
Fig. 2.
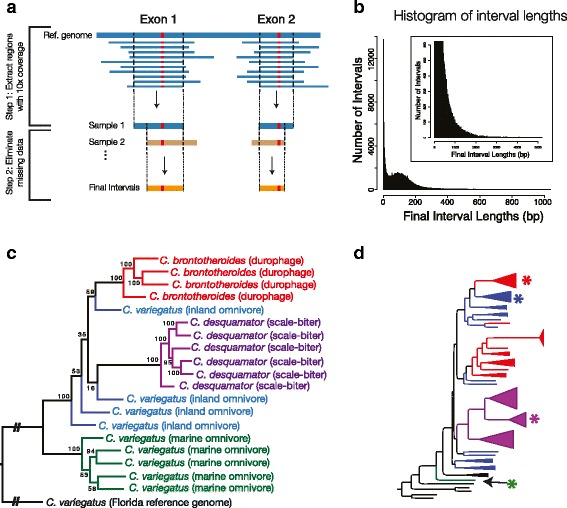
Phylogenetic relationships among species of pupfish from San Salvador Island. a Overview of bioinformatic methods to generate sequence data for phylogenetic analysis. Consensus genome sequences were generated for each parental pair by substituting SNP (red bars) allele assignments. Genomic intervals for use in phylogenetic analysis were selected based on contiguous regions that had a minimum coverage of 10 reads at every nucleotide position. We then further selected intervals across all parental pairs such that there was no missing data in our dataset. b The length distribution of genomic intervals used for phylogenetic reconstruction (from a). Inset shows intervals exceeding 1 kb in length. Many intervals are less than 10 bp long and some intervals are over 1 kb in length. We restricted our final interval set to regions that were longer than 50 bp and less than 1 kb. c. A maximum likelihood tree built from expressed exonic sequence data produced from the current study, and d. a maximum likelihood tree built from RAD-seq data, reproduced here from Martin and Feinstein [31, 50]. Note the overall congruence in the inferred phylogenetic relationships among taxa from both studies. In particular, both studies find a monophyletic San Salvador Island clade and that the marine omnivore population diverges prior to the speciation of endemic San Salvador taxa. Stars in d. indicate samples from the same populations as those of the current study