgl2 antibody was then added, followed by H2O2 and tetramethylbenzidine, and plates incubated for 10 min at 37°C. The reaction was stopped by adding 50 µl 1 M sulfuric acid and absorbance measured at 450 nm with a microplate reader (Bio-Rad, Hercules, CA, USA).
SOD and CAT assays
The remaining supernatants were used to measure levels of SOD and CAT. These were determined using the respective assay kits, according to the manufacturer’s instructions.
Quantification of mtDNA copy number
mtDNA copy number was measured by qPCR using an Applied Biosystems 7500 Sequence Detection System (Applied Biosystems, Foster, CA, USA). Two pairs of primers, one primer pair specific for mtDNA (ND1) and another specific for nuclear DNA (18s), were designed for relative quantification of mtDNA copy number. The DNA concentrations in all samples were adjusted to 30 µg/ml, measured using a BioPhotometer (Eppendorf, Hamburg, Germany). The 2-ΔΔCT method was used to calculate relative mtDNA copy number, as described previously [28-30]. Primer sequences for the mitochondrial ND1 gene were: forward primer (ND1-F), 5‘-TTTTATCTGCATCTGAGTTTAATCCTGT-3’; reverse primer (ND1-R), 5’-CCACTTCATCTTACCATTTATTATCGC-3’. Primer sequences for the nuclear gene 18s was: forward primer (18s-F), 5’-TAGAGGGACAAGTGGCGTTC-3’; reverse primer (18s-R), 5’-CGCTGAGCCAGTCAGTGT-3’. The PCR mixture, in a total volume of 20 µl, contained 2×Go Taq® qPCR Master Mix, 250 nM ND1-R (or 18s-R) primer, 250 nM ND1-F (or 18s-F) primer and 60 ng genomic DNA for ND1 and 18s. The thermal cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min for ND1 (or 18s).
Isolation of mitochondria
Mitochondria from rat heart samples were isolated with the Mitochondria Isolation Kit for Tissue, according to the manufacturer’s instructions. Briefly, fresh cardiac tissue was immersed in ice-cold Reagent A and cut into pieces of approximately 1 mm3 and homogenized using a Teflon pestle. Reagent C was added to the homogenate and the sample centrifuged at 700×g for 10 min. The supernatant was stored on ice and the pellet resuspended in isolation buffer and centrifuged at 700×g to increase the yield of mitochondria. Both supernatants were pooled and centrifuged at 3000×g for 15 min and the resulting pellet of isolated mitochondria was resuspended in the isolation buffer and used immediately for analysis of mPTP. All procedures were performed at 0-4°C.
mPTP opening assay
Mitochondrial protein concentrations were quantified using the BCA kit with bovine serum albumin (BSA) as a standard. Isolated mitochondria were diluted in swelling buffer (120 Mm KCl, 10 mM Tris-HCl, 20 mM MOPS and 5 mM KH2PO4, pH 7.4) at a concentration of 1 mg/ml [31,32]. A Ca2+-induced mitochondria swelling assay was performed to detect mPTP opening. With addition of 200 µM CaCl2, mPTP opening was increased, resulting in a stable decline in optical density of the mitochondrial suspension. Moreover, Atr, a mPTP opener, was added and effects of PGE1 assessed. Absorbance at 520 nm was continuously measured to monitor mPTP opening.
Heart mitochondrial suspensions were divided into 8 groups: (1) sham (n = 5); (2) sham+Atr 100 µM (n = 5); (3) CME (n = 5); (4) CME+Atr 100 µM (n = 5); (5) PGE1(H) (n = 5); (6) PGE1(H)+Atr 100 µM (n = 5); (7) PGE1(L) (n = 5); (8) PGE1(L)+Atr 100 µM (n = 5). The relative change in A520 was calculated by dividing each A520 measurement by the initial value for the sample. The value of minA520 (the last relative A520, 10 min after CaCl2 or CaCl2+Atr addition) normalized to the maxA520 (the initial relative A520, 1.5 min before adding CaCl2 or CaCl2+Atr) was used for statistical analysis.
Western blotting
Myocardial samples were homogenized in RIPA lysis buffer containing protease (1×), and a phosphatase (2×) inhibitors. The homogenized tissues were centrifuged at 14,000×g for 5 min at 4°C and protein concentration determined with the BCA assay. The supernatant containing myocardial protein was transferred to a tube and SDS-PAGE loading buffer (5×) was added. Proteins were denatured in a heating block at 100°C for 10 min. The samples were electrophoretically separated by SDS-PAGE and transferred to polyvinylidene fluoride sheets using a transblot apparatus. Membranes were blocked for 90 min at room temperature with 5% BSA dissolved in TBS-T buffer (25 mM Tris, 0.8% NaCl, 0.02% KCl and 0.1% Tween-20, pH 7.4). Samples were analyzed for phosphorylated Akt (p-Akt Ser473 and Thr308) and phosphorylated GSK-3β (p-GSK-3β Ser9) using the appropriate primary and secondary antibodies. Subsequently, membranes were stripped and re-probed for total Akt (t-Akt) and total GSK-3β (t-GSK-3β), to normalize p-Akt(Ser473), p-Akt(Thr308) and p-GSK-3β(Ser9) band intensities, respectively.
Statistical analysis
All values were expressed as means ± S.E.M. Statistical analysis was performed with SPSS 19 software (SPSS Inc., Chicago, IL, USA). Significant differences were assessed by one-way analysis of variance followed by a Student-Newman-Keuls or Dunnett’s post hoc test. Correlation analysis was carried out using linear regression. Differences among groups were considered significant at P<0.05.
Results
Histopathology
At 24 h after injection of sodium laurate, the CME group showed severe microthrombi obstructing coronary arterioles and significant myocardial ischemia with inflammatory cell infiltration (P<0.01 for all, Table 1; Figure 1A, 1B). Coronary microthrombi and myocardial ischemia were decreased in the PGE1(H), as compared with the CME, group (P<0.01). The PGE1(L) group also showed protection against myocardial ischemia and coronary microthrombi (P<0.01), but the PGE1(H) showed greater reduction of microthrombi than did the PGE1(L) group (P<0.05). By transmission electron microscopy, in contrast to the sham group, the CME model group showed endothelial damage and fibrin (Figure 1Ca), with mitochondria exhibiting markedly decreased internal complexity and irregular arrangements and morphologies (Figure 1Cb). These effects were partially ameliorated in PGE1 pretreated groups. These morphological observations indicated that PGE1 pretreatment alleviated damage to myocardial organelles, particularly mitochondria, induced by CME.
Table 1.
Effects of PGE1 on histology in a CME rat model
| Microthrombosis | Early myocardial ischemia | |
|---|---|---|
| sham | 0±0 | 0±0 |
| CME | 3.26±0.08949b | 3.80±0.20000b |
| PGE1(H) | 1.56±0.17606d | 1.60±0.31623d |
| PGE1(L) | 2.16±0.21857d,e | 1.80±0.42426d |
Both PGE1(H) and PGE1(L) administered 20 min prior to CME reduced the number of microthrombosis and myocardial ischemia. However, PGE1(H) had a better effect in anti-microthrombosis than PGE1(L). All values are the mean ± S.E.M. (n = 5/group or 50 coronary arterioles/group).
P<0.01 vs Sham group.
P<0.01 vs CME model group.
P<0.05 vs PGE1(H) group.
Figure 1.

Histological analysis of the hearts. A. Myocardium from the four groups with hematoxylin and eosin staining (magnification, 1 = ×100, 2 = ×400; bar = 100 µm). Green arrows indicate coronary arterioles (vessels <100 µm in diameter). Two treatment groups exhibited a lower incidence of thromboembolism, compared with the other groups. B. Myocardium from the four groups with Heidenhain’s hematoxylin staining (magnification, 1 = ×100, 2 = ×400; bar = 100 µm). Red arrows indicate early myocardial ischemia. Both PGE1(H) and PGE1(L) groups exhibited a lower incidence of early myocardial ischemia than did the CME group. C. Transmission electron microscopy of coronary arterioles and mitochondria. En, endothelium; Er, erythrocyte; F, fibrin; and M, mitochondria (magnification ×10000 for a and ×25000 for b; bar = 2 µm for a and 1 µm for b). These pathological changes in the CME group were partially inhibited by PGE1.
Expression of Fgl2
We confirmed high levels of fgl2 in the CME group by immunohistochemistry and sandwich ELISA. Immunohistochemistry results showed higher levels of fgl2 in microvascular endothelium of the CME groups, compared with the others (Figure 2A). ELISA results also indicated higher fgl2 expression in the CME group, compared with sham and PGE1 groups (P<0.01 for all comparisons, Figure 2B). To investigate the relationship between oxidative stress and fgl2 expression, we conducted a correlation analysis of SOD and CAT levels with those of fgl2. Levels of fgl2 were inversely correlated with those of SOD (Figure 2C, R2 = 0.508, P<0.01, n = 20) and CAT (Figure 2D, R2 = 0.433, P<0.01, n = 35).
Figure 2.

Expression of fgl2. A. Myocardium from the four groups showing immunohistochemical staining for fgl2 (magnification, ×400; bar = 100 µm). Blue arrows indicate coronary arterioles (vessels <100 µm in diameter). B. Effects of PGE1 showing decreased expression of fgl2. Both PGE1(H) and PGE1(L) pretreatments prevented the increased fgl2 expression induced by CME. All values are means ± S.E.M. (n = 10 per group). **P<0.01 vs Sham group. ##P<0.01 vs CME group. C. Correlation analysis between serum fgl2 and SOD levels. A total of 20 data points was analyzed, showing a significant correlation (P<0.01, linear regression, n = 20). D. Correlation analysis between serum fgl2 and CAT levels, showing a significant correlation (P<0.01, linear regression, n = 35).
Determining SOD and CAT levels in serum
To test whether cardioprotection by PGE1 in our CME model was mediated by its antioxidant effects, serum SOD and CAT levels were measured. SOD and CAT levels were significantly lower in the CME group than in the sham, PGE1(H) and PGE1(L) groups (P<0.05, P<0.01 and P<0.01, respectively, Figure 3).
Figure 3.
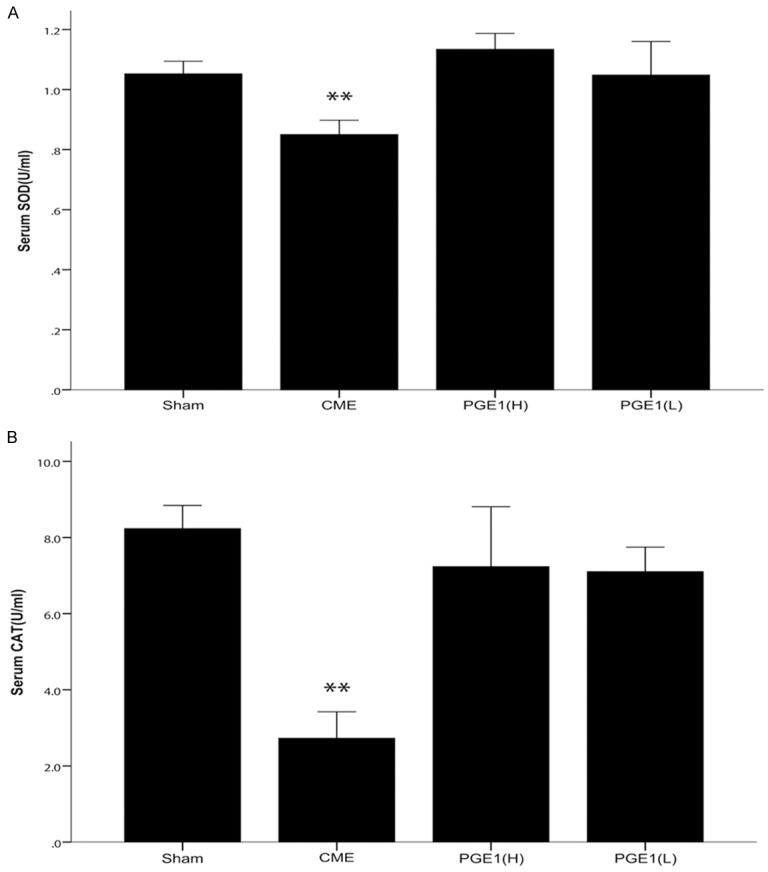
Effects of PGE1 on inhibition of oxidative stress. A. Serum SOD levels, an index of antioxidant protection (n = 5 per group). In the CME group, SOD levels were significantly decreased, while this effect was prevented in the two pretreatment groups. B. Serum CAT levels, also reflecting resistance to oxidative stress (n = 8-10 per group). Consistent with the SOD results, both PGE1(H) and PGE1(L) pretreatments prevented the decrease in CAT levels. All values are means ± S.E.M. **P<0.01 vs Sham, PGE1(H) or PGE1(L) groups.
Analysis of mtDNA copy numbers
To test the hypothesis that the cardioprotective effects of PGE1 involved the mitochondria, mtDNA copy number was determined by qPCR. The mtDNA copy number was higher in the CME than in the sham group (P<0.01, Figure 4A). Compared with the CME and PGE1(L) groups, the PGE1(H) group had a lower mtDNA copy number (P<0.01). To confirm the relationship between oxidative stress and mtDNA copy number, we conducted a correlation analysis of SOD and CAT levels with mtDNA copy number. The mtDNA copy number was inversely correlated with both SOD and CAT levels (Figure 4B, 4C, R2 = 0.513 and 0.499, n = 20 and 35, respectively, P<0.01 for all comparisons).
Figure 4.

Functional effects on mitochondria. A. Effects of PGE1 on relative mtDNA copy number. Both PGE1(H) and PGE1(L) pretreatments prevented the CME-induced increase in relative mtDNA copy number. Moreover, the PGE1(H) group had a significantly lower mtDNA copy number (n = 10 per group). **P<0.01 vs Sham group. ##P<0.01 vs CME or PGE1(L) group. ++ P<0.01 vs CME or Sham group. B. Correlation analysis between relative mtDNA copy number and serum SOD content. A total of 20 data points was analyzed, showing a significant correlation (P<0.01, linear regression, n = 20). C. Correlation analysis between relative mtDNA copy number and serum CAT content, also showing a significant correlation (P<0.01, linear regression, n = 35). D. Downward sloping curves indicating mPTP opening induced by Ca2+ or Ca2++Atr. E. A520 change (min/max A520), indicating mPTP opening. Both PGE1(H) and PGE1(L) pretreatments prevented CME-induced mPTP opening. PGE1(H) appeared to be more effective than PGE1(L), but the difference was not statistically significant (P>0.05, n = 5 per group). All values are means ± S.E.M. **P<0.01 vs Sham group. ##P<0.01 vs CME group. ++ P<0.01 vs PGE1(H) group. × P<0.05 vs PGE1(L) group.
mPTP opening assay
Because mPTP opening is recognized as a major determinant in reperfusion injury, we examined whether it was increased in CME and whether the cardioprotective effects of PGE1 were mediated by mPTP. The CME group exhibited a significant increase in the degree of mPTP opening, as compared with the sham group (P<0.01, Figure 4D, 4E). In both the PGE1(H) and PGE1(L) groups, mPTP opening in response to CME was prevented (P<0.01 for all comparisons). The higher dose of PGE1 appeared to be more effective than the lower dose, but this difference was not statistically significant (P>0.05). A mPTP opening agent, Atr, added together with CaCl2, completely abolished inhibition of mPTP opening by PGE1 (P<0.05 and P<0.01 versus PGE1(H) and PGE1(L), respectively).
Western blotting analysis of the Akt-GSK-3β pathway
To test whether the cardioprotective effects of PGE1 were mediated by the Akt-GSK-3β signaling pathway, phosphorylation of Akt and GSK-3β were assessed by western blotting for p-Akt(Ser473), p-Akt(Thr308) and p-GSK-3β(Ser9), respectively. CME decreased relative levels of p-GSK-3β(Ser9), normalized to the t-GSK-3β band intensity. Compared with the CME group, PGE1 pretreated groups had higher relative p-GSK-3β(Ser9) levels (P<0.01 for all, Figure 5). However, As compared with sham and CME groups, the relative amount of p-Akt(Ser473), normalized to the t-Akt signal, was decreased in the PGE1(H) group (P<0.05 for all).
Figure 5.

Effects of PGE1 on Akt and GSK-3β phosphorylation. A. Representative immunoreactive bands for phosphorylated Akt (p-Akt Ser473 and Thr308), total Akt (t-Akt), phosphorylated GSK-3β (p-GSK-3β Ser9) and total GSK-3β (t-GSK-3β). B. The average of p-Akt Ser473/t-Akt. *P<0.05 vs Sham or CME. Akt phosphorylation at Ser473 was decreased in the PGE1(H) group. C. The average of p-Akt Thr308/t-Akt. D. The average of p-GSK-3β/t-GSK-3β. *P<0.05 vs Sham group. ##P<0.01 vs CME group. Compared with in the sham group, GSK-3β expression was lower in the CME group, while this effect was prevented in the two pretreatment groups. All values are means ± S.E.M. (n = 10 per group).
Discussion
Clinically, there is no drug for specific treatment of coronary microvascular dysfunction. The major conclusion of our study is that PGE1 is cardioprotective, based on its attenuation of early myocardial ischemia and microthrombosis induced by CME. Although some patients present at hospitals with existing coronary microvascular dysfunction, decreasing the potential clinical usefulness of PGE1 pretreatment, this treatment could effectively prevent iatrogenic coronary microvascular dysfunction in patients who require coronary interventions.
PGE1 is known to be anti-thrombotic [6-9]. Therefore, it was important to determine whether its cardioprotective effects involved inhibition of fgl2 expression. We found increased fgl2 expression in the CME, compared with the sham, group and prevention of this increase in the PGE1 pretreated groups. Fgl2 is reportedly localized in impaired endothelial cells, suggesting that PGE1 pretreatment may have decreased fgl2 levels because it protected endothelial cells from injury. Hence, we propose that PGE1 decreased oxidative stress-associated injury, preventing endothelial damage, in the CME model. Although many studies [12,13] showed that PGE1 decreased oxidative stress, it was unknown whether PGE1 pretreatment could confer resistance to oxidative stress in CME. To address this, we tested levels of two major antioxidant enzymes, SOD and CAT, to assess oxidative stress resistance. As expected, we found that PGE1 was beneficial against loss of these antioxidant defense enzymes in CME rats and that there was an inverse correlation between fgl2 levels and those of SOD and CAT.
To further explore effects of PGE1 against oxidative stress, we investigated mitochondria functionally and structurally, because of their importance in resistance to oxidative stress [33-36]. By transmission electron microscopy, compared with in the sham group, the arrangement and morphology of mitochondria were irregular and their internal complexity was decreased in the CME group. The changes in mitochondrial structure were partially inhibited by PGE1. mtDNA copy number, as an indicator of mitochondrial damage, is closely related to mitochondrial function [37,38], with severe mitochondrial damage inducing decreased mtDNA copy number. Interestingly, we found that mtDNA copy number was increased in the CME group, with no change in mtDNA copy number in the PGE1(H) group. One possible explanation is that the mitochondrial respiratory chain suffered mild oxidative stress-induced injury in CME. Mild oxidative stress can stimulate production of mtDNA molecules, thus compensating for reduced mitochondrial respiratory function, preserving protein synthesis and ensuring cell survival. However, with high dose PGE1 pretreatment, these compensatory changes did not occur because stimulation by mild injury was prevented. This idea would be consistent with the characteristic microvascular injury in the CME model. Therefore, there was an inverse correlation between the mtDNA copy number and SOD and CAT levels, but this correlation would represent a qualitative change caused by the degree of mitochondrial damage [37].
We also investigated which part of the mitochondria was affected during cardioprotection by PGE1, hypothesizing that mPTP was its site of action. mPTP is a large-conductance megachannel, including voltage-dependent anion channels in the outer membrane, the adenine nucleotide transporter in the inner membrane and cyclophilin D in the matrix, and serves as an important gating valve for mitochondrial function. Under physiological conditions, mPTP is predominantly in a closed state but opens in response to high Ca2+, reactive oxygen species (ROS) or decreased inner membrane potential [23]. mPTP opening results in depolarization of the mitochondrial membrane and matrix swelling, leading to rupture of the outer membrane and cytochrome C release. This decreases ATP generation, ultimately affecting cellular function. Moreover, several studies [16-20] showed that putative signaling pathways for protecting coronary microvascular function ultimately involved mPTP blockade. To verify our hypothesis, we detected mPTP opening based on mitochondrial swelling. We demonstrated that mPTP opening was increased in the CME model and prevented by PGE1 pretreatment. However, the inhibition of mPTP opening by PGE1 was completely abolished by the mPTP opener Atr, further proving that the cardioprotective effects of PGE1 were mediated by mPTP.
To further support involvement of mPTP blockade in cardioprotection by PGE1, we examined the role of the Akt-GSK-3β pathway. Previous studies [21-23] identified this pathway as the upstream signal of mPTP, playing an important role in ischemic preconditioning for treating myocardial ischemia-reperfusion injury. So, we hypothesized that the cardioprotective effects of PGE1 pretreatment in CME were mediated by the Akt-GSK-3β pathway. Ours is the first evidence suggesting that PGE1 pretreatment can phosphorylate GSK-3β. Unexpectedly, the PGE1(H) pretreated group had decreased phosphorylation of Akt at Ser473. We identified two mechanisms potentially explaining this observation: 1) GSK-3β signaling is mediated not only by Akt, but also by ERK1/2, p38MAPK, JNK, Wnt and so forth [39-42], which might also have cardioprotective efficacy, with crosstalk between them. So PGE1 pretreatment might have activated the others, leading to GSK-3β phosphorylation and decreased Akt expression because of negative feedback [39,40]. The negative feedback could also explain why this phenomenon occurred only in the PGE1(H) group; 2) Our results also suggested that PGE1 seemed to prevent the reduction of phosphorylation of Akt at Thr308, as compared with CME, group. Akt phosphorylation of two main residues, Thr308 and Ser473, has been established as being equivalent to Akt activation [43]. So it is difficult to get a definite conclusion whether the total activation of Akt decreased or not. Therefore, we speculate that the protective effects of PGE1 against coronary microcirculation dysfunction involved activation of the GSK-3β-mPTP pathway and preservation of mitochondrial function and structure. Furthermore, PGE1 increased resistance to oxidative stress injury, protecting endothelial cells from damage, decreasing the conditions favoring fgl2 production and infiltration of inflammatory cells and, ultimately, preventing early myocardial ischemia and CME (Figure 6).
Figure 6.
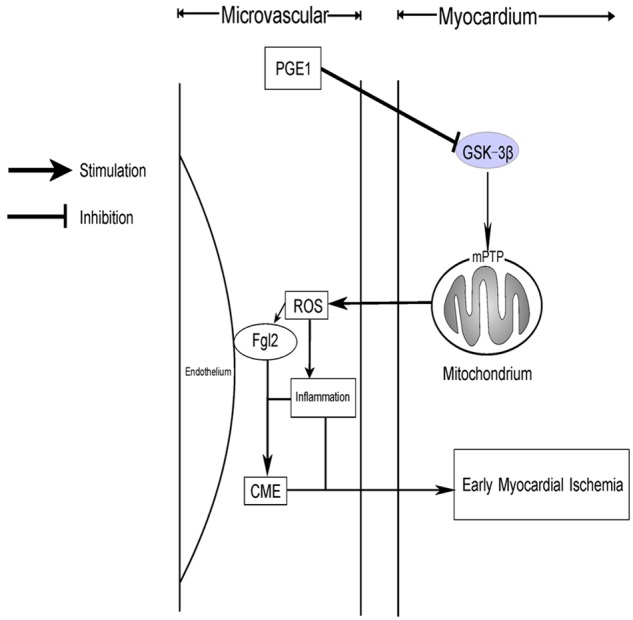
Schematic model showing the role of PGE1 in improving coronary microvascular dysfunction. Sodium laurate induces CME, causing decreased GSK-3β phosphorylation and mPTP opening, leading to damaged mitochondrial function and structure. The low anti-oxidative stress facilitates the expression of fgl2 and inflammatory infiltration. Finally, coronary microcirculation dysfunction induced early myocardial ischemia. However, PGE1 can prevent these molecular effects at the site of GSK-3β phosphorylation.
In our study, we used PGE1 concentrations comparable to both normal and doubled clinical doses, converted for SD rats using a transformation formula [44]. Our results also suggested that some cardioprotective effects of PGE1 against CME were partial and dose-dependent, including effects on mtDNA copy number and formation of microemboli. Adjusting dosages of PGE1 might be an effective and simple way to improve prognosis of CME patients. We did not test a specific inhibitor or agonist of GSK-3β in this study. Hence, we cannot exclude the possibility that our findings regarding GSK-3β phosphorylation by PGE1 resulted from an epiphenomenon. However, previous studies [21-23] showed that GSK-3β was an upstream signal for mPTP and, in our study, we confirmed the role of mPTP in the cardioprotective effects of PGE1 in CME. In addition, we do not yet have definitive evidence to explain the downregulation of Akt at 473 observed in this study. It would be useful to further elucidate the specific mechanisms for protection of coronary microvascular function by PGE1 by, for example, employing specific inhibitors or observing PGE1 effects at earlier time points.
We conclude that PGE1 is an effective cardioprotective agent. Its protective mechanism involves activation of the GSK3-β-mPTP pathway, protection of mitochondria, increasing resistance to oxidative stress-induced injury, preventing formation of microthrombi and suppressing inflammation. PGE1 may, therefore, improve coronary microvascular dysfunction in the clinic.
Acknowledgements
This study was supported by Beijing Lisheng Cardiovascular Health Foundation of China (LSG1501132) and Zhejiang Bureau of Science and Technology (2017C37130).
Disclosure of conflict of interest
None.
References
- 1.Crea F, Camici PG, Bairey MC. Coronary microvascular dysfunction: an update. Eur Heart J. 2014;35:1101–1111. doi: 10.1093/eurheartj/eht513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herrmann J, Kaski JC, Lerman A. Coronary microvascular dysfunction in the clinical setting: from mystery to reality. Eur Heart J. 2012;33:2771–2782. doi: 10.1093/eurheartj/ehs246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Ma XJ, Guo CY, Wang MM, Kou N, Qu H, Mao HM, Shi DZ. Pretreatment with a combination of ligustrazine and berberine improves cardiac function in rats with coronary microembolization. Acta Pharmacol Sin. 2016;37:463–472. doi: 10.1038/aps.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen C, Liang C, Chen L, Qian J, Wang K, Chen H, Ge C. The establishment of a new model of rat coronary microthrombosis by Coronary microthrombosis by coronary sodium laurate injection. Chinese Journal of Arteriosclerosis. 2005;13:447–450. [Google Scholar]
- 6.Weiss T, Fischer D, Hausmann D, Weiss C. Endothelial function in patients with peripheral vascular disease: influence of prostaglandin E1. Prostaglandins Leukot Essent Fatty Acids. 2002;67:277–281. doi: 10.1054/plef.2002.0429. [DOI] [PubMed] [Google Scholar]
- 7.Schutte H, Lockinger A, Seeger W, Grimminger F. Aerosolized PGE1, PGI2 and nitroprusside protect against vascular leakage in lung ischaemia-reperfusion. Eur Respir J. 2001;18:15–22. doi: 10.1183/09031936.01.00070001. [DOI] [PubMed] [Google Scholar]
- 8.Umemura K, Nakashima M. Effect of prostaglandin E1 on the rat inner ear microvascular thrombosis. Gen Pharmacol. 1997;28:221–224. doi: 10.1016/s0306-3623(96)00192-9. [DOI] [PubMed] [Google Scholar]
- 9.Khanna V, Armstrong PC, Warner TD, Curzen N. Prostaglandin E1 potentiates the effects of P2Y12 blockade on ADP-mediated platelet aggregation in vitro: insights using short thromboelastography. Platelets. 2015;26:689–692. doi: 10.3109/09537104.2014.1001832. [DOI] [PubMed] [Google Scholar]
- 10.Shao L, Wu D, Zhang P, Li W, Wang J, Su G, Liao Y, Wang Z, Liu K. The significance of microthrombosis and fgl2 in no-reflow phenomenon of rats with acute myocardial ischemia/reperfusion. Clin Appl Thromb Hemost. 2013;19:19–28. doi: 10.1177/1076029612437577. [DOI] [PubMed] [Google Scholar]
- 11.Ding Y, Liu K, Wang Y, Su G, Deng H, Zeng Q, Liao Y, Wang Z. Expression and significance of fgl2 prothrombinase in cardiac microvascular endothelial cells of rats with type 2 diabetes. J Huazhong Univ Sci Technolog Med Sci. 2010;30:575–581. doi: 10.1007/s11596-010-0545-y. [DOI] [PubMed] [Google Scholar]
- 12.Gezginci-Oktayoglu S, Orhan N, Bolkent S. Prostaglandin-E1 has a protective effect on renal ischemia/reperfusion-induced oxidative stress and inflammation mediated gastric damage in rats. Int Immunopharmacol. 2016;36:142–150. doi: 10.1016/j.intimp.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 13.Fang WT, Li HJ, Zhou LS. Protective effects of prostaglandin E1 on human umbilical vein endothelial cell injury induced by hydrogen peroxide. Acta Pharmacol Sin. 2010;31:485–492. doi: 10.1038/aps.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura TY, Nakao S, Wakabayashi S. Neuronal Ca2+ sensor-1 contributes to stress tolerance in cardiomyocytes via activation of mitochondrial detoxification pathways. J Mol Cell Cardiol. 2016;99:23–34. doi: 10.1016/j.yjmcc.2016.08.013. [DOI] [PubMed] [Google Scholar]
- 15.Fouque A, Lepvrier E, Debure L, Gouriou Y, Malleter M, Delcroix V, Ovize M, Ducret T, Li C, Hammadi M, Vacher P, Legembre P. The apoptotic members CD95, BclxL, and Bcl-2 cooperate to promote cell migration by inducing Ca(2+) flux from the endoplasmic reticulum to mitochondria. Cell Death Differ. 2016;23:1702–1716. doi: 10.1038/cdd.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiao S, Olson JM, Paterson M, Yan Y, Zaja I, Liu Y, Riess ML, Kersten JR, Liang M, Warltier DC, Bosnjak ZJ, Ge ZD. MicroRNA-21 mediates isoflurane-induced cardioprotection against ischemia-reperfusion injury via Akt/nitric oxide synthase/mitochondrial permeability transition pore pathway. Anesthesiology. 2015;123:786–798. doi: 10.1097/ALN.0000000000000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Fu H, Zhang H, Xu F, Zou Z, Liu M, Wang Q, Miao M, Shi X. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3beta signaling and inhibiting mitochondrial permeability transition. PLoS One. 2013;8:e74422. doi: 10.1371/journal.pone.0074422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3beta pathway after ischemia/reperfusion. PLoS One. 2015;10:e135988. doi: 10.1371/journal.pone.0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ong SG, Lee WH, Theodorou L, Kodo K, Lim SY, Shukla DH, Briston T, Kiriakidis S, Ashcroft M, Davidson SM, Maxwell PH, Yellon DM, Hausenloy DJ. HIF-1 reduces ischaemia-reperfusion injury in the heart by targeting the mitochondrial permeability transition pore. Cardiovasc Res. 2014;104:24–36. doi: 10.1093/cvr/cvu172. [DOI] [PubMed] [Google Scholar]
- 20.Jaffe R, Dick A, Strauss BH. Prevention and treatment of microvascular obstruction-related myocardial injury and coronary no-reflow following percutaneous coronary intervention: a systematic approach. JACC Cardiovasc Interv. 2010;3:695–704. doi: 10.1016/j.jcin.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Donato M, Goyeneche MA, Garces M, Marchini T, Perez V, Del MJ, Hocht C, Rodriguez M, Evelson P, Gelpi RJ. Myocardial triggers involved in activation of remoteischaemic preconditioning. Exp Physiol. 2016;101:708–716. doi: 10.1113/EP085535. [DOI] [PubMed] [Google Scholar]
- 22.Gonzalez AL, Ciocci PA, Fantinelli JC, Mosca SM. Cyclosporine-A mimicked the ischemic pre- and postconditioning-mediated cardioprotection in hypertensive rats: role of PKCepsilon. Exp Mol Pathol. 2016;100:266–275. doi: 10.1016/j.yexmp.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Chen H, Su Q, Zhou Y, Liu T, Li L. The PTEN/Akt signaling pathway mediates myocardial apoptosis in swine after coronary microembolization. J Cardiovasc Pharmacol Ther. 2016;21:471–477. doi: 10.1177/1074248415624158. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, Zhou Y, Liu YC, Wang JY, Su Q, Tang ZL, Li L. Coronary microembolization induces cardiomyocyte apoptosis through the LOX-1-dependent endoplasmic reticulum stress pathway involving JNK/P38 MAPK. Can J Cardiol. 2015;31:1272–1281. doi: 10.1016/j.cjca.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Chen ZW, Qian JY, Ma JY, Chang SF, Yun H, Jin H, Sun AJ, Zou YZ, Ge JB. TNF-alpha-induced cardiomyocyte apoptosis contributes to cardiac dysfunction after coronary microembolization in mini-pigs. J Cell Mol Med. 2014;18:1953–1963. doi: 10.1111/jcmm.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toshima Y, Satoh S, Ikegaki I, Asano T. A new model of cerebral microthrombosis in rats and the neuroprotective effect of a Rho-kinase inhibitor. Stroke. 2000;31:2245–2250. doi: 10.1161/01.str.31.9.2245. [DOI] [PubMed] [Google Scholar]
- 28.Schmittgen TD. Real-time quantitative PCR. Methods. 2001;25:383–385. doi: 10.1006/meth.2001.1260. [DOI] [PubMed] [Google Scholar]
- 29.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 30.Lee EJ, Schmittgen TD. Comparison of RNA assay methods used to normalize cDNA for quantitative real-time PCR. Anal Biochem. 2006;357:299–301. doi: 10.1016/j.ab.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Guo Z, Liao Z, Huang L, Liu D, Yin D, He M. Kaempferol protects cardiomyocytes against anoxia/reoxygenation injury via mitochondrial pathway mediated by SIRT1. Eur J Pharmacol. 2015;761:245–253. doi: 10.1016/j.ejphar.2015.05.056. [DOI] [PubMed] [Google Scholar]
- 32.Wu L, Shen F, Lin L, Zhang X, Bruce IC, Xia Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett. 2006;402:184–189. doi: 10.1016/j.neulet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Ghanta S, Tsoyi K, Liu X, Nakahira K, Ith B, Coronata AA, Fredenburgh LE, Englert JA, Piantadosi CA, Choi AM, Perrella MA. Mesenchymal stromal cells deficient in autophagy proteins are susceptible to oxidative injury and mitochondrial dysfunction. Am J Respir Cell Mol Biol. 2017;56:300–309. doi: 10.1165/rcmb.2016-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehmood T, Maryam A, Zhang H, Li Y, Khan M, Ma T. Deoxyelephantopin induces apoptosis in HepG2 cells via oxidative stress, NF-kappaB inhibition and mitochondrial dysfunction. Biofactors. 2017;43:63–72. doi: 10.1002/biof.1324. [DOI] [PubMed] [Google Scholar]
- 35.Powell RD, Goodenow DA, Mixer HV, Mckillop IH, Evans SL. Cytochrome c limits oxidative stress and decreases acidosis in a rat model of hemorrhagic shock and reperfusion injury. J Trauma Acute Care Surg. 2017;82:35–41. doi: 10.1097/TA.0000000000001257. [DOI] [PubMed] [Google Scholar]
- 36.Davuluri G, Allawy A, Thapaliya S, Rennison JH, Singh D, Kumar A, Sandlers Y, Van Wagoner DR, Flask CA, Hoppel C, Kasumov T, Dasarathy S. Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J Physiol. 2016;594:7341–7360. doi: 10.1113/JP272796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada K, Kang D, Suomalainen A, Sunagawa K. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10:e119687. doi: 10.1371/journal.pone.0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Kafaji G, Golbahar J. High glucose-induced oxidative stress increases the copy number of mitochondrial DNA in human mesangial cells. Biomed Res Int. 2013;2013:754946. doi: 10.1155/2013/754946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Padhan N, Nordling TE, Sundstrom M, Akerud P, Birgisson H, Nygren P, Nelander S, Claesson-Welsh L. High sensitivity isoelectric focusing to establish a signaling biomarker for the diagnosis of human colorectal cancer. BMC Cancer. 2016;16:683. doi: 10.1186/s12885-016-2725-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bliksoen M, Rutkovskiy A, Vaage J, Stenslokken KO. Mode of perfusion influences infarct size, coronary flow and stress kinases in the isolated mouse heart. Acta Physiol (Oxf) 2017;220:36–46. doi: 10.1111/apha.12773. [DOI] [PubMed] [Google Scholar]
- 41.Xu WD, Wang J, Yuan TL, Li YH, Yang H, Liu Y, Zhao Y, Herrmann M. Interactions between canonical Wnt signaling pathway and MAPK pathway regulate differentiation, maturation and function of dendritic cells. Cell Immunol. 2016;310:170–177. doi: 10.1016/j.cellimm.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 42.Xu P, Zhang WB, Cai XH, Qiu PY, Hao MH, Lu DD. Activating AKT to inhibit JNK by troxerutin antagonizes radiation-induced PTEN activation. Eur J Pharmacol. 2017;795:66–74. doi: 10.1016/j.ejphar.2016.11.052. [DOI] [PubMed] [Google Scholar]
- 43.Risso G, Blaustein M, Pozzi B, Mammi P, Srebrow A. Akt/PKB: one kinase, many modifications. Biochem J. 2015;468:203–214. doi: 10.1042/BJ20150041. [DOI] [PubMed] [Google Scholar]
- 44.Tang JM, Chen ML. Medical Experimental Zoology: China Traditional Medicine Press. 2014 [Google Scholar]