

Abstract
RNA metabolism is tightly controlled across different tissues and developmental stages, and its dysregulation is one of the molecular hallmarks of cancer. Through direct binding to specific sequence element(s), RNA binding proteins (RBPs) play a pivotal role in co- and post-transcriptional RNA regulatory events. We have recently demonstrated that, in pancreatic cancer cells, acquisition of a drug resistant (DR)-phenotype relied on upregulation of the polypyrimidine tract binding protein (PTBP1), which in turn is recruited to the pyruvate kinase pre-mRNA and favors splicing of the oncogenic PKM2 variant. Herein, we describe a step-by-step protocol of the ultraviolet (UV) light cross-linking and immunoprecipitation (CLIP) method to determine the direct binding of an RBP to specific regions of its target RNAs in adherent human cell lines.
Keywords: CLIP, Protein-RNA interaction, Protein-RNA-immunoprecipitation, RNA processing
Background
While being transcribed in the nucleus, nascent RNAs are immediately assembled with trans-acting factors collectively named RNA binding proteins (RBPs). These factors interact directly with specific cis-acting regulatory sequences in RNA molecules, thus forming ribonucleoprotein (RNP) complexes ( Dreyfuss et al., 2002 ; Singh et al., 2015 ). These complexes control co-transcriptional RNA processing events as well as post-transcriptional mechanisms involved in RNA metabolism, such as subcellular localization and translation. For instance, spliceosomal and cleavage/polyadenylation complex components recognize specific RNA elements in the pre-mRNA, permitting introns removal (Black, 2003) and coordination between 3’-end processing and transcription termination (Proudfoot, 2016). A large number of RBPs functions as splicing factors, by assisting recognition of constitutively and alternatively spliced exons by the spliceosome (Chen and Manley, 2009) or by improving usage of alternative polyadenylation signals (Tian and Manley, 2016). Likewise, RBP-mediated recognition of zip code localization elements allows transport and local translation of mRNA in the cytoplasm (Martin and Ephrussi, 2009).
Eukaryotic genomes encode a wide array of RBPs to fine-tune cell-specific gene expression programs in a time- and space-sensitive manner, thus contributing to tissue homeostasis. RNPs are highly dynamic structures, which remodel under the influence of specific cell signaling pathways that influence the fate of the RNA transcript (Naro and Sette, 2013; Fu and Ares, 2014). By precisely integrating co- and post-transcriptional RNA regulatory events, RBPs ensure the physiological adaptation in response to environmental constraints. It follows that the precise arrangement of RNP complexes must be highly coordinated and that deregulation of these complexes can be harmful for cells. Indeed, dysregulation of each aspect of RNA metabolism is involved in a large number of pathological conditions, such as neurodegenerative disease and cancer (Mayr and Bartel, 2009; Cooper et al., 2009 ; Silvera et al., 2010 ; Pagliarini et al., 2015 ). In cancer, aberrant alternative splicing regulation often yields splice variants that confer a selective advantage to the tumor, in terms of proliferation, metabolism, invasion, drug resistance and survival ( David et al., 2010 ; Olshavsky et al., 2010 ; Paronetto et al., 2010 ; Valacca et al., 2010 ; Anczuków et al., 2012 ; Cappellari et al., 2014 ; Bielli et al., 2014 ; Calabretta et al., 2016 ). Moreover, specific splicing signatures correlate with cancer progression, and alteration of RBPs expression and/or of cis-regulatory elements can contribute to tumorigenesis ( Cooper et al., 2009 ; Danan- Gotthold et al., 2015 ). High-throughput next-generation sequencing technologies now allow genome-wide identification of alternative splicing events associated with pathological processes (Chen and Weiss, 2015; Byron et al., 2016 ). Furthermore, they might help understanding the global complexity of RNA regulation and the correlation between binding sites for RBPs and the splicing outcome in health and disease (Wang and Burge, 2008). Thus, understanding alternative splicing changes in pathological conditions requires deciphering the regulatory network between RBPs and cis-regulatory elements and the identification of RBP binding sites is a key step in this direction.
In a recent study, we investigated the role of alternative splicing and RBPs in the acquisition of a drug-resistant (DR) phenotype in pancreatic ductal adenocarcinoma cells (PDAC) ( Calabretta et al., 2016 ). We demonstrated that acquisition of the DR-phenotype relied on upregulation of the polypyrimidine tract binding protein (PTBP1), which is recruited to the PKM pre-mRNA and favors splicing of the oncogenic PKM2 variant. To investigate the recruitment of PTBP1 on PKM pre-mRNA in vivo, we used the UV cross-linking and immunoprecipitation (CLIP) experimental approach modified from Wang et al. (2009) protocol. Herein, we describe a step-by-step protocol to investigate the direct binding of a specific factor to its RNA target(s), which can be extended to most adherent human cell lines.
Materials and Reagents
100 mm dish
PDAC cells or other adherent cells
Cold PBS (Sigma-Aldrich, catalog number: D8537)
Liquid nitrogen
Turbo DNAse (Thermo Fisher Scientific, AmbionTM, catalog number: AM2239)
Protein G Dynabeads (Thermo Fisher Scientific, NovexTM, catalog number: 10004D)
RBP polyclonal hnRNP I antibody for immunoprecipitation (Santa Cruz Biotechnology, catalog number: sc-16547)
RNAse I (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM2295)
Bradford solution (Bio-Rad Laboratories, catalog number: 500-0006)
Phenol:chloroform:isoamyl alcohol 25:24:1 (Sigma-Aldrich, catalog number: P3803)
Ethanol (VWR, catalog number: 20821.321)
Trizol (Thermo Fisher Scientific, AmbionTM, catalog number: 15596018)
Chloroform (Honeywell International, Riedel-de Haen, catalog number: 32211)
Isopropanol (CARLO ERBA Reagents, catalog number: 415156)
Agarose (Lonza, catalog number: 50004)
Water (Sigma-Aldrich, catalog number: W4502)
cDNA synthesis kit (Promega, catalog number: M1705)
Tris (VWR, catalog number: 0826)
Sodium chloride (NaCl) (VWR, catalog number: 27810.364)
Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M2670)
Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C1016)
NP-40 (Alfa Aesar, Affymetrix/USB, catalog number: J19628)
Sodium deoxycholate (Sigma-Aldrich, catalog number: D6750)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771)
DL-dithiothreitol (DTT) (Sigma-Aldrich, catalog number: D9779)
Protease inhibitor cocktail (Sigma-Aldrich, catalog number: P8340)
RNase inhibitor (Promega, catalog number: N2511)
EDTA (Sigma-Aldrich, catalog number: E5134)
Glycerol (Sigma-Aldrich, catalog number: G9012)
β-mercaptoethanol (Sigma-Aldrich, catalog number: M3148)
Bromphenol blue (Sigma-Aldrich, catalog number: 114391)
Proteinase K (Roche Molecular Systems, catalog number: 3115836001)
DEPC (Sigma-Aldrich, catalog number: D5758)
XQuantitative Real-Time PCR Kit (LightCycler®480 SYBR Green I Master) (Roche Molecular Systems, catalog number: 04887352001)
Sodium acetate (Sigma-Aldrich, catalog number: S2889)
Sodium metavanadate (Sigma-Aldrich, catalog number: 72060)
Lysis buffer (see Recipes)
High-salt buffer (see Recipes)
3 M sodium acetate (pH 5.2) (see Recipes)
Laemmli buffer (2x) (see Recipes)
Proteinase K buffer (see Recipes)
Equipment
Centrifuge
UV crosslinker (Uvitec, model: CL 508)
SDS-PAGE and PVDF/nitrocellulose transfer apparatus (Bio-Rad Laboratories)
Denaturating agarose gel apparatus (Bio-Rad Laboratories)
Magnetic stand (Thermo Fisher Scientific, Invitrogen)
Thermoblock
Quantitative Real-Time PCR (Roche Molecular Systems, model: LightCycler® 480)
Sonicator (Hielscher ultrasonics, model: UP200S)
Procedure
-
Cell culture preparation and UV cross-linking
Grow adherent PDAC cells until they reach 70-80% confluence. Usually, 5-10 x 106 cells are used for each immunoprecipitation (IP).
Discard the medium and wash once with cold PBS. Add 4-5 ml of PBS in 100 mm cell culture Petri dish (~1 ml/cm2).
-
Place the dish on ice without lid. Irradiate once with 400 mJ/cm2 (see Figure 1).
Note: UV irradiation produces a covalent bound between RNA and protein that are in contact. The energy level to use depends on availability of aromatic acids. This covalent bound allows purification of RBP/RNA complex under stringent condition. The choice of UV irradiation should be set as the minimum irradiation that allows purification of a control RNA.
-
Remove the PBS and add 0.5 ml of lysis buffer into the 100 mm dish.
Note: Do not exceed with lysis buffer.
Alternatively, gently scrape off the cells in cold PBS. Centrifuge for 5 min at 300 × g, at 4 °C. Discard the supernatant, snap freeze the pellets in liquid nitrogen and store at -80 °C.
-
Cell lysis
-
Gently resuspend the cells in lysis buffer and transfer the suspended cells to a 1.5 ml centrifuge tube and sonicate on ice [Set: Amplitude (%) at 100 and Cycle at 1] (see Figure 2).
Note: Cells tend to aggregate in lysis buffer. Sonication is required to completely dissolve cells aggregate. Usually 5 sec is enough.
Incubate the cells for 10 min on ice.
Add 5 µl per ml of sample of Turbo DNAse. Incubate for 3 min at 37 °C.
Centrifuge at 15,000 × g at 4 °C for 3 min.
-
Collect the supernatant (cell extract), dilute sample to 1 mg/ml in lysis buffer and store in ice.
Note: Lysis buffer composition interferes with Bradford assay. No more than 1 µl of cell extract should be added to 1 ml of Bradford solution.
-
-
RNA fragmentation and RBP immunoprecipitation
For each IP use 10 µl of Protein G Dynabeads.
Wash Protein G Dynabeads with 500 µl of lysis buffer and mixing the suspension. Place the tube containing the suspension in the magnetic stand and remove the supernatant. Repeat this step 3 times.
Resuspend the Protein G Dynabeads in 100 µl of lysis buffer and add 3-5 µg of specific polyclonal antibody. Use IgG isotype as a negative control.
-
Collect 3 aliquots of 10% of cell extract (100 µg) (Input) before adding RNase I.
Note: Aliquots of inputs are required for (i) quantitative analysis of RBP binding (see steps D1a and F3), (ii) evaluation of RBP IP (see step C10a), and (iii) for evaluation of RNase I fragmentation (see step D3, and Figure 4).
Add 1 ml of cell extract (1 mg) to 100 µl Protein G Dynabeads (see step C2). Store on ice.
Prepare a dilution of RNase I 1:1,000 in lysis buffer.
Add to each IP 10 µl of RNase I 1:1,000.
Rotate at 4 °C for 2 h.
-
After incubation place the samples in the magnetic stand, collect supernatant aliquots (100 µl) from each IPs and discard the residual supernatant. Store aliquots on ice.
Note: Aliquots are required for evaluation of RNAse I fragmentation (see step D3, and Figure 4).
Wash IPs twice with 1 ml of high salt buffer and 2 times with 1 ml proteinase K buffer.
-
Resuspend the Dynabeads in 100 µl of proteinase K buffer. Collect an aliquot of 10% of suspension (10 µl) from each IP. Add 10 µl of Laemmli buffer (2x) to the aliquots and boil aliquot for 5-10 min at 100 °C (see step C10a).
Note: Aliquots are required for evaluation of RBP IP by Western-blot analysis (see C10a).
-
Control of RBPs IP
Run an SDS-PAGE following Western blot analysis using aliquots from step C2 (input) and aliquots from step C10 (IP) samples (Figure 3).
-
-
RNA isolation
-
From IPs:
RNA is eluted by adding 50 µg of proteinase K (2.5 µl of proteinase K solution) to 90 µl of the remaining IP suspensions (step C10).
Incubate for 1 h at 55 °C.
Place samples in the magnetic stand, collect the supernatants and add water up to 300 µl.
Add at each supernatant 1 volume of phenol:chloroform:isoamilic alcohol (25:24:1).
Mix vigorously for 10 sec. Incubate for 10 min at room temperature.
Centrifuge at 15,000 × g for 10 min. Collect the supernatant.
Add 1/10 volume of 3 M sodium acetate (pH 5.2).
Mix and add 2.5 volume of ethanol absolute.
Mix and incubate overnight at -20 °C.
After incubation, centrifuge at 15,000 × g for 15 min.
Discard the supernatant and wash the RNA pellet with 1 ml of 70% ethanol.
Centrifuge at 15,000 × g for 10 min.
Discard the supernatant and air dry the pellet.
-
From Input:
50 µg of proteinase K (2.5 µl of proteinase K solution) was added to one of 10% input aliquot (step C2).
Incubate for 1 h at 55 °C.
RNA is recovered by adding 1 ml Trizol to the samples. Incubate for 5 min at RT.
Add 200 µl of chloroform and mix vigorously.
Centrifuge at 12,000 × g for 15 min.
-
After centrifugation, collect half of aqueous phase (≈ 250 µl) to avoid DNA contamination.
Note: In our experience, treatment of RNA input sample with DNAse is not sufficient to remove DNA contamination. We prevent DNA contamination by collecting only half of the aqueous phase and avoiding to touch the interphase layer during RNA isolation with Trizol reagent.
Add 1 volume of 100% isopropanol. And incubate for 10 min at RT.
Centrifuge at 12,000 × g for 15 min and discard the supernatant.
Wash RNA pellet with 1 ml of 70% ethanol.
Centrifuge at 12,000 × g for 10 min.
Discard the supernatant and air dry the pellet.
-
Control of RNAse I fragmentation
Add 50 µg of proteinase K (2.5 µl of proteinase K solution) to one aliquot from step C2 and step C7.
Incubate for 1 h at 55 °C. Add 1 volume of phenol:chloroform:isoamilic alcohol.
Proceed as described in steps D1e-D1m.
Resuspend the RNA in 20 µl of nuclease-free water. After quantification at 260 nm, load the samples on 1.5% denaturating agarose gel for evaluation of RNA fragmentation (Figure 4).
-
-
Reverse transcription (cDNA synthesis)
-
Resuspend the RNA pellets from steps D1m and D2k in the same volume of nuclease free water.
Note: Volume of water depends on the cDNA synthesis kit used.
Heat for 5 min at 55 °C and store on ice.
-
Perform cDNA synthesis according to manufacturer’s instructions using half volume of RNA samples. Use the other half to perform cDNA synthesis reaction in absence of reverse transcriptase enzyme (negative control). cDNA synthesis has to be performed using random hexamer primers.
Note: Negative control is required to evaluate DNA contamination in all samples.
-
-
Real-time quantitative PCR (qRT-PCR)
-
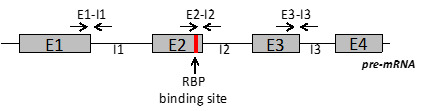
Design pairs of primer located in different regions of the pre-mRNA, including the region embedding the RBP binding site (Figure 5).
Notes:
Optimal PCR product length is 100 nt.
If RBP binding site is not known, it is possible to identify the region in which this is embedded by using several primers pair designed to span the entire pre-mRNA.
2. Perform qRT-PCR according to manufacturer’s instructions.
Note: Quantification cycle (Cq) of negative control should be at least 6 cycles less than relative sample (i.e., Cq negative control = 36; Cq sample = 30).
-
Figure 1. UV cross-linking.

UV cross-linking was performed to bind RNA covalently with proteins. During irradiation the dish is kept on ice to minimize heating.
Figure 2. Cells sonication.

Sonication was performed to dissolve cells aggregate. During sonication the tube is kept in ice to minimize protein denaturation.
Figure 4. Control of RNA fragmentation.
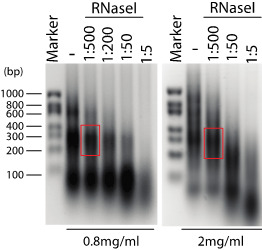
Denaturating agarose gel (1.5%) showing RNA fragmentation in presence of different RNAse I dilutions (1:500 corresponds to 2 U/ml of RNase I), and in presence of different extract concentration (0.8 and 2 mg/ml, left and right panel respectively). The optimal RNA fragmentation is indicated (red box).
Figure 3. Control of RBP immunoprecipitation in HEK293T cells.
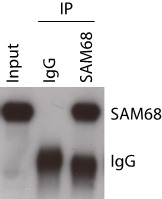
Western blot analysis of aliquotes (10%) from step C2 (Input) and step C10 (IPs).
Figure 5. Schematic representation of primers position along the pre-mRNA for evaluation of RBP binding.
Data analysis
Binding of RBP is reported as % of Input in different regions of the RNA target, using the comparative Cq method as follow (Figure 6).
Figure 6. Data analysis of RBP association to RNA target.
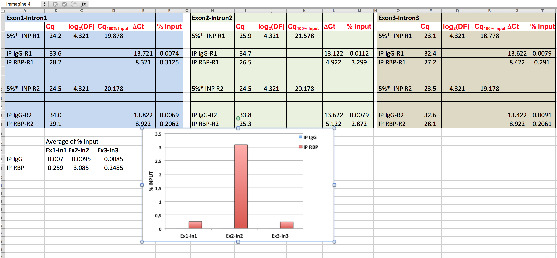
Using hypothetical Cq values, data analysis of RBP binding to the indicated pre-mRNA regions (Exon1-Intron1; Exon2-Intron2; Exon3-Intron3) is shown. * = % of Input is 5% see steps C2 and D2f; DF (dilution factor) = 20; R = replicate.
-
Cq of Input (Cq5% input; 5%; see steps C2 and D2f) need to be adjusted to 100% (Cq100% Input) according to dilution: Cq100% Input = Cq5% input - [log2(DF)]
Note: 5% input (see steps C2 and D2f) correspond to a dilution factor (DF) of 20. Moreover, Thus, Cq100% Input = Cq5% input - log2(10).
-
Cq value of IPs (CqIP) is compared (Cq) to Cq value of 100% of Input (Cq100% input):
Cq = CqIP - Cq100% input
Note: For IgG IP: CqIgG = CqIgG IP - Cq100% input. For RBP IP: CqRBP = CqRBP IP - Cq100% input.
-
Association of RBP is reported as % input: % input = 100 x 2-Cq
Note: For IgG: % InputIgG = 100 x 2- CqIgG. For RBP: % InputRBP = 100 x 2- CqRBP
Notes
To ensure reproducibility, biological experiments are usually performed in replicates (triplicate). We also recommend to perform each CLIP IPs in duplicate (technical duplicates) to increase the reliability of each experiment (Figure 7). Statistical analysis is performed by t-test procedure.
Figure 7. Data analysis of RBP association to RNA target from three biological replicate experiments.

Using hypothetical values, data analysis of RBP binding to pre-mRNA from three biological experiments is shown. By performing technical duplicates for each experiment is possible to eliminate value(s) that deviate from the others. In this example, the value highlighted in red clearly deviates from the other five. Thus, it is conceivable to eliminate it from further analysis, as it may result from a mistake.
Recipes
Note: Make sure to prepare all the following solution in DEPC-treated water.
-
Lysis buffer
50 mM Tris-HCl (pH 7.4)
100 mM NaCl
1 M MgCl2
0.1 mM CaCl2
1% NP-40
0.5% sodium deoxycholate
0.1% SDS
Supplemented with fresh 1 mM DTT, 0.5 mM sodium metavanadate, protease inhibitor cocktail, RNase inhibitor (1 µl/ml)
-
High-salt buffer
50 mM Tris-HCl (pH 7.4)
1 M NaCl
1 mM EDTA
1% NP-40
0.5% sodium deoxycholate
0.1% SDS
Supplemented with fresh RNase inhibitor (0.5 µl/ml)
-
Proteinase K buffer
100 mM Tris-HCl (pH 7.4)
50 mM NaCl
10 mM EDTA
Supplemented with fresh RNase inhibitor (0.5 µl/ml)
-
3 M sodium acetate (pH 5.2)
For preparing this solution, please refer to ( Iglesias et al., 2017 , http://en.bio-protocol.org/e2180)
-
Leammli buffer (2x)
0.125 M Tris-HCl, pH 6.8
20% glycerol
4% SDS
2% β-mercaptoethanol
0.02% bromphenol blue
-
Proteinase K solution (20 mg/ml)
Dissolve 20 mg of lyophilized proteinase K in 1 ml of double distilled water
Acknowledgments
The CLIP method presented herein is a modified protocol from Wang et al. (2009). This method was employed to identify the binding sites of PTBP1 on the PKM pre-mRNA in Calabretta et al. (2016). The research in our laboratory was supported by the Associazione Italiana Ricerca sul Cancro (AIRC; IG18790), by Telethon Foundation (GGP14095) and by Italian Ministry of Health ‘Ricerca Finalizzata 2011’ (GR-2011-02348423) and ‘5x1000 Anno 2014’ to Fondazione Santa Lucia.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Anczuków O., Rosenberg A. Z., Akerman M., Das S., Zhan L., Karni R., Muthuswamy S. K. and Krainer A. R.(2012). The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19(2): 220-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielli P., Busa R., Di Stasi S. M., Munoz M. J., Botti F., Kornblihtt A. R. and Sette C.(2014). The transcription factor FBI-1 inhibits SAM68-mediated BCL-X alternative splicing and apoptosis. EMBO Rep 15(4): 419-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black D. L.(2003). Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291-336. [DOI] [PubMed] [Google Scholar]
- 4.Byron S. A., Van Keuren-Jensen K. R., Engelthaler D. M., Carpten J. D. and Craig D. W.(2016). Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat Rev Genet 17(5): 257-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calabretta S., Bielli P., Passacantilli I., Pilozzi E., Fendrich V., Capurso G., Fave G. D. and Sette C.(2016). Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells . Oncogene 35(16): 2031-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cappellari M., Bielli P., Paronetto M. P., Ciccosanti F., Fimia G. M., Saarikettu J., Silvennoinen O. and Sette C.(2014). The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 33(29): 3794-3802. [DOI] [PubMed] [Google Scholar]
- 7.Chen J. and Weiss W. A.(2015). Alternative splicing in cancer: implications for biology and therapy. Oncogene 34(1): 1-14. [DOI] [PubMed] [Google Scholar]
- 8.Chen M. and Manley J. L.(2009). Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10(11): 741-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper T. A., Wan L. and Dreyfuss G.(2009). RNA and disease. Cell 136(4): 777-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danan-Gotthold M., Golan-Gerstl R., Eisenberg E., Meir K., Karni R. and Levanon E. Y.(2015). Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 43(10): 5130-5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.David C. J., Chen M., Assanah M., Canoll P. and Manley J. L.(2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463(7279): 364-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreyfuss G., Kim V. N. and Kataoka N.(2002). Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3(3): 195-205. [DOI] [PubMed] [Google Scholar]
- 13.Fu X. D. and Ares M., Jr (2014). Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15(10): 689-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iglesias R., Citores L. and Ferreras J. M.(2017). Ribosomal RNA N-glycosylase activity assay of ribosome-inactivating proteins. Bio-protocol 7(6): e2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin K. C. and Ephrussi A.(2009). mRNA localization: gene expression in the spatial dimension. Cell 136(4): 719-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayr C. and Bartel D. P.(2009). Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138(4): 673-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naro C. and Sette C.(2013). Phosphorylation-mediated regulation of alternative splicing in cancer. Int J Cell Biol 2013: 151839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olshavsky N. A., Comstock C. E., Schiewer M. J., Augello M. A., Hyslop T., Sette C., Zhang J., Parysek L. M. and Knudsen K. E.(2010). Identification of ASF/SF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res 70(10): 3975-3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pagliarini V., Naro C. and Sette C.(2015). Splicing regulation: A molecular device to enhance cancer cell adaptation. Biomed Res Int 2015: 543067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paronetto M. P., Cappellari M., Busa R., Pedrotti S., Vitali R., Comstock C., Hyslop T., Knudsen K. E. and Sette C.(2010). Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer Res 70(1): 229-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Proudfoot N. J.(2016). Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science 352(6291): aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silvera D., Formenti S. C. and Schneider R. J.(2010). Translational control in cancer. Nat Rev Cancer 10(4): 254-266. [DOI] [PubMed] [Google Scholar]
- 23.Singh G., Pratt G., Yeo G. W. and Moore M. J.(2015). The clothes make the mRNA: Past and present trends in mRNPfashion. Annu Rev Biochem 84: 325-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian B. and Manley J. L.(2016). Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valacca C., Bonomi S., Buratti E., Pedrotti S., Baralle F. E., Sette C., Ghigna C. and Biamonti G.(2010). Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 191(1):87-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z. and Burge C. B.(2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14(5): 802-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z., Tollervey J., Briese M., Turner D. and Ule J.(2009). CLIP: construction of cDNA libraries for high-throughput sequencing from RNAs cross-linked to proteins in vivo . Methods 48(3): 287-293. [DOI] [PubMed] [Google Scholar]