

Abstract
The molecular mechanisms by which the endothelial barrier becomes compromised during lipopolysaccharide (LPS) mediated acute lung injury (ALI) are still unresolved. We have previously reported that the disruption of the endothelial barrier is due, at least in part, to the uncoupling of endothelial nitric oxide synthase (eNOS) and increased peroxynitrite-mediated nitration of RhoA. The purpose of this study was to elucidate the molecular mechanisms by which LPS induces eNOS uncoupling during ALI. Exposure of pulmonary endothelial cells (PAEC) to LPS increased pp60Src activity and this correlated with an increase in nitric oxide (NO) production, but also an increase in NOS derived superoxide, peroxynitrite formation and 3-nitrotyrosine (3-NT) levels. These effects could be simulated by the over-expression of a constitutively active pp60Src (Y527FSrc) mutant and attenuated by over-expression of dominant negative pp60Src mutant or reducing pp60Src expression. LPS induces both RhoA nitration and endothelial barrier disruption and these events were attenuated when pp60Src expression was reduced. Endothelial NOS uncoupling correlated with an increase in the levels of asymmetric dimethylarginine (ADMA) in both LPS exposed and Y527FSrc over-expressing PAEC. The effects in PAEC were also recapitulated when we transiently over-expressed Y527FSrc in the mouse lung. Finally, we found that the pp60-Src-mediated decrease in DDAH activity was mediated by the phosphorylation of DDAH II at Y207 and that a Y207F mutant DDAH II was resistant to pp60Src-mediated inhibition. We conclude that pp60Src can directly inhibit DDAH II and this is involved in the increased ADMA levels that enhance eNOS uncoupling during the development of ALI.
Keywords: Signal transduction, eNOS uncoupling, Acute lung injury
1. Introduction
In acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) the integrity of the separation between the alveolus and the pulmonary circulation is compromised either by endothelial or epithelial injury or more commonly both. This damage leads to increased vascular permeability, alveolar flooding, and surfactant abnormalities [1]. ALI/ARDS can occur in response to a number of insults that either directly, or indirectly, produce lung injury. The most common indirect insult leading to ALI is the release of lipopolysaccharide (LPS) from the outer bacterial cell wall producing gram negative sepsis [2]. However, despite significant investigations, the mechanisms underlying the development of ALI/ARDS are still unresolved and the therapies are predominantly supportive.
Recent studies have suggested that endothelial injury in a variety of cardiovascular diseases is linked to increased levels of the amino acid, asymmetric dimethylarginine (ADMA) [3–7]. ADMA is derived from hydrolysis of methylated proteins [3–7]. The synthesis and degradation of ADMA can become dysregulated and can result in increased levels of ADMA [3–7]. ADMA is metabolized via hydrolytic degradation to L-citrulline and dimethylamine by the enzyme dimethylarginine dimethy-laminohydrolase (DDAH) [7]. There are two isoforms of DDAH: I and II [8,9]. The important isoform in the endothelium is thought to be DDAH II [6,10]. We have previously shown that during the development of LPS-induced ALI there is a significant increase in ADMA in the mouse lung, and this is due to decreased activity, but not expression, of DDAH II [11]. Further, increasing the expression of DDAH II in the pulmonary endothelium of the lung reduces the development of ALI in the LPS-exposed mouse [12]. ADMA increases superoxide generation from eNOS and higher levels of peroxynitrite and protein nitration [11–13]. Peroxynitrite appears to play a role in the pathogenesis of LPS-lung injury and elevated levels of 3-NT have been identified in the lung and BAL fluid in ALI [11,14,15]. The importance of eNOS and protein nitration in the development of ALI has also been shown by the protection afforded eNOS knockout mice that correlated with a reduction in the nitration-mediated activation of RhoA [16]. RhoA and Rho-associated kinase directly catalyze myosin light chain (MLC) phosphorylation, or act indirectly via inactivation of MLC, phosphatase to induce cell contraction and endothelial barrier disruption [17,18].
Despite data demonstrating the involvement of the DDAH/ADMA axis in the development of ALI in the mouse lung, the mechanism by which LPS attenuates DDAH activity is unresolved. One of the earliest effects of LPS binding to its receptor, TLR4 is the activation of the subclass A of the Src Family Kinases (SFK), pp60Src. LPS-mediated activation of pp60Src increases endothelial paracellular permeability [19]. In addition, pp60Src inhibition attenuates LPS-induced endothe-lial barrier disruption [20]. We have previously shown a link between ADMA, eNOS uncoupling, and the nitration-mediated activation of RhoA [21] while prior studies have shown that pp60Src can activate RhoA [22]. However, pp60Src has also been shown to stimulate eNOS activity through an Akt1-mediated phosphorylation at Ser1177 [23]. Ser1177 phosphorylation is thought to stimulate eNOS catalytic activity by increasing electron flow through the protein [24]. Based on these two opposing facts we developed a hypothesis that pp60Src signaling induces eNOS uncoupling by both increasing ADMA levels, through the inhibition of DDAH II, and stimulating eNOS catalytic cycling through Ser1177 phosphorylation. Our data indicate that pp60Src does inhibit DDAH II activity through a single phosphorylation at Y207 and that this increases cellular ADMA levels and eNOS derived superoxide. In addition, the phosphorylation of eNOS at Ser1177 is stimulated and NO levels also increase, leading to an increase in peroxynitrite generation and protein nitration. Thus, the role of pp60Src in protein nitration involved in the development of ALI requires both the activation of eNOS and its uncoupling.
2. Materials and methods
2.1. Cell culture
Primary cultures of ovine PAEC were isolated as described previously [25]. Cells were maintained in DMEM containing phenol red supplemented with 10% fetal calf serum (Hyclone, Logan, UT), antibiotics, and antimycotics (MediaTech, Herndon, VA) at 37 °C in a humidified atmosphere with 5% CO2–95% air. Cells were utilized between passages 8 and 12. Between 7 and 8 isolates of PAEC were used to carry out the experimental procedures.
2.2. Animal studies
Adult male C57BL/6NHsd mice (7–8 weeks; Harlan, Indianapolis, IN) were used in all experiments. All animal care and experimental procedures were approved by the Committee on Animal Use in Research and Education of the Augusta University. Mice were injected intraperitoneally with Escherichia coli 0111:B4 lipopolysaccharide (LPS; 6.75×104 EU/gm body wt, Sigma-Aldrich, St. Louis, MO) prepared in 0.9% saline. The control mice received vehicle (0.9% saline), as previously described [11]. Mice were euthanized 12 h after LPS injection, and the lungs were flushed with ice-cold EDTA-PBS, excised, snap-frozen in liquid nitrogen, and stored at −80 °C until used.
2.3. In vivo over-expression of constitutively active pp60Src
In vivo, polyethyleneimine derivative transfection reagent (in vivo-jetPEI) was used to deliver the plasmids, pAd/CMV/V5-DEST-Y527FSrc cDNA or pDST-luciferase, to the mouse lung endothelium as described previously [12]. Briefly, 40 μg of each plasmid were incubated with glucose and the jetPEI reagent (Polyplus-transfection Inc, New York, NY), as per manufacture's instruction for 15–30 min following which the cDNA-jetPEI complexes were injected into the tail vein.
2.4. Measurement of peroxynitrite and protein nitration levels
The formation of peroxynitrite was determined by the peroxynitrite dependent oxidation of dihydrorhodamine (DHR) 123 to rhodamine 123 in the presence of PEG-catalase (100U, 30 min), as described previously [12]. Protein nitration was measured via a dot blot procedure, as previously described [12].
2.5. DDAH activity assay
Total DDAH activity was determined using a radioactive assay to measure the conversion of L-[3H]-NMMA to [3H]-L-citrulline. Briefly, 20 mg of tissue samples in 125 μl of ice cold 0.1 M sodium phosphate buffer (SPB, pH 6.5) were sonicated and centrifuged at 10,000g for 10 min at 4 °C. Similarly, ovine PAEC grown on 10 cm dishes were harvested in 1 ml of ice cold SPB and centrifuged at 10,000g for 10 min at 4 °C. The supernatant was removed and 125 μl of ice cold fresh SPB was added to the pellet followed by sonication and centrifugation at 10,000g for 10 min at 4 °C. The homogenates were analyzed in duplicate (50 μl), while the remainder was used for protein concentration using the BCA protein assay. To the supernatant, a reaction mixture was added containing 0.1 M SPB and 0.1 μCi/ml of L-[3H]-NMMA (specific activity: 1.48–2.96 TBq/mmol) (PerkinElmer, Santa Clara, CA) in a final volume of 100 μl and incubated for 1 h at 37 °C. The reaction was terminated by placing the tubes on ice for 5 min and diluting the reaction with 2 ml of ice cold SPB. The samples were then passed through 1 ml of activated Dowex AG50W-8X cation exchange resin to remove un-metabolized L-[3H]-NMMA followed by a rinse with 1 ml SPB. The eluted fractions were mixed with 10 ml of scintillation fluid (ScintiVerse BD Cocktail, Fisher Scientific, Pittsburgh, PA) and quantified using a liquid scintillation counter. A reaction mixture containing L-[3H]-NMMA in the absence of enzyme was added to the Dowex column to determine background counts. DDAH activity is defined as the amount of L-[3H]-NMMA degraded per hour per mg protein.
2.6. Measurement of ADMA levels
ADMA levels were analyzed by high-performance liquid chromato-graphy (HPLC) as published previously [11,12]. The crude fraction of cell lysate or lung lysate were isolated using a solid phase extraction column and subsequently, ADMA was separated using pre-column derivatization with ortho-phthaldialdehyde (OPA) reagent (4.5 mg/mL in borate buffer, pH 8.5, containing 3.3 μl/mL β-mercaptoethanol) prior to injection. HPLC was performed using a Shimadzu UFLC system with a Nucleosil phenyl reverse phase column (4.6×250 mm; Supelco, Bellefonte, PA), equipped with an RF-10AXL fluorescence detector (Shimadzu USA Manufacturing Corporation). ADMA levels were quantified by fluorescence detection at 450 nm (emission) and 340 nm (excitation). Mobile phase A was composed of 95% potassium phosphate (50 mM, pH 6.6), 5% methanol and mobile phase B was composed of 100% methanol. ADMA was separated using a pre-gradient wash of 25% mobile phase B (flow rate 0.8 mL/min), followed by a linear increase in mobile phase B concentration from 20% to 25% over 7 min followed by a constant flow at 25% for 10 min and another linear increase from 25% to 27% mobile phase B over 5 min followed by constant flow at 27% mobile phase B for another 7 min. Retention time for ADMA was approximately 28 min. ADMA concentrations were calculated using standards and an internal homoarginine standard. The detection limit of the assay was 0.1 μmol/L.
2.7. Expression and purification of endothelial NOS
Wildtype human eNOS and an S1177D eNOS mutant were expressed and purified from E.coli as previously described [26,27]. NO and superoxide generation from these proteins was determined as previously described [28].
2.8. Detection of NOX
Nitric oxide (NO) generation were measured in the media collected from PAEC over-expressing dominant negative (K295M) or constitu-tively active (Y527F) pp60Src in response to LPS using an NO-sensitive electrode with a 2-mm diameter tip (ISO-NOP sensor, WPI) connected to an NO meter (ISO-NO Mark II, WPI) as described previously [29]. NO levels in the lung lysate were determined by indirect method of measuring the levels of nitrite as previously described [30]. Briefly, samples were deproteinized by adding cold ethanol to the sample (1:4 v:v) and then concentrated using a speed-vac. Potassium iodide/ acetic acid reagent was prepared fresh daily. This reagent was added to a septum sealed purge vessel and bubbled with nitrogen gas. The gas stream was connected via a trap containing 1N NaOH, to a Sievers 280i Nitric Oxide Analyzer (GE). Deproteinized samples were injected with a syringe through a silicone/Teflon septum. Results were analyzed by measuring the area under curve of the chemiluminescence signal using the Liquid software (GE).
2.9. Determination of superoxide levels
NOS-derived superoxide levels were estimated by electronic paramagnetic resonance (EPR) assay using the spin-trap compound 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine HCl (CMH , Axxora LLC, Farmingdale, NY) in the presence of ethylisothiourea (ETU, 100 μm, 30 min, Sigma-Aldrich), as previously described [11].
2.10. Generation of a specific DDAH II pY207 antibody
The DDAH2 phospho-tyrosine Y207 specific antibody was raised against a synthetic peptide antigen (MAV LTD HPY(PO4) ASL TLP DDA), where Y(PO4) represent phosphotyrosine. The peptide was used to immunize rabbits. DDAH2 tyrosine phosphorylation-reactive rabbit antiserum was first purified by affinity chromatography. Further purification was carried out using immunodepletion using the non-phosphorylated peptide, MAVLTDHPYASLTLPDDA resin chromato-graphy, after which the resulting eluate was tested for antibody specificity by ELISA, and immunoblotting. Relative phosphorylation was determined by stripping and reprobing blots with an antibody specific for DDAH II.
2.11. Western blot analysis
PAEC were treated with LPS for 4 h and solubilized with a lysis buffer containing 1% Triton X-100, 20 mM Tris. pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail (Pierce). Insoluble proteins were precipitated by centrifugation at 13,000 rpm for 10 min at 4 °C, and the supernatants were then subjected to SDS-PAGE on 4–20% polyacrylamide gels and transferred to a PVDF membrane (Biorad). The membranes were blocked with 5% nonfat dry milk or 5% BSA in Tris-buffered saline containing 0.1% Tween (TBST). The primary antibodies used for immunoblotting were anti-pp60Src Cell Signaling (1:1000), (Danvers, MA), anti-phospho Y416 pp60Src (1:1000; Cell Signaling Technology), β-actin (1:5000, Sigma), phospho-Ser1177 eNOS, and anti-eNOS (1:1000, BD Biosciences) and custom made DDAH II (1:500) and 3NT-Y34-RhoA (1:1000) [16] antibodies. Membranes were then washed with TBST three times for 10 min, incubated with the appropriate secondary antibody coupled to horseradish peroxidase, washed again with TBST as described above, and the protein bands visualized with ECL reagent (Pierce) using a Kodak 440CF image station.
2.12. Adenoviral-mediated overexpression of pp60Src mutants
Constitutively active (Y527F) and dominant negative (K295M) pp60Src mutants were overexpressed using an adenoviral construct as we have previously described [31]. For these studies, an MOI of 200:1 was used. An adenoviral construct expressing green fluorescent protein (GFP) was used as a transduction control.
2.13. siRNA-mediated down-regulation of pp60Src
PAEC were transfected with the appropriate small interfering RNA (siRNA) using HiPerFect transfection reagent (Qiagen, Valencia, CA) as described previously [32]. Briefly, the day before transfection, 1.5×105 cells were seeded in each well of a six-well plate and fresh DMEM containing serum and antibiotics added. On the day of transfection, the medium was changed to one without antibiotics. For each well, 6 μl of a 10-μM siRNA stock of pp60Src (Santa Cruz) or the control (a scrambled siRNA with no known homology to any human gene) was diluted into 100 μl of DMEM without serum (to give a final siRNA concentration of 30 nM). To this was added 12 μl of HiPerFect transfection reagent. The solution was vortexed, incubated for 10 min at room temperature, and added drop-wise to the cells. As the siRNA's utilized were designed against human mRNA sequences their ability to silence pp60Src was validated by Western blot analysis 48 h after transfection (Fig. 2A). The cell culture medium was changed for fresh complete DMEM. LPS was then added at final concentration 500 EU/ml, the cells were lysed 2– 4 h later, and the lysates were analyzed by immunoblotting.
Fig. 2.
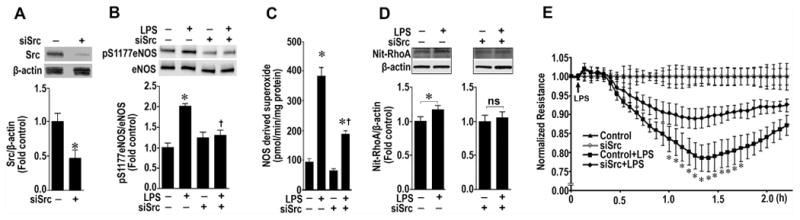
Decreasing pp60Src expression attenuated RhoA nitration and preserves the endothelial barrier. PAEC were transiently transfected with a siRNA for pp60Src or a control siRNA for 48 h to decrease pp60Src expression (A). Cells were then exposed to LPS (500 EU/ml) for 2–4 h. Whole-cell lysates (20 μg) were prepared, and pS1177eNOS levels determined by immunoblotting with anti-pS1177eNOS. Loading was normalized by re-probing the membranes with an antibody specific to total eNOS. The depletion of pp60Src significantly reduces the LPS-mediated p-S1177 eNOS levels (B). Decreasing pp60Src expression also attenuated the LPS mediated increase in NOS-derived superoxide, as estimated using EPR (C). LPS also increased the nitration of RhoA as determined by Western blot analysis using an antibody that specifically recognizes the nitration of Y34 in RhoA (Nit-RhoA, D). Again the depletion of pp60Src significantly reduces the LPS-mediated nitration of RhoA (D). Loading was normalized by re-probing the membranes with an antibody specific to β-actin. Depleting pp60Src also attenuates the barrier disruption associated with LPS (50 EU/ml, E). Values are mean ± SEM, n=4–6, *P < 0.05 vs. scr siRNA; †P < 0.05 vs. LPS+Scr siRNA.
2.14. Immunoprecipitation analysis
For each immunoprecipitation, cell lysates or tissue lysates were subjected to a pre-clearing step in which the lysates were incubated with a protein G Plus/Protein A agarose suspension (Calbiochem) for 30 min at 4 °C. The agrose beads were then pelleted and the lysate removed to a fresh tube and incubated with anti-DDAH II or pp60Src antibodies overnight at 4 °C followed by the addition of the protein G Plus/Protein A agarose suspension for 1 h at 4 °C. The immune complexes were washed three times with the lysis buffer and boiled in SDS-PAGE sample buffer for 5 min. Agarose beads were pelleted by centrifugation, and the protein supernatants were loaded and run on 4–20% polyacrylamide gels, followed by transfer of the proteins to nitrocellulose membranes. The membranes were blocked with 2% BSA in Tris-buffered saline containing 0.05% Tween 20 (TBST) for 2 h at room temperature, incubated with anti-phosphotyrosine antibody (Calbiochem) for 2 h at room temperature, washed three times with TBST (room temperature, 10 min), and then incubated with a horseradish peroxidase-conjugated secondary antibody (Pierce). The reactive bands were visualized with the SuperSignal West Femto maximum sensitivity substrate kit (Pierce) using a Kodak 440CF image station. The same blot was reprobed with anti-DDAH II or pp60Src antibody to normalize for the levels of catalase immunoprecipitated in each sample.
2.15. Measurement of endothelial barrier function
The integrity of PAEC monolayers was characterized using an electrical cell-substrate impedance sensing (ECIS) instrument (Applied BioPhysics, Troy, NY) as previously described [21]. Cells were plated in 8-well ECIS arrays (Applied BioPhysics) in DMEM supplemented with 10%-FBS (50,000 cells per well). Eighteen hours later, cells were transiently transfected with control or c-Src-specific siRNA's using HiPerFect (Qiagen) transfection reagent. Sixty hours post-transfection, the cell culture medium was changed for fresh complete DMEM, and the cells were used in Transendothelial Electrical Resistance (TER) assay. Initial resistance at the onset of our experiments was 1400–1700 in array wells, and then all wells were normalized to 1. 4000-Hz AC signal with 1-V amplitude was applied to the EC monolayers through a 1-M-Ω resistor, creating an approximate constant-current source (1 μA). After a baseline measurement, the cells were treated with E. coli LPS (50 EU/ml) or vehicle. Changes in TER were recorded in real time. Control curves (LPS-untreated cells) were normalized to 1 in order to eliminate any temporal effects. To take into account the deviations from control curves, data obtained from LPS-treated cells were normalized using the original control curves to adjust for the temporal effects that may be present in controls.
2.16. Statistical analysis
Statistical calculations were performed using the GraphPad Prism V.4.01 software. The mean ± SE was calculated for all samples, and significance was determined by either the unpaired or paired t-test or ANOVA. For ANOVA, Newman–Keuls post hoc testing was also utilized. A value of P < 0.05 was considered significant.
3. Results
3.1. Effect of pp60Src on NO signaling in pulmonary arterial endothelial cells
The exposure of PAEC to LPS for 4 h significantly increased pp60Src activity as measured by increase in its phosphorylation at Y416 (Fig. 1A). LPS also increased nitric oxide (NO) generation in PAEC, which was attenuated by the over-expression of a dominant negative pp60Src mutant (AdK295MSrc, Fig. 1B). The increase in NO generation correlated with an LPS-mediated increase in the phosphorylation of eNOS at Ser 1177, which was suppressed by overexpression of AdK295MSrc (Fig. 1C). The increase in pSer1177 appeared to be Akt-dependent as LPS-induced the phosphorylation of Akt at Ser473, indicative of Akt activation and this was blocked by AdK295MSrc over-expression (Fig. 1D). However, LPS also increased eNOS-derived superoxide suggesting that the enzyme was becoming uncoupled (Fig. 1E). Again, AdK295MSrc over-expression attenuated the LPS mediated increased in NOS-derived superoxide (Fig. 1E). These changes in NO and superoxide led to an increase in cellular peroxyni-trite levels (Fig. 1F) and total protein nitration (Fig. 1G) which were reduced by the over-expression of AdK295MSrc (Fig. 1F and G). When pp60Src expression was attenuated using an siRNA approach (Fig. 2A) this reduced both the LPS-mediated increase in pS1177eNOS (Fig. 2B) and NOS-derived superoxide levels (Fig. 2C). Silencing pp60Src expression also prevented the LPS-mediated increase in RhoA nitration (Fig. 2D) and attenuated the disruption of the endothelial barrier (Fig. 2E).
Fig. 1.
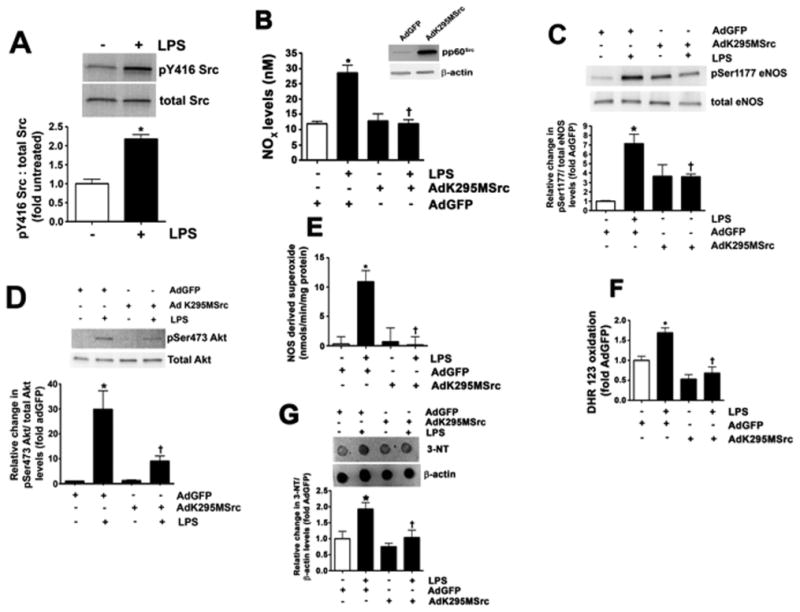
The over-expression of a dominant negative pp60Src stimulates eNOS uncoupling in pulmonary arterial endothelial cells. PAEC were exposed to LPS (1000 EU/ml; 4 h). LPS increased pp60Src activity as measured by increase in its phosphorylation at Y416 (A). PAEC were also transduced with an adenovirus containing either a dominant negative pp60Src (AdK295MSrc) or GFP. After 48 h the cells were challenged with LPS (1000 EU/ml; 4 h). LPS increased NO generation (B) and this correlated with an increase in p-Ser1177 eNOS (C) mediated via Akt1 as indicated by increased p-Ser473 Akt1 levels (D). All these effects were attenuated by AdK295MSrc over-expression (insert, panel B). AdK295MSrc also attenuated the LPS mediated increase in NOS-derived superoxide as estimated using EPR (E). LPS also increased peroxynitrite generation (F) and 3-nitrotyrosine (3-NT) levels (G). These effects were again attenuated by overexpression of AdK295MSrc. Values are mean ± SEM, n=3–6, *P < 0.05 vs. untreated; †P < 0.05 vs. LPS.
3.2. Stimulating pp60Src activity in the mouse lung induces eNOS uncoupling
To verify in vivo our data in PAEC, we transiently over-expressed a constitutively active pp60Src mutant (Y527FSrc) mutant in the mouse lung endothelium using the jet-PEI technique we have recently described [12]. This resulted in a ∼2-fold increase in pp60Src protein levels (Fig. 3A) and a significant increase in lung pp60Src activity as determined by increased pY416Src levels (Fig. 3B). The increase in pp60Src activity resulted in a significant increase in NO levels (Fig. 3C) and p-Ser1177 eNOS (Fig. 3D). Similar to PAEC, not only did Y527FSrc over-expression in the mouse lung increase NO levels it also appeared to cause an increased in uncoupling of eNOS as indicated by an increase in NOS-derived superoxide (Fig. 3E). As with PAEC, this increase in NO and superoxide resulted in increased peroxynitrite levels (Fig. 3F) and protein nitration (Fig. 3G).
Fig. 3.
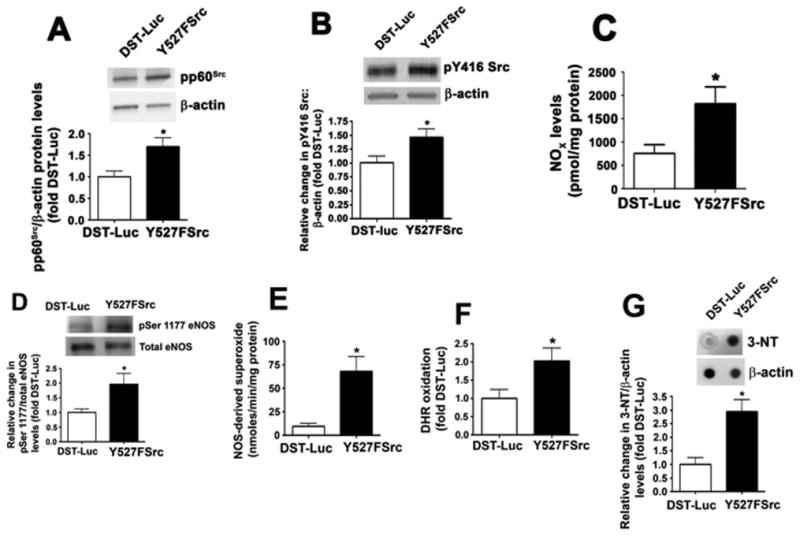
Enhancing pp60Src activity in the mouse lung induces eNOS uncoupling. Mice were injected with either DST-luciferase (DST) or pAD/CMV/V5-DEST-CApp60Src (Y527FSrc) plasmids via a tail vein injection. After 72 h, the animals were sacrificed and peripheral lung lysates prepared. Mice injected with Y527FSrc had a significant increase in pp60Src protein levels (A) and increase in pp60Src activity, as determined by increase in phospho-Y416 Src (B). The increase in pp60Src activity resulted in an increase in NO generation (C) and p-Ser1177 eNOS levels (D). Over-expression of Y527FSrc in the mouse lung also significantly increased the levels NOS-derived superoxide (E), peroxynitrite (F), and 3-NT protein (G). Values are mean ± SEM, n=4–16, *P<0.05 vs. DST.
3.3. An S1177D eNOS mutant protein is uncoupled
To further explore the role of eNOS phosphorylation at S1177 in regulating NO and superoxide generation we utilized purified wildtype and a phospho-mimic S1177D eNOS mutant. NO and superoxide generation was monitored under conditions that stimulate maximal velocity of the enzyme [28]. The S1177D mutant generates significantly more NO than wildtype eNOS (Fig. 4A). However, this mutant also produces significantly more superoxide as well (Fig. 4B) and overall the coupling index, determined by dividing the levels of NO by the amount of superoxide generated in each protein, is actually lower in the S1177D mutant. This suggests that the activation of eNOS by phos-phorylation at S1177 leads to increased catalytic cycling but also enhances eNOS uncoupling.
Fig. 4.
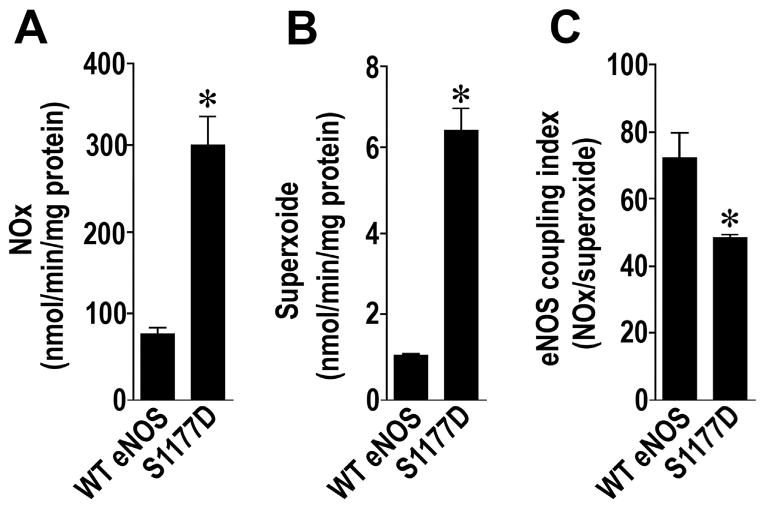
An S1177D eNOS mutant is catalytically more active but uncoupled. NO (A) and superoxide (B) generation are both significantly higher in the p-S1177 eNOS mimic protein, S1177D eNOS compared to the wildtype enzyme. The eNOS coupling index, determined as the ratio between NO and superoxide generation, was significantly decreased in the S1177D eNOS mutant suggesting the protein is more uncoupled (C). Values are mean ± SEM, n=3, *P < 0.05 vs. wildtype eNOS.
3.4. LPS increases ADMA levels by attenuating DDAH activity
We have previously shown that LPS increases the endogenous eNOS uncoupler, ADMA in the mouse lung by attenuating DDAH activity [13]. As in the mouse lung [11], LPS did not change DDAH II protein levels in PAEC either in the absence or presence of AdK295MSrc (Fig. 5A). However, LPS did significantly reduce DDAH activity (Fig. 5B) and increase cellular ADMA levels (Fig. 5C). These changes were attenuated by the over-expression of AdK295MSrc implicating pp60Src in the mechanism by which LPS inhibits DDAH activity. To further investigate the mechanism by which pp60Src inhibits DDAH activity we treated PAEC with LPS and used immuno-precipitation analyses to determine if there was a specific interaction of pp60Src with DDAH II, the predominant endothelial isoform [6,10]. Our data indicate that in PAEC, LPS stimulates the interaction of pp60Src with DDAH II (Fig. 6A) and this correlates with an increase in pY-DDAH II (Fig. 6B). Similarly, in the LPS exposed mouse lung, there is a significant increase in the interaction of pp60Src with DDAH II (Fig. 6C) and pYDDAH II levels (Fig. 6D).
Fig. 5.
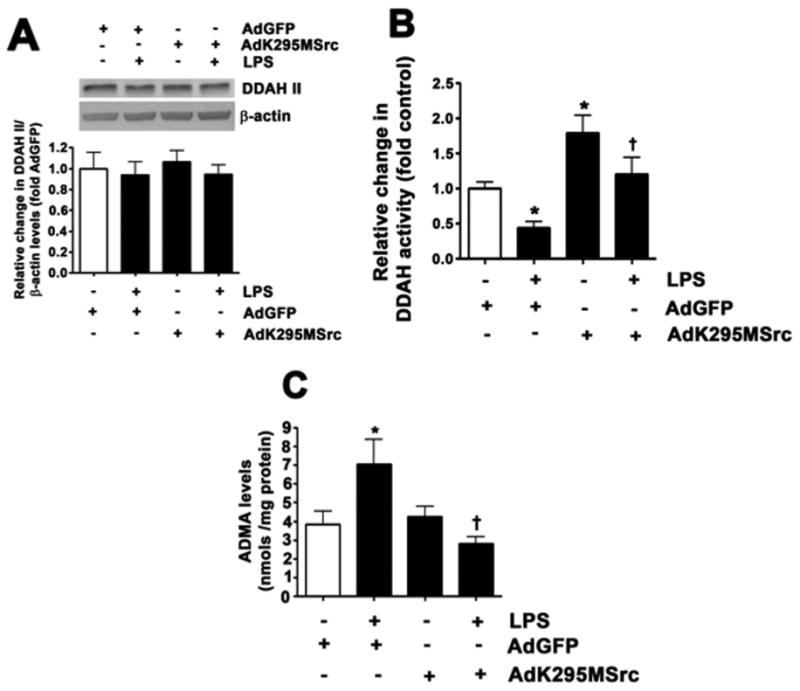
LPS decreases DDAH activity and increases ADMA levels in pulmonary arterial endothelial cells. PAEC were transduced with AdK295MSrc or AdGFP for 48 h then challenged with LPS (1000 EU/ml; 4 h). Although DDAH II protein levels were unchanged (A), LPS attenuated DDAH (B) activity and increased ADMA levels (B). AdK295MSrc prevented both the decrease in DDAH activity (B) and the increase in ADMA (C). Values are mean ± SEM, n=4–11, * P < 0.05 vs. untreated; †P < 0.05 vs. LPS.
Fig. 6.
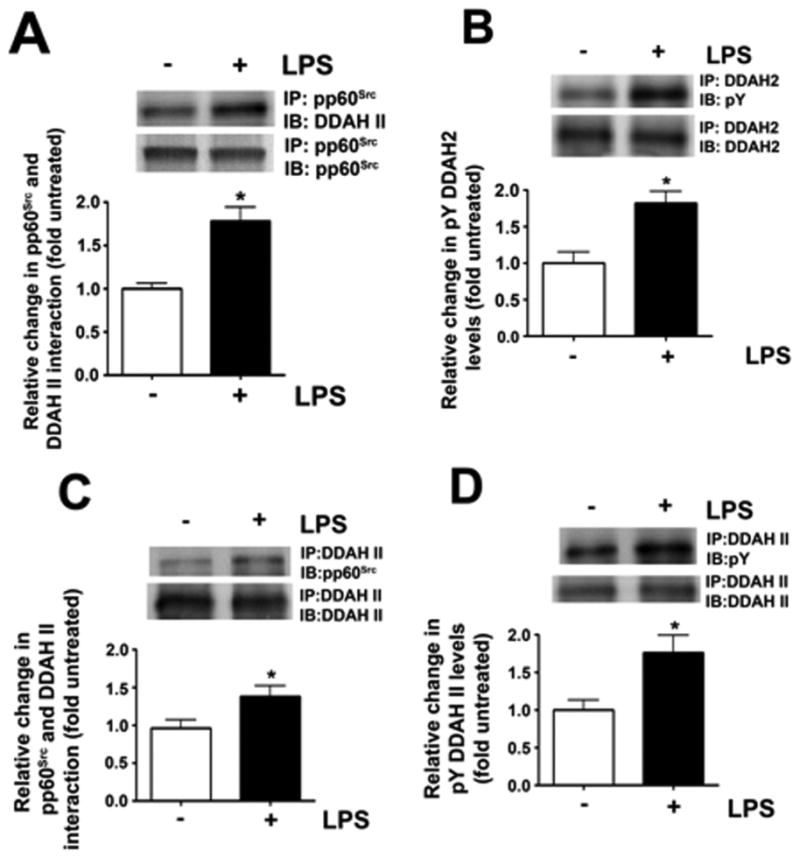
The decrease in DDAH activity corresponds to an increase in pp60Src-mediated phosphorylation of DDAH II. PAEC were exposed to LPS (1000 EU/ml; 4 h) then lysates were subjected to immunoprecipitation and immunoblotting using pp60Src with DDAH II specific antibodies. LPS stimulates the interaction of interaction of pp60Src and DDAH II (A) and this correlates with an increase in the tyrosine phosphorylation of DDAH II (B). Mice were also injected with LPS (6.75×104 EU/gm body wt.) intraperitoneally. After 12 h lung lysates were prepared and subjected to immunoprecipitation and immuno-blotting. LPS treatment increases both the interaction of pp60Src and DDAH II in the moue lung (D) and the tyrosine phosphorylation of DDAH II (E). Values are mean ± SEM, n=4–12, * P < 0.05 vs. untreated. LPS.
3.5. Over-expression of a constitutively active pp60Src mutant mimics the effects of LPS on DDAH activity
To further demonstrate the functional significance of pp60Src in attenuating DDAH activity, we overexpressed a constitutively active pp60Src mutant in PAEC using an adenovirus containing Y527FSrc (AdY527FSrc, Fig. 7A). This over-expression increased pY416 pp60Src levels, indicating increased pp60Src activity (Fig. 7B) but again did not change DDAH II protein levels (Fig. 7C). As with LPS, AdY527FSrc over-expression increased the interaction of pp60Src with DDAH II (Fig. 7D), pY-DDAH II levels (Fig. 7E), decreased DDAH activity (Fig. 7F) and increased ADMA levels (Fig. 7G). Thus, the stimulation of pp60Src signaling is sufficient to mimic the effects of LPS on the DDAH/ ADMA axis. Similarly, overexpression of Y527FSrc in the mouse lung did not significantly alter DDAH II protein levels (Fig. 8A). However, the interaction of DDAH II with pp60Src was significantly increased (Fig. 8B) as was pY-DDAH II (Fig. 8C). These changes correlated with a decrease in DDAH activity (Fig. 8D) and an increase in ADMA levels (Fig. 8E).
Fig. 7.
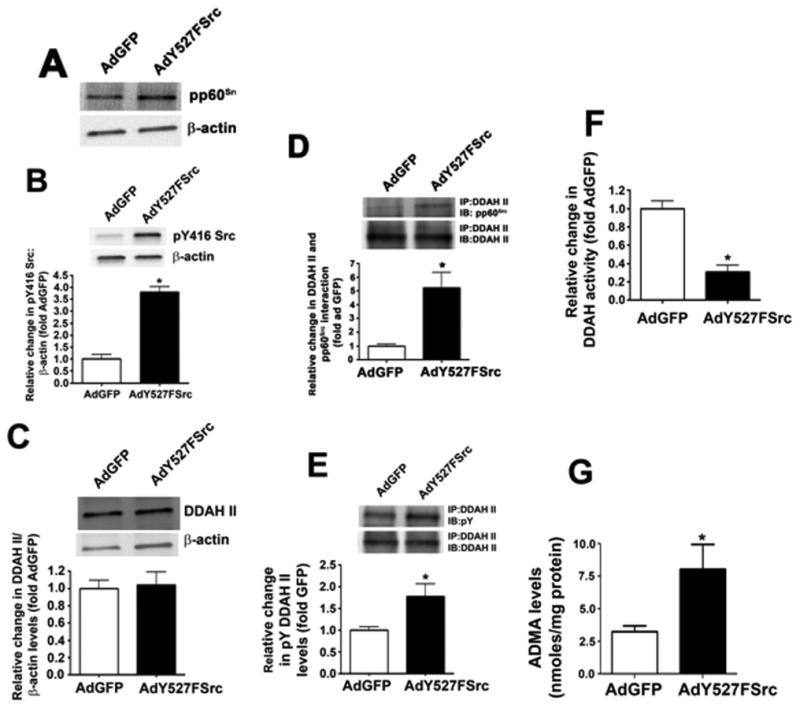
The over-expression of a constitutively active pp60Src mutant mimics the effects of LPS on DDAH activity in pulmonary arterial endothelial cells. PAEC were transduced with AdY527FSrc or AdGFP for 48 h. Immunoblot analysis identified a significant increase in pp60Src protein levels (A) and an increase in pp60Src activity, as determine by increase in phospho-Y416 pp60Src levels (B) in PAEC transduced with AdY527FSrc. AdY527FSrc did not alter DDAH II protein levels (C). However, AdY527FSrc transduction significantly increased the both the interaction of pp60Src with DDAH II (D) and the tyrosine phosphorylation of DDAH II (E). DDAH activity was significantly decreased (F) while ADMA levels were significantly increased (G). Values are mean ± SEM, n=4–6, * P < 0.05 vs. AdGFP.
Fig. 8.
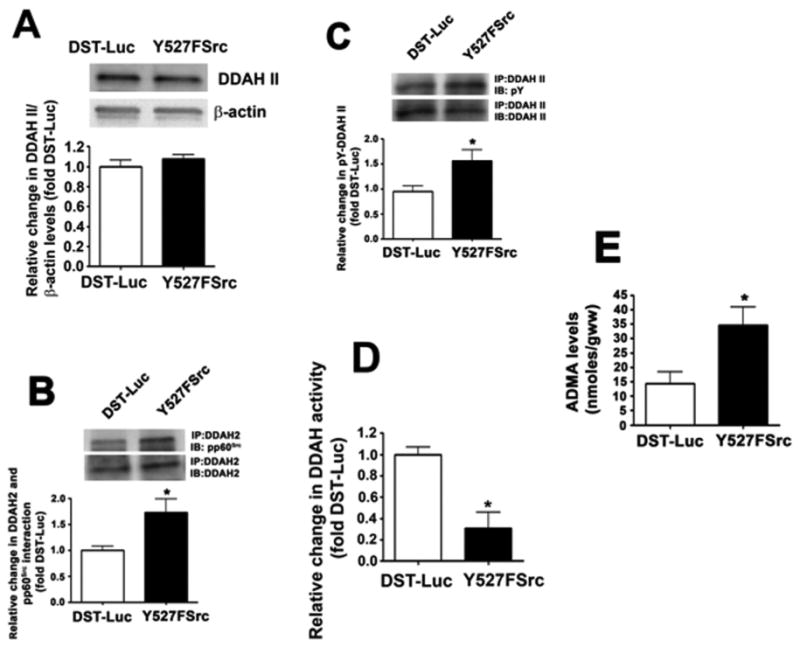
The over-expression of a constitutively active pp60Src mutant in decreases DDAH activity and increases ADMA levels in the mouse lung. Mice were injected with either DST-luciferase (DST) or pAD/CMV/V5-DEST- Y527FSrc (Y527FSrc) plasmids via the tail vein. After 72 h, the animals were sacrificed and peripheral lung lysates prepared. Y527FSrc over-expression dis not alter DDAH II protein levels (A). However, the interaction of pp60Src with DDAH II was significantly increased (B) as was the tyrosine phosphorylation of DDAH II (C). DDAH activity (D) was significantly reduced and ADMA levels were significantly increased (E). Values are mean ± SEM, n=5–6, *P < 0.05 vs. DST-Luc.
3.6. pp60Src mediates its inhibitory effect on DDAH II through Y207
An analysis of the human DDAH II protein revealed the presence of a single tyrosine residue located at amino acid position 207. To determine if phosphorylation of this residue was responsible for mediating the pp60Src-dependent decrease in DDAH activity we generated a phospho-antibody that recognizes pY207 in DDAH II. Using this antibody we were able to demonstrate an increase in the phosphorylation of Y207 in DDAH II in PAEC over-expressing Y527FSrc (Fig. 9A). Then, using side-directed mutagenesis we replaced the tyrosine at 207 with phenylalanine to generate a Y207F DDAH II mutant. We then co-expressed wild-type and Y207F-DDAH II with Y527FSrc in HEK293 cells (Fig. 9B and C). The over-expression of Y527FSrc decreased DDAH activity (Fig. 9D) and increased ADMA levels (Fig. 9E) in cells expressing wild-type DDAH II. However, Y527FSrc did not alter DDAH activity (Fig. 9D) and ADMA levels (Fig. 9E) in cells over-expressing the Y207F DDAH II mutant. These results confirm that pp60Src attenuates DDAH II activity through a specific phosphorylation at Y207.
Fig. 9.
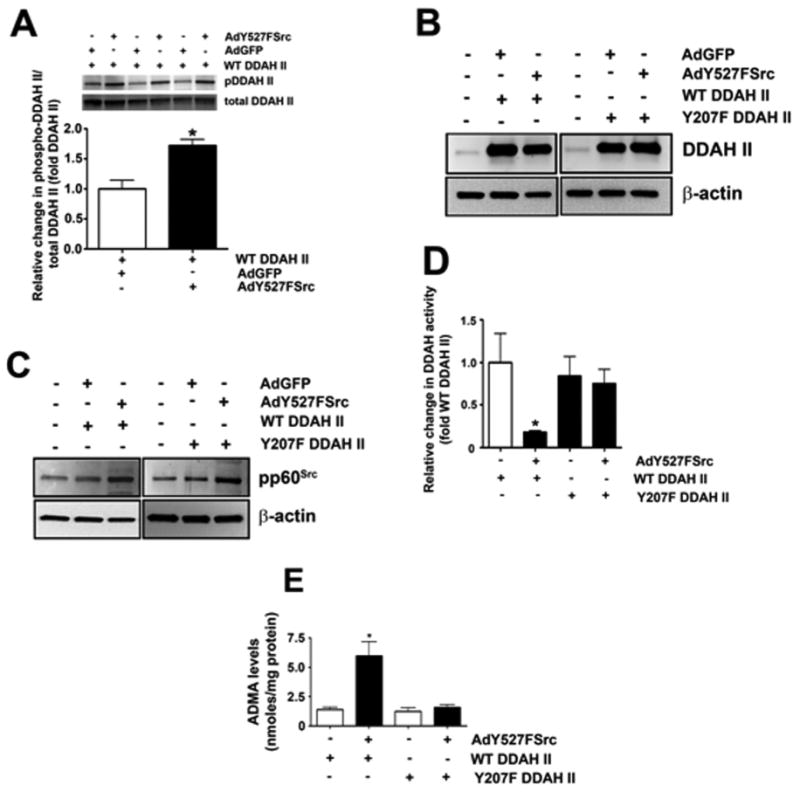
pp60Src inhibits DDAH II activity through Y207. HEK293 cells were transduced AdY527FSrc or AdGFP along with wild-type DDAH II for 48 h. Over-expression of Ad527FSrc increased pY207 DDAH II levels as measured by Western blot analysis using a phospho-antibody that recognizes pY207 in DDAH II (A). HEK293 were transduced with (AdY527FSrc) or AdGFP and transfected with either wildtype (WT) DDAH II or a DDAH II mutant in which Y207 was replaced by phenlyalanine (Y207FDDAH II). Immunoblot analysis identified a similar increase in DDAH II levels in WT DDAH II or Y207F DDAH II transfected cells (B). HEK293 cells transduced with AdY527FSrc exhibited a significant increase in pp60Src expression (C). The over-expression of AdY527FSrc decreased DDAH II activity (D) and increased ADMA levels (E) in cells expressing WT DDAH II. However, AdY527FSrc did decrease DDAH activity (D) or increase ADMA levels (E) levels in cells expressing the Y207F DDAH II mutant. Values are mean± SEM, n=4–6, *P< 0.05 vs. WT DDAH II alone.
4. Discussion
The most significant finding in this study is the identification of pp60Src as a kinase capable of binding to, and directly inhibiting the activity of, DDAH II and resulting in an increase in cellular ADMA. As ADMA is an endogenous NOS inhibitor, this attenuates NO generation for eNOS by enhancing its uncoupling [13]. This occurs despite a pp60Src–mediated increase in eNOS phosphorylation at Ser1177. Phosphorylation of eNOS at Ser1177 is thought to stimulate enzymatic activity by making the enzyme calcium/calmodulin independent [33], or more likely, it makes the enzyme more responsive to lower levels of calcium. Although the phosphorylation of eNOS at Thr495 is more often associated with eNOS uncoupling [34], Ser1177 phosphorylation has also been associated with the uncoupling of eNOS [24]. In addition, a mutant eNOS protein that has an aspartic acid introduced at Ser1177, to mimic phosphorylation, in combination with an alanine at Thr495, to prevent its phosphorylation, is uncoupled [35]. Our S1177D eNOS mutant data which appears to have increased catalytic activity, at the cost of increased superoxide generation, is thus in agreement with prior studies. However, we speculate that the LPS-mediated increases in ADMA likely synergize with the phosphorylation of eNOS at Ser1177 to further stimulate eNOS uncoupling and together this results in increased peroxynitrite production and protein nitration we observe in both cultured PAEC and the mouse lung. Further, as our prior studies suggest that eNOS rather than iNOS may play a more important role in the development of ALI [11,16,21], attenuating eNOS uncoupling could be a potential therapeutic target for treating a disease that, besides low tidal mechanical ventilation, has no viable therapy.
In the pulmonary vasculature, and other vascular beds, eNOS utilizes L-arginine to produce L-citrulline and NO [36]. Although a simple reaction, a complex regulatory network is required to maintain sufficient NO generation and a healthy endothelium. Besides, its substrate, L-arginine, the generation of NO requires the binding of Ca2+/calmodulin, flavin adenine dinucleotide, flavin mononucleotide, NADPH, and tetrahydrobiopterin (BH4) [37]. If the levels of L-arginine or BH4 decrease, electrons donated by NADPH are utilized to convert molecular oxygen to superoxide, a process known as uncoupling [38]. Another factor that can induce eNOS uncoupling is ADMA. ADMA competes with L-arginine for the substrate binding site in eNOS, and the ratio between L-arginine and ADMA is a key component in the regulation of endothelial NOS activity. Elevated ADMA levels have been shown to antagonize the endothelium-dependent vasodilation in humans [3–7]. Increased levels of ADMA have been identified in a number of pulmonary diseases including pulmonary hypertension [39], congenital heart disease (CHD) [40], and ALI [11,12]. ADMA appears to increase secondary to a reduction in the activity of the ADMA metabolizing enzyme, DDAH II. In humans with PH [41] and piglets with persistent pulmonary hypertension of the newborn [42] this correlates with a reduction in DDAH II expression. While in lambs with CHD [40] and mice exposed to LPS [11,12] DDAH II protein levels are unchanged but activity is decreased. Similarly, in vitro studies have shown that oxidized LDL [43], TNF-α [43], and hypergly-cemia [44] decrease DDAH activity, but not its expression. The molecular mechanism by which the post-translational inhibition of DDAH II occurs has been unresolved. Thus, our data add significantly to our understanding of how this process occurs by identifying a pp60Src-mediated phosphorylation of DDAH II on Y207 as the inhibitory phosphorylation. Further, our data fit well with the prior studies that identified oxidative stress [43,44] as being important in DDAH II inhibition as pp60Src can be activated by reactive oxygen species [45–47]. However, further studies will be required to determine if pp60Src activation is a common mechanism responsible for the inhibition of DDAH II activity in other pulmonary diseases such as CHD [40].
It is well established that pp60Src plays a major role in the endothelial barrier disruption associated with ALI [48–50] and pp60Src inhibition attenuates the severity of injury [20]. However, pp60Src has also been shown to indirectly enhance eNOS activity and NO generation through the sequential signaling of PI3 kinase and Akt1 [23]. NO signaling appears to be involved in maintaining the endothe-lial barrier under physiologic conditions [51–54] and can protect against its disruption by oxidative stress [54,55]. These findings are apparently contradictory. However, we show that pp60Src activation leads to both NO and superoxide generation and the formation of peroxynitrite. A number of studies have shown that peroxynitrite, generated by the interaction of NO with superoxide, plays an important role in the disruption of the endothelial barrier during the development of ALI/ARDS [11,14,15]. Peroxynitrite can lead to protein tyrosine nitration forming 3-nitrotyrosine (3-NT). 3-NT is widely used as a marker of peroxynitrite formation and elevated levels have been found in animal models of ALI [11,56–59] and in patients with ALI [60]. We have shown that scavenging peroxynitrite can attenuate the development of ALI [11]. However, the key protein targets for nitration in ALI are only poorly understood. Indeed, to our knowledge, the only verified nitration target in ALI is the Rho (Ras homologous) GTP-binding protein, RhoA [21]. The canonical pathway by which RhoA is stimulated is via the activation of G-protein-coupled receptors and/or tyrosine kinases that results in the activation of guanine nucleotide exchange factors (GEFs) inducing the exchange of GDP for GTP and translocation of GTP-RhoA to the plasma membrane. Upon transloca-tion to the plasma membrane RhoA is able to activate one or more of its effector proteins, particularly Rho-associated kinases (ROCKs). Rho A and Rho-associated kinase can directly catalyze myosin light chain (MLC) phosphorylation, or act indirectly via inactivation of MLC phosphatase [17,18] inducing cell contraction and endothelial barrier disruption. However, when nitrated at Y34, the need for GEFs are minimized as the nitration of Y34 enhances GDP release mimicking the effect of GEFs [21]. Our data confirm that pp60Src is required for RhoA nitration in LPS-exposed PAEC, likely due to increased eNOS uncoupling and that decreasing RhoA nitration correlates with reduced endothelial barrier disruption in response to LPS. Another potential target for nitration is nuclear factor (NF)-κB. The NF-κB family of transcription factors regulate the generation of cytokines/chemokines in response to various injurous states including oxidative and nitrative stress. The activation of NF-κB is well established in ALI [61,62]. Under basal conditions, NF-κB is sequestered in the cytoplasm in a complex with IκBα. In a previous study, we have demonstrated that the nitration of IκBα at Y181 triggers its dissociation from NF-κB leading to its nuclear accumulation [63]. In addition, our prior studies have shown that reducing protein nitration, by over-expressing either DDAH II in the pulmonary endothelium of the mouse [12] or using eNOS-/mice [16], reduces the inflammatory cytokines normally induced by LPS via NF-κB again implicating eNOS rather than iNOS in the progression of ALI. Thus, although correlative, this is highly suggestive that NF-κB nitration may be involved in its non-canonical activation during ALI. However, further studies will be required to test this hypothesis.
In conclusion our data indicate that during ALI, the activation of pp60Src through the uncoupling of eNOS drives the generation of peroxynitrite and enhances protein nitration, with at least one of the nitration targets being the barrier disruptive small GTPase, RhoA. This is mediated by increasing ADMA levels via the inhibition of DDAH II and through an increase in Ser1177 eNOS phosphorylation. These data again suggest that treatments that target the increased generation of ADMA during ALI may have therapeutic value.
Acknowledgments
This research was supported in part by grants P01HL0101902, HL60190, and HL67841 from the National Institutes of Health and 14SDG20480354. Christine M. Gross was funded in part by a pre-doctoral Fellowship award, 12PRE12060224, from the American Heart Association.
References
- 1.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, LeGall JR, Morris A, Spragg R. Report of the American-European consensus conference on acute respiratory distress syndrome: definitions, mechanisms, relevant outcomes, and clinical trial coordination. Consensus committee. J Crit Care. 1994;9:72–81. doi: 10.1016/0883-9441(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 2.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 3.Boger RH, Bode-Boger SM. Asymmetric dimethylarginine, derangements of the endothelial nitric oxide synthase pathway, and cardiovascular diseases. Semin Thromb Hemost. 2000;26:539–545. doi: 10.1055/s-2000-13210. [DOI] [PubMed] [Google Scholar]
- 4.Boger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. 2003;59:824–833. doi: 10.1016/s0008-6363(03)00500-5. [DOI] [PubMed] [Google Scholar]
- 5.Boger RH. Association of asymmetric dimethylarginine and endothelial dysfunction. Clin Chem Lab Med. 2003;41:1467–1472. doi: 10.1515/CCLM.2003.225. [DOI] [PubMed] [Google Scholar]
- 6.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl. 2003;4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 7.Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004;134:2842S–2847S. doi: 10.1093/jn/134.10.2842S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- 8.Boger RH, Vallance P, Cooke JP. Asymmetric dimethylarginine (ADMA): a key regulator of nitric oxide synthase. Atheroscler. 2003;(Suppl 4):1–3. doi: 10.1016/s1567-5688(03)00027-8. [DOI] [PubMed] [Google Scholar]
- 9.Miyazaki H, Matsuoka H, Cooke JP, Usui M, Ueda S, Okuda S, Imaizumi T. Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis. Circulation. 1999;99:1141–1146. doi: 10.1161/01.cir.99.9.1141. [DOI] [PubMed] [Google Scholar]
- 10.Jones LC, Tran CT, Leiper JM, Hingorani AD, Vallance P. Common genetic variation in a basal promoter element alters DDAH2 expression in endothelial cells. Biochem Biophys Res Commun. 2003;310:836–843. doi: 10.1016/j.bbrc.2003.09.097. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Smith A, Kumar S, Aggarwal S, Rehmani I, Snead C, Harmon C, Fineman J, Fulton D, Catravas JD, Black SM. Mechanisms of nitric oxide synthase uncoupling in endotoxin-induced acute lung injury: role of asymmetric dimethylarginine. Vasc Pharmacol. 2010;52:182–190. doi: 10.1016/j.vph.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aggarwal S, Gross CM, Kumar S, Dimitropoulou C, Sharma S, Gorshkov BA, Sridhar S, Lu Q, Bogatcheva NV, Jezierska-Drutel AJ, Lucas R, Verin AD, Catravas JD, Black SM. Dimethylarginine dimethylaminohydrolase II over-expression attenuates LPS-mediated lung leak in acute lung injury. Am J Respir Cell Mol Biol. 2014;50:614–625. doi: 10.1165/rcmb.2013-0193OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am J Physiol Cell Physiol. 2008;294:C1407–C1418. doi: 10.1152/ajpcell.00384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okamoto T, Gohil K, Finkelstein EI, Bove P, Akaike T, van der Vliet A. Multiple contributing roles for NOS2 in LPS-induced acute airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L198–L209. doi: 10.1152/ajplung.00136.2003. [DOI] [PubMed] [Google Scholar]
- 15.Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Tuder R, Garcia JG, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am J Respir Crit Care Med. 2005;172:470–479. doi: 10.1164/rccm.200411-1547OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross CM, Rafikov R, Kumar S, Aggarwal S, Ham PB, 3, Meadows ML, Cherian-Shaw M, Kangath A, Sridhar S, Lucas R, Black SM. Endothelial nitric oxide synthase deficient mice are protected from lipopolysaccharide induced acute lung injury. PLoS One. 2015;10:e0119918. doi: 10.1371/journal.pone.0119918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell. 1998;9:2639–2653. doi: 10.1091/mbc.9.9.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 19.Gong P, Angelini DJ, Yang S, Xia G, Cross AS, Mann D, Bannerman DD, Vogel SN, Goldblum SE. TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J Biol Chem. 2008;283:13437–13449. doi: 10.1074/jbc.M707986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HS, Moon C, Lee HW, Park EM, Cho MS, Kang JL. Src tyrosine kinases mediate activations of NF-kappaB and integrin signal during lipopolysaccharide-induced acute lung injury. J Immunol. 2007;179:7001–7011. doi: 10.4049/jimmunol.179.10.7001. [DOI] [PubMed] [Google Scholar]
- 21.Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, Sharma S, Jezierska-Drutel A, Patel V, Snead C, Lucas R, Verin A, Fulton D, Catravas JD, Black SM. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem. 2014;289:4710–4722. doi: 10.1074/jbc.M114.547596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manukyan M, Nalbant P, Luxen S, Hahn KM, Knaus UG. RhoA GTPase activation by TLR2 and TLR3 ligands: connecting via Src to NF-kappaB. J Immunol. 2009;182:3522–3529. doi: 10.4049/jimmunol.0802280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas S, Chen K, Keaney J., Jr Hydrogen peroxide activates endothelial nitric oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem. 2001 doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- 24.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–27047. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wedgwood S, Bekker JM, Black SM. Shear stress regulation of endothelial NOS in fetal pulmonary arterial endothelial cells involves PKC. Am J Physiol Lung Cell Mol Physiol. 2001;281:L490–L498. doi: 10.1152/ajplung.2001.281.2.L490. [DOI] [PubMed] [Google Scholar]
- 26.Rafikov R, Kumar S, Aggarwal S, Pardo D, Fonseca FV, Ransom J, Rafikova O, Chen Q, Springer ML, Black SM. Protein engineering to develop a redox insensitive endothelial nitric oxide synthase. Redox Biol. 2014;2:156–164. doi: 10.1016/j.redox.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rafikov R, Fonseca FV, Kumar S, Pardo D, Darragh C, Elms S, Fulton D, Black SM. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun X, Fratz S, Sharma S, Hou Y, Rafikov R, Kumar S, Rehmani I, Tian J, Smith A, Schreiber C, Reiser J, Naumann S, Haag S, Hess J, Catravas JD, Patterson C, Fineman JR, Black SM. C-terminus of heat shock protein 70-interacting protein-dependent GTP cyclohydrolase I degradation in lambs with increased pulmonary blood flow. Am J Respir Cell Mol Biol. 2011;45:163–171. doi: 10.1165/rcmb.2009-0467OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S, Sud N, Fonseca FV, Hou Y, Black SM. Shear stress stimulates nitric oxide signaling in pulmonary arterial endothelial cells via a reduction in catalase activity: role of protein kinase C delta. Am J Physiol Lung Cell Mol Physiol. 2010;298:L105–L116. doi: 10.1152/ajplung.00290.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMullan DM, Bekker JM, Parry AJ, Johengen MJ, Kon A, Heidersbach RS, Black SM, Fineman JR. Alterations in endogenous nitric oxide production after cardiopulmonary bypass in lambs with normal and increased pulmonary blood flow. Circulation. 2000;102:III172–178. doi: 10.1161/01.cir.102.suppl_3.iii-172. [DOI] [PubMed] [Google Scholar]
- 31.Brennan LA, Wedgwood S, Black SM. The overexpression catalase reduces NO-mediated inhibition of endothelial NO synthase. IUBMB Life. 2002;54:261–265. doi: 10.1080/15216540215679. [DOI] [PubMed] [Google Scholar]
- 32.Sharma S, Sun X, Kumar S, Rafikov R, Aramburo A, Kalkan G, Tian J, Rehmani I, Kallarackal S, Fineman JR, Black SM. Preserving mitochondrial function prevents the proteasomal degradation of GTP cyclohydrolase I. Free Radic Biol Med. 2012;53:216–229. doi: 10.1016/j.freeradbiomed.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phos-phorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 34.Sun X, Kumar S, Sharma S, Aggarwal S, Lu Q, Gross C, Rafikova O, Lee SG, Dasarathy S, Hou Y, Meadows ML, Han W, Su Y, Fineman JR, Black SM. Endothelin-1 induces a glycolytic switch in pulmonary arterial endothelial cells via the mitochondrial translocation of endothelial nitric oxide synthase. Am J Respir Cell Mol Biol. 2014;50:1084–1095. doi: 10.1165/rcmb.2013-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, Jr, Sessa WC. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- 36.Parviz M, Bousamra M, 2, Chammas JH, Birks EK, Presberg KW, Jacobs ER, Nelin LD. Effects of chronic pulmonary overcirculation on pulmonary vasomotor tone. Ann Thorac Surg. 1999;67:522–527. doi: 10.1016/s0003-4975(98)01141-2. [DOI] [PubMed] [Google Scholar]
- 37.Yokoyama M, Hirata K. Endothelial nitric oxide synthase uncoupling: Is it a physiological mechanism of endothelium-dependent relaxation in cerebral artery? Cardiovasc Res. 2007;73:8–9. doi: 10.1016/j.cardiores.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Investig. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shao Z, Wang Z, Shrestha K, Thakur A, Borowski AG, Sweet W, Thomas JD, Moravec CS, Hazen SL, Tang WH. Pulmonary hypertension associated with advanced systolic heart failure: dysregulated arginine metabolism and importance of compensatory dimethylarginine dimethylaminohydrolase-1. J Am Coll Cardiol. 59:1150–1158. doi: 10.1016/j.jacc.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun X, Fratz S, Sharma S, Hou Y, Rafikov R, Kumar S, Rehmani I, Tian J, Smith A, Schreiber C, Reiser J, Naumann S, Haag S, Hess J, Catravas JD, Patterson C, Fineman JR, Black S. M.C-terminus of heat shock protein 70-interacting protein-dependent GTP cyclohydrolase I degradation in lambs with increased pulmonary blood flow. Am J Respir Cell Mol Biol. 45:163–171. doi: 10.1165/rcmb.2009-0467OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pullamsetti S, Kiss L, Ghofrani HA, Voswinckel R, Haredza P, Klepetko W, Aigner C, Fink L, Muyal JP, Weissmann N, Grimminger F, Seeger W, Schermuly RT. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. Faseb J. 2005;19:1175–1177. doi: 10.1096/fj.04-3223fje. [DOI] [PubMed] [Google Scholar]
- 42.Arrigoni FI, Vallance P, Haworth SG, Leiper JM. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. 2003;107:1195–1201. doi: 10.1161/01.cir.0000051466.00227.13. [DOI] [PubMed] [Google Scholar]
- 43.Ito A, Tsao PS, Adimoolam S, Kimoto M, Ogawa T, Cooke JP. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine di-methylaminohydrolase. Circulation. 1999;99:3092–3095. doi: 10.1161/01.cir.99.24.3092. [DOI] [PubMed] [Google Scholar]
- 44.Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylami-nohydrolase. Circulation. 2002;106:987–992. doi: 10.1161/01.cir.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- 45.Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J Biol Chem. 1997;272:20389–20394. doi: 10.1074/jbc.272.33.20389. [DOI] [PubMed] [Google Scholar]
- 46.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 47.Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 48.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vasc Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 49.Tinsley JH, Ustinova EE, Xu W, Yuan SY. Src-dependent, neutrophil-mediated vascular hyperpermeability and beta-catenin modification. Am J Physiol Cell Physiol. 2002;283:C1745–C1751. doi: 10.1152/ajpcell.00230.2002. [DOI] [PubMed] [Google Scholar]
- 50.Hu G, Place AT, Minshall RD. Regulation of endothelial permeability by Src kinase signaling: vascular leakage versus transcellular transport of drugs and macromolecules. Chem Biol Interact. 2008;171:177–189. doi: 10.1016/j.cbi.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunduz D, Thom J, Hussain I, Lopez D, Hartel FV, Erdogan A, Grebe M, Sedding D, Piper HM, Tillmanns H, Noll T, Aslam M. Insulin stabilizes microvascular endothelial barrier function via phosphatidylinositol 3-kinase/Akt-mediated Rac1 activation. Arterioscler Thromb Vasc Biol. 2010;30:1237–1245. doi: 10.1161/ATVBAHA.110.203901. [DOI] [PubMed] [Google Scholar]
- 52.Schutte H, Witzenrath M, Mayer K, Weissmann N, Schell A, Rosseau S, Seeger W, Grimminger F. The PDE inhibitor zaprinast enhances NO-mediated protection against vascular leakage in reperfused lungs. Am J Physiol Lung Cell Mol Physiol. 2000;279:L496–L502. doi: 10.1152/ajplung.2000.279.3.L496. [DOI] [PubMed] [Google Scholar]
- 53.Moldobaeva A, Welsh-Servinsky LE, Shimoda LA, Stephens RS, Verin AD, Tuder RM, Pearse DB. Role of protein kinase G in barrier-protective effects of cGMP in human pulmonary artery endothelial cells. Am J Physiol Lung Cel Mol Physiol. 2006;290:L919–L930. doi: 10.1152/ajplung.00434.2005. [DOI] [PubMed] [Google Scholar]
- 54.Pearse DB, Shimoda LA, Verin AD, Bogatcheva N, Moon C, Ronnett GV, Welsh LE, Becker PM. Effect of cGMP on lung microvascular endothelial barrier dysfunction following hydrogen peroxide. Endothel: J Endothel Cell Res. 2003;10:309–317. doi: 10.1080/10623320390272307. [DOI] [PubMed] [Google Scholar]
- 55.Stephens RS, Rentsendorj O, Servinsky LE, Moldobaeva A, Damico R, Pearse DB. cGMP increases antioxidant function and attenuates oxidant cell death in mouse lung microvascular endothelial cells by a protein kinase G-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2010;299:L323–L333. doi: 10.1152/ajplung.00442.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szabo C, Cuzzocrea S, Zingarelli B, O'Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Investig. 1997;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabo C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br J Pharmacol. 1997;120:259–267. doi: 10.1038/sj.bjp.0700872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen LW, Wang JS, Chen HL, Chen JS, Hsu CM. Peroxynitrite is an important mediator in thermal injury-induced lung damage. Crit Care Med. 2003;31:2170–2177. doi: 10.1097/01.CCM.0000079605.28852.D0. [DOI] [PubMed] [Google Scholar]
- 59.Tsuji C, Shioya S, Hirota Y, Fukuyama N, Kurita D, Tanigaki T, Ohta Y, Nakazawa H. Increased production of nitrotyrosine in lung tissue of rats with radiation-induced acute lung injury. Am J Physi Lung Cell Mol Physiol. 2000;278:L719–L725. doi: 10.1152/ajplung.2000.278.4.L719. [DOI] [PubMed] [Google Scholar]
- 60.Thomson L, Christie J, Vadseth C, Lanken PN, Fu X, Hazen SL, Ischiropoulos H. Identification of immunoglobulins that recognize 3-nitrotyrosine in patients with acute lung injury after major trauma. Am J Respir Cell Mol Biol. 2007;36:152–157. doi: 10.1165/rcmb.2006-0288SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007;9:1991–2002. doi: 10.1089/ars.2007.1785. [DOI] [PubMed] [Google Scholar]
- 62.Ali S, Mann DA. Signal transduction via the NF-kappaB pathway: a targeted treatment modality for infection, inflammation and repair. Cell Biochem Funct. 2004;22:67–79. doi: 10.1002/cbf.1082. [DOI] [PubMed] [Google Scholar]
- 63.Yakovlev VA, Barani IJ, Rabender CS, Black SM, Leach JK, Graves PR, Kellogg GE, Mikkelsen RB. Tyrosine nitration of IkappaBalpha: a novel mechanism for NF-kappaB activation. Biochemistry. 2007;46:11671–11683. doi: 10.1021/bi701107z. [DOI] [PMC free article] [PubMed] [Google Scholar]