

Abstract
Mitochondria comprise both nuclear and mitochondrially encoded proteins requiring precise stoichiometry for their integration into functional complexes. The augmented protein synthesis associated with mitochondrial biogenesis results in the accumulation of unfolded proteins, thus triggering cellular stress. As such, the unfolded protein responses emanating from the endoplasmic reticulum (UPRER) or the mitochondrion (UPRMT) are triggered to ensure correct protein handling. Whether this response is necessary for mitochondrial adaptations is unknown. Two models of mitochondrial biogenesis were used: muscle differentiation and chronic contractile activity (CCA) in murine muscle cells. After 4 days of differentiation, our findings depict selective activation of the UPRMT in which chaperones decreased; however, Sirt3 and UPRER markers were elevated. To delineate the role of ER stress in mitochondrial adaptations, the ER stress inhibitor TUDCA was administered. Surprisingly, mitochondrial markers COX-I, COX-IV, and PGC-1α protein levels were augmented up to 1.5-fold above that of vehicle-treated cells. Similar results were obtained in myotubes undergoing CCA, in which biogenesis was enhanced by ~2–3-fold, along with elevated UPRMT markers Sirt3 and CPN10. To verify whether the findings were attributable to the terminal UPRER branch directed by the transcription factor CHOP, cells were transfected with CHOP siRNA. Basally, COX-I levels increased (~20%) and COX-IV decreased (~30%), suggesting that CHOP influences mitochondrial composition. This effect was fully restored by CCA. Therefore, our results suggest that mitochondrial biogenesis is independent of the terminal UPRER. Under basal conditions, CHOP is required for the maintenance of mitochondrial composition, but not for differentiation- or CCA-induced mitochondrial biogenesis.
Keywords: CHOP, mitochondria, chronic contractile activity, muscle differentiation, TUDCA
muscle differentiation from the myoblast to the myotube stage requires a large increase in the synthesis of new proteins. This increase in protein synthesis can perturb the proteostasis of the cell via an accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) or in the mitochondrial matrix (27, 43). Unfolded proteins trigger the activation of a quality control system termed the unfolded protein response (UPR). The UPR involves a transcriptional program that feeds back to decrease global protein translation, while increasing the synthesis of selected chaperones and proteases involved in protein folding in order to restore homeostasis (33). In the ER lumen, the UPR (UPRER) is triggered by aggregated unfolded proteins binding to the BiP chaperone. This results in the release of BiP’s inhibition upon UPR transmembrane sensors, and the activation of ATF6α, IRE1α, and PERK. Subsequently, a series of transcription factors are upregulated to attenuate the originating ER stress (31). A similar, yet independent, mechanism lies within the mitochondrial matrix and intermembrane space (29). This mitochondrial UPR (UPRMT) also induces organelle-specific chaperones and proteases to restore interorganelle homeostasis (3). If the unfolded proteins exceed the capacity of the folding machinery and proteostasis cannot be attained, autophagy, or ultimately, apoptosis is triggered (29, 37).
Although the UPR functions to attenuate cellular stress, it appears that a certain amount of stress is required for optimal muscle differentiation (27, 42). Myotube fusion has been shown to be defective when ER stress is inhibited and reversed with the induction of stress via a chemical inducer, thapsigargin (42). ER stress was also found to enhance myofiber formation due to triggered apoptosis, eliminating vulnerable cells (26). Hence, the UPR can be selectively activated to control cell growth and proper tissue differentiation, which may have implications for myogenesis and muscle fiber size (6, 27, 42).
The UPR is also activated with exercise-induced mitochondrial biogenesis (19, 23, 44). Skeletal muscle has a relatively low mitochondrial content under basal conditions, but muscle mitochondria are highly adaptive during physiological processes, such as myogenesis or exercise, in which the cellular energy demands are increased (8, 11, 14, 15). Both of these conditions trigger signal transduction pathways that activate the master transcriptional regulator PGC-1α, leading to an increased expression of nuclear and mitochondrial genes (32). This mitochondrial biogenesis requires signaling to transcription and translation for the production of additional proteins for import into the organelle. However, the signals preceding mitochondrial biogenesis remain enigmatic. Is the UPR required for adequate mitochondrial biogenesis, as it is for muscle differentiation? It is known that an increase in ER stress and UPR activation is associated with exercise, particularly, in untrained subjects during acute contractile activity (19, 23, 44). This may serve as a precursor signaling system for exercise-induced mitochondrial biogenesis. Despite this, CHOP deletion, a stress-induced transcription factor involved in both the UPRER and the UPRMT, has been found to ameliorate phenotypic exercise intolerance (44), inferring a potential role of UPR components in mitochondrial adaptations. Therefore, it is of interest to us to investigate whether mitochondrial biogenesis relies upon intracellular communication with the UPR.
To investigate this, recent work has employed the chemical chaperone mimetic drug, tauroursodeoxycholic acid (TUDCA) to attenuate stress-induced CHOP expression (23). TUDCA functions to diminish the terminal UPRER via assisting in protein folding in the ER lumen, and, thus, reducing overall stress induction and UPR activation (9, 23, 40). Recently, it was observed that the mitochondrial biogenesis induced in TUDCA-injected rats undergoing chronic contractile activity (CCA) did not significantly differ from those injected with vehicle control (23). Therefore, this would suggest that the exercise-induced mitochondrial adaptations occurred independently of ER stress-induced CHOP expression. Thus, the purposes of the present study were to investigate the necessity of the UPR during mitochondrial biogenesis induced during muscle cell differentiation, as well as subsequent chronic contractile activity, with a specific focus on the role of CHOP protein.
METHODS
Cell culture.
C2C12 murine skeletal muscle cells were proliferated on six-well cell culture plates in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (P/S) [growth medium (GM)]. The cells were incubated at 37°C in 5% CO2. Upon reaching 95–100% confluency, the medium was switched to DMEM supplemented with 5% heat-inactivated horse serum and 1% P/S to induce differentiation of the myoblasts into myotubes. Cells were harvested either immediately before the GM being switched to differentiation medium (DM; day 0), or 4 days after differentiation in DM. The DM was changed daily. To investigate the role of the UPR in myoblast differentiation, we aimed to attenuate UPR activation before cells exiting the cell cycle. Thus, tauroursodeoxycholic acid (TUDCA; Millipore) was used for the partial inhibition of the UPRER (23). Cells were grown to subconfluence and then pretreated with 500 µg/ml of either water or TUDCA in GM for 24 h. Thereafter, cells were switched to DM in the absence of TUDCA to differentiate for 4 days before subsequent collection. In the figure legends, each experiment within one N refers to cells acquired from the same cell passage, which came from same cells grown in one flask and collected at the same time.
Fusion index.
For quantification of myoblast fusion, cells were washed twice with ice-cold phosphate-buffered saline (PBS; Sigma-Aldrich) and fixed in 100% methanol at −20°C for 5 min before being left to air dry for 10 min. Wells were then incubated in 1 ml of Giemsa stain (Electron Microscopy Sciences, Hatfield, PA) diluted 1:20 in 1 mM sodium phosphate buffer (pH 5.6) for 10 min at room temperature. Thereafter, the solution was aspirated and rinsed with distilled water. Phase-contrast images were captured using a Canon Powershot G5 camera adapted to a light microscope at ×10 magnification. Nuclei were also manually counted from three randomly chosen regions per experimental group. The fusion index was calculated by determining the fraction of total nuclei found in myotubes. Myotubes were defined as cells with two or more nuclei (43). All treatments were performed in triplicate.
Fluorescence microscopy.
C2C12 muscle cells were grown and differentiated on custom-made glass bottom six-well dishes that were precoated with gelatin. On days 0 and 4 of differentiation, myotubes were treated with 100 nM of MitoTracker Green FM (Life Technologies) and incubated at 37°C for 45 min. Following incubation, the media were aspirated, and the cells were washed with PBS and reincubated in fresh DM. Fluorescence was visualized using the Nikon Eclipse TE2000-U microscope. Comparisons among images were made using the same magnification (×20) and camera exposure to minimize fluorescence variability among samples.
Electrical stimulation.
Myotubes were stimulated on the 5th day of differentiation, as done previously (7, 39). In brief, customized six-well plate lids outfitted with two platinum wires per well were submerged into the media of plates. The DM was changed 1 h before stimulation and immediately after. For TUDCA experiments, fully differentiated myotubes were treated with 500 µg/ml of either TUDCA or sterile water in DM on day 5, 1 h before chronic contractile activity (CCA) and was replenished following stimulation. Electrically induced CCA was conducted for 3 h/day (9 V, 5 Hz) with 21-h recovery periods for a total of 4 days. The presence of TUDCA had no impact on the quality of contractile activity induced by the stimulation, when contracting cells were visualized under the microscope. Cells were harvested on the 5th day, 21 h after the last stimulation.
Transfection.
On day 3 of myotube differentiation, cells were incubated in pretransfection media (5% HS in DMEM). The following day, myotubes were transfected for 6 h with 30 nM of scrambled (Silencer select negative control; 4390846; Life Technologies) or CHOP siRNA (Life Technologies; s201245 and s64888), using 10 µl of Lipofectamine 2000 in 2 ml of DMEM. The media were then changed back to DMEM supplemented with 5% HS and 1% P/S. On day 5, cells were kept under control conditions or were subjected to CCA, as described previously (7, 39). On day 6, cells were incubated in pretransfection media for another siRNA treatment of 6 h of transfection at day 7 before subsequent stimulation. Cells were collected 21 h after the 4th day of CCA.
Protein extraction.
Cells were rinsed twice with ice-cold PBS and then trypsinized at 37°C for collection on days 0 and 4 of differentiation and on day 9 for cells that underwent CCA treatment. Protein extracts were prepared by suspending the collected cells in lysis buffer supplemented with protease and phosphatase inhibitors. Thereafter, the cells were frozen in liquid N2 and subsequently thawed in a 37°C water bath for five freeze-thaw cycles. Following centrifugation at 16,000 g at 4°C for 10 min, the pellets were discarded, and the supernatant fractions were collected and stored at −80°C for subsequent immunoblotting analysis.
Immunoblotting.
The protein content of samples was measured using the Bradford method. Equal amounts of protein (25–50 µg) were separated by electrophoresis on 8–15% SDS-polyacrylamide gels. The proteins were then wet-transferred (Mini Trans-Blot electrophoretic transfer cell, Bio-Rad, Mississauga, Ontario, Canada) onto nitrocellulose membranes. After blocking the membranes for 1 h in 5% skim milk, they were then probed overnight at 4°C with primary antibodies. For a full list of proteins probed and antibodies, see Table 1. Blots were washed (3 × 5 min) in 1 × TBST [Tris-buffered saline-Tween-20, 25 mM Tris·HCl (pH 7.5), 1 mM NaCl, and 0.1% Tween-20] solution and incubated for 1 h at room temperature with the appropriate anti-mouse or anti-rabbit secondary antibody, followed by 1 × TBST wash (3 × 5 min). Membranes were visualized with enhanced chemiluminescence using Clarity Western ECL Substrate (Bio-Rad) and exposed to film. Signals were quantified with ImageJ Software (NIH, Bethesda, MD). Values were normalized to the appropriate loading controls: α-tubulin, β-actin, or GAPDH.
Table 1.
List of primary antibodies used for immunoblotting
| Antibody | Manufacturer | Product Number |
|---|---|---|
| α-Tubulin | Millipore | CPO6 |
| ATF4 | Santa Cruz Biotechnology | SC-200 |
| ATF6 | Santa Cruz Biotechnology | SC-22799 |
| β-Actin | Santa Cruz Biotechnology | SC-47778 |
| BiP | Cell Signaling | 3183S |
| CHOP | Cell Signaling | 2895 |
| Santa Cruz Biotechnology | SC-7351 | |
| COX I | Abcam | Ab14705 |
| COX IV | Abcam | Ab14744 |
| CPN10 | Enzo Life Sciences | ADI-SPA-110 |
| GAPDH | Abcam | Ab8245 |
| MHC-II (MY-32) | Abcam | Ab51263 |
| mtHSP60 | Enzo Life Sciences | ADI-SPA-806 |
| mtHSP70 | Enzo Life Sciences | ADI-SPA-825 |
| PGC-1α | Millipore | AB3242 |
| Tfam | In house | n/a |
| Sirt3 | Cell Signaling | 5490S |
n/a, not applicable.
Statistical analysis.
All data are represented as means ± SE. Comparisons between days 0 and 4 were made with Student’s paired t-tests. Similar statistics were performed for the differentiated TUDCA-treated vs. vehicle cells, as well as for control vs. CCA myotubes. For experiments involving CCA in combination with TUDCA treatment or CHOP siRNA, two-way ANOVA was performed followed by Bonferroni’s post hoc tests when appropriate. Analyses were made with GraphPad Prism 7.0. Differences were considered significant if P < 0.05.
RESULTS
Differentiation model of C2C12 induces mitochondrial biogenesis.
C2C12 cells were stained with MitoTracker Green (Life Technologies) on days 0 and 4 of differentiation to identify the mitochondrial network. As depicted in Fig. 1A, myotubes had greater green fluorescence representing a higher mitochondrial content than myoblasts. To confirm this qualitative analysis, C2C12 whole cell lysates were collected at days 0 and 4 of differentiation and mitochondrial markers were probed as a measure of mitochondrial content. COX-I, a mitochondrially encoded subunit of complex IV, increased by five-fold from days 0 to 4 (P < 0.05; Fig. 1C). Similarly COX-IV, a nuclear encoded subunit of complex IV increased three-fold by day 4 of differentiation (P < 0.05; Fig. 1D). During the same timeframe, a twofold increase in mitochondrial transcription factor, Tfam (P < 0.05; Fig. 1E), was also observed. These data strongly confirm the increase in mitochondrial biogenesis during myocyte differentiation, similar to previous studies (8, 21).
Fig. 1.

C2C12 differentiation induces mitochondrial biogenesis. A: MitoTracker green fluorescent images captured at ×20 magnification of C2C12 myoblasts vs. myotubes on days (D) 0 and 4 of differentiation. Representative Western blots (B) and graphical quantifications of COX-I (C), COX-IV (D), and Tfam (E) at days 0 and day 4 of differentiation. Data are represented as means ± SE and are measured in arbitrary units (AU). *P < 0.05 vs. day 0; n = 3 experiments.
UPR is selectively activated during differentiation.
UPR protein markers of both the ER and mitochondria were analyzed in whole cell lysates at days 0 and 4 of differentiation by immunoblotting. The intramitochondrial chaperones, mtHSP70, mtHSP60, and CPN10 were all decreased significantly by day 4 (P < 0.05; Fig. 2, B–D). In contrast, the antioxidant protein Sirt3 increased by three-fold during differentiation (Fig. 2E). In relation to the UPRER markers, there was no change in CHOP protein between days 0 to 4 of differentiation (Fig. 2F); however, transcription factor ATF4 increased by 3.4-fold at day 4 (P < 0.05; Fig. 2G). BiP chaperone was at undetectable levels in the immunoblots at day 0 but increased significantly by day 4, which indicates increased ER stress (P < 0.05; Fig. 2H). Taken together, the UPRER is activated, while there is a decrease in UPRMT activity, with a very selective UPRMT protein induction in response to differentiation in muscle cells.
Fig. 2.

Skeletal muscle differentiation induces ER stress, while the UPRMT is largely inactivated. A: representative Western blots of UPRMT markers. Graphical quantifications of muscle cells during differentiation on days (D) 0 and 4: mtHSP70 (B), mtHSP60 (C), CPN10 (D), and Sirt3 (E). F: representative blots of UPRER markers. Graphical quantification of ATF4 (G) and BiP (H). Data are represented as means ± SE and are measured in arbitrary units (AU). *P < 0.05 vs. day 0; n = 3 experiments. n.d., not detectable.
UPRER inhibition with TUDCA augments mitochondrial content, despite reduced differentiation.
Cultured murine myoblasts were pretreated with one dose of TUDCA for 24 h before subsequent differentiation for 4 days. Phase-contrast images of stained cells were captured at day 4 of differentiation (Fig. 3A) to assess the fusion index (FI) as a measure of myotube formation. In vehicle-treated cells, differentiation was accompanied by enhanced myotube formation and fusion, as expected. In comparison to vehicle, cells treated with TUDCA had a greater number of nuclei dispersed outside of the myotubes remaining as unfused myoblasts (Fig. 3A). The percentage of nuclei in myotubes [fusion index (FI)] on day 4 of differentiation in TUDCA-treated cells (73%) was less than the vehicle (89%; P < 0.05, Fig. 3B). The FI of the control cells was high, and similar to the values found by Wiles et al. (43). In addition, significantly less MHC-II was present in TUDCA-treated cells than in the vehicle-treated cells, due to fewer myotube formations by day 4 (P < 0.05; Fig. 3C). Thus, partial inhibition of the UPR with TUDCA compromises myoblast fusion and the differentiation of muscle cells into myotubes.
Fig. 3.

TUDCA treatment increases mitochondrial biogenesis markers despite inhibiting differentiation. C2C12 cells were pretreated once for 24 h at subconfluence with TUDCA (T) or water [vehicle (V)] before either immediate collection (day 0) or differentiation (for 2 or 4 days). A: phase-contrast imaging was captured on day (D) 4 of differentiation. Lightly stained dots represent nuclei. B: fusion index (FI) was calculated as % of nuclei within myotubes. C: representative blot and quantification of MHC-II during differentiation D0, D2, and D4 of pretreated cells. Representative Western blots and their graphical summaries in TUDCA-treated cells mitochondrial markers: COX-I (D), COX-IV (E), Tfam (F), and PGC-1α (G) level measured in arbitrary units (AU). Data are represented as means ± SE. *P < 0.05 vs. vehicle (V); n = 3 to 10 experiments; n.d., not detectable.
To gain a better insight into the significance of the UPR in differentiation-induced mitochondrial biogenesis, mitochondrial biogenesis markers were measured on day 4 of differentiation in vehicle- and TUDCA-treated cells. While Tfam was unaffected by TUDCA treatment, PGC-1α, COX-1, and COX-IV were all ~1.3 to 1.5 times higher than control (P < 0.05; Fig. 3, D–G). Our results suggest that inhibition of the UPRER during myogenesis augments mitochondrial content, despite reduced differentiation.
TUDCA pretreatment in myoblasts decreased BiP induction but had no impact upon the UPRMT after 4 days of skeletal muscle differentiation.
UPR protein markers were assessed at day 4 of differentiation after 24 h of TUDCA or vehicle treatment at the myoblast stage. UPRMT markers mtHSP70, mtHSP60, and its cochaperone CPN10, as well as Sirt3, were not affected by TUDCA, as protein levels remained elevated at day 4 of differentiation, 4 days after the treatment (Fig. 4A). Additionally, there was no difference in UPRER markers CHOP and ATF4 protein content between treated and vehicle cells on day 4 of differentiation (Fig. 4B). However, levels of the ER chaperone BiP were attenuated by 45% in TUDCA-treated cells (P < 0.05; Fig. 4C).
Fig. 4.
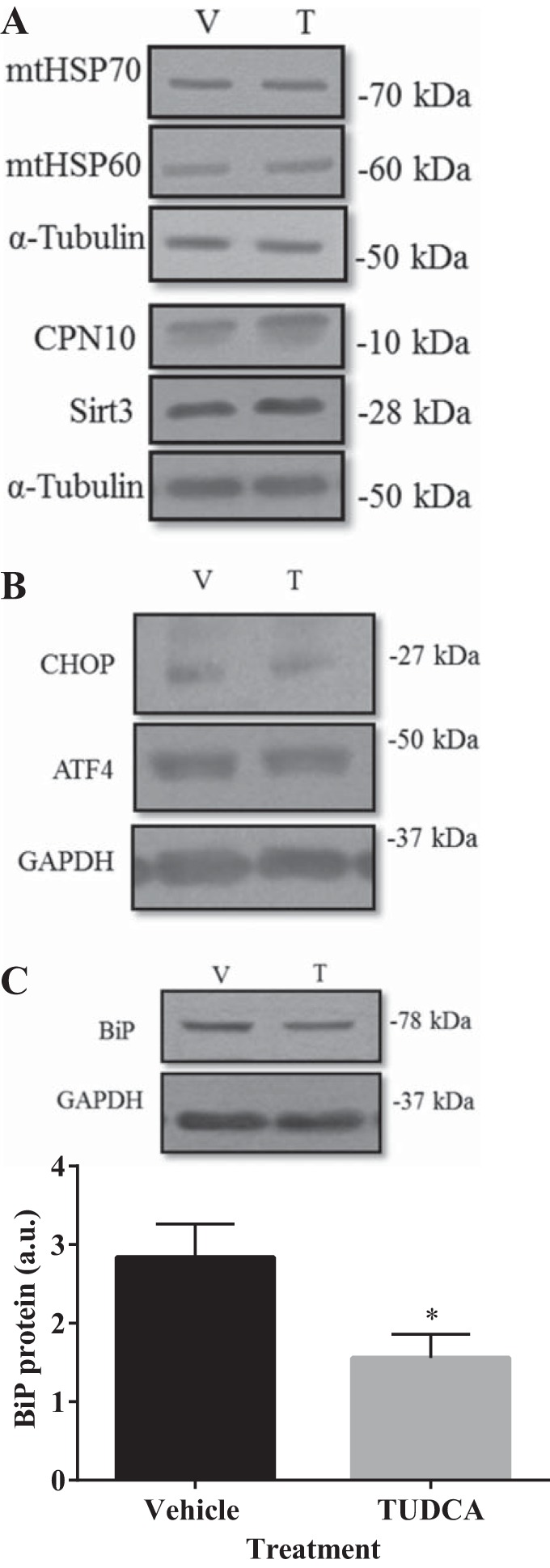
TUDCA treatment decreased BiP protein but had no impact upon the UPRMT after 4 days of skeletal muscle differentiation. Cells were pretreated for 24 h in 500 µg/ml of TUDCA (T) or water [vehicle (V)] before differentiation for 4 days. Representative Western blots of UPRMT proteins mtHSP70, mtHSP60, CPN10, and Sirt3 (A) were all similar in protein expression between vehicle- or TUDCA-treated cells. No effect of TUDCA was observed for UPRER markers CHOP and ATF4 (B). C: representative blot and graphical quantification of BiP protein. α-Tubulin served as the loading control. Data are represented as means ± SE and are measured in arbitrary units (AU). *P < 0.05 vs. vehicle; n = 3 or 4 experiments.
TUDCA treatment attenuates the increase in CHOP, while augmenting CCA-induced mitochondrial biogenesis.
To confirm whether our CCA model induces mitochondrial biogenesis, murine skeletal muscle cells were electrically stimulated on day 5 of differentiation for four consecutive days. As expected, CCA augmented COX-I and COX-IV by ~2.7-fold, and PGC-1α levels were augmented by 1.9-fold (P < 0.05; Fig. 5, A–C). Incubating cells with TUDCA from day 5 of differentiation increased mitochondrial markers in both resting (control) and CCA conditions, with a larger effect of TUDCA on COX-I and PGC-1α levels under CCA-stimulated conditions relative to the vehicle-treated myotubes (P < 0.05; Fig. 5, D and F). Interestingly, the nuclear encoded subunit COX-IV was increased by 2.6-fold with CCA as expected, but this increase was dependent on the presence or absence of TUDCA (P < 0.05; Fig. 5E). These findings suggest that CCA-induced mitochondrial biogenesis can be augmented under conditions of potent TUDCA treatment in skeletal muscle cells.
Fig. 5.

Chronic contractile activity induces mitochondrial biogenesis and is further augmented with TUDCA treatment. C2C12 cells were differentiated for 5 days before chronic contractile activity (CCA) for four successive days. Representative blots and graphical representations of mitochondrial markers COX-I (A), COX-IV (B), and PGC-1α (C). Blots in A, B, and C were reordered within the same blot to eliminate irrelevant lanes for direct comparison. Representative blots and graphical representations of cells treated with either TUDCA (T) or water [vehicle (V)] before and after CCA: COX-1 (D), COX-IV (E), and PGC-1α (F). Data are represented as means ± SE and are measured in arbitrary units (AU). πP < 0.05 vs. control (CON); *P < 0.05 main effect of CCA; φP < 0.05 main effect of TUDCA treatment; #P < 0.05 interaction effect of CCA and TUDCA treatment; n = 5 to 7 experiments.
With respect to UPR markers, TUDCA inhibited CCA-induced CHOP on both day 2 and 4 of stimulation by 64% and 30%, respectively, relative to control, thus attenuating this branch of the UPRER during CCA (P < 0.05; Fig. 6A). Of note, CHOP levels were unaltered by TUDCA under control conditions. In accordance with previously conducted studies, TUDCA inhibition of CHOP was not significant at basal levels unless an additional stressor was included (23). Furthermore, TUDCA had no impact on upstream general UPRER signaling evident by unchanged ATF4 and BiP content (Fig. 6, C and D).
Fig. 6.

TUDCA attenuates ER stress-induced CHOP associated with CCA and increases UPRMT antioxidant marker Sirt3 and CPN10 chaperone. C2C12 cells were differentiated for 5 days 1 h before chronic contractile activity (CCA), and cells were treated with either TUDCA (T) or water [vehicle (V)] for four consecutive days before collection. A: UPRER marker CHOP representative blot and graphical representation. B: representative blots of other UPR markers and their graphical representations: ATF4 (C), BIP (D), mtHSP70 (E), mtHSP60 (F), CPN10 (G), and Sirt3 (H). Data are expressed as means ± SE and are measured in arbitrary units (AU). πP < 0.05 vs. control (CON); *P < 0.05 main effect of CCA; φP < 0.05 main effect of TUDCA treatment; #P < 0.05 interaction effect of CCA and TUDCA treatment; n = 4 to 6 experiments.
TUDCA enhances the UPRMT antioxidant response and CPN10 induction with CCA.
As discussed above, UPRER markers CHOP, ATF4, and BiP remained unaffected with TUDCA treatment under basal conditions (Fig. 6, A, C, and D). Similar results were obtained basally with UPRMT chaperones mtHSP70, mtHSP60, and CPN10 (Fig. 6, E–G). However, Sirt3, the mitochondrial antioxidant marker of the UPR, increased by 30% (P < 0.05; Fig. 6G) with TUDCA treatment compared with vehicle control.
UPRER transcription factor ATF4 was elevated by an average of ~48% with CCA-induced stress, regardless of treatment (P < 0.05; Fig. 6C). BiP levels were augmented with CCA as well, particularly in vehicle-treated cells (66% vs. 8%; P < 0.05; Fig. 6D). Likewise, UPRMT markers increased with CCA, as displayed by the elevated levels of mtHSP70, mtHSP60, and Sirt3 (P < 0.05; Fig. 6, E, F, and H). Moreover, similar to basal levels, Sirt3 was 44% greater with in TUDCA-treated cells (P < 0.05; Fig. 6H), putatively as an antioxidant response accompanying the augmented mitochondrial content. Interestingly, CPN10 was augmented by 1.7-fold with CCA only upon TUDCA treatment (P < 0.05; Fig. 6G).
CHOP is required basally for the correct stoichiometry of electron transport chain subunits, and is compensated with CCA.
Because TUDCA attenuated CCA-induced CHOP expression but led to an increase in mitochondrial markers during differentiation, and as a result of CCA, it was of interest to investigate whether knocking down CHOP via siRNA would reproduce similar results of augmenting mitochondrial biogenesis. One day before CCA on day 4 of differentiation, C2C12 cells were transfected with CHOP or scrambled siRNA. To prolong the knockdown during the subsequent 4-day CCA treatment, cells were transfected a second time on day 7. CHOP knockdown was confirmed at the protein level. Basally, CHOP levels in knockdown cells were reduced by 42% relative to control. Under CCA conditions, CHOP protein was induced by three-fold in scrambled siRNA-treated cells, but there was no significant effect of CCA in knockdown cells (P < 0.05; Fig. 7A).
Fig. 7.

CHOP influences mitochondrial content basally and is not required for CCA-induced mitochondrial biogenesis. Cells were transfected with either 30 nM of scrambled (Scr) or CHOP siRNA (si-CHOP) on days 4 and 7 of differentiation. Myotubes underwent 4 days of successive chronic contractile activity (CCA) starting on day 5. A: CHOP representative blot and graphical representation. B: representative blots and graphical representations of mitochondrial markers COX-I (C), COX-IV (D), and Tfam (E). Data are expressed as means ± SE and are measured in arbitrary units (AU). πP < 0.05 vs. control (CON); *P < 0.05 main effect of CCA; φP < 0.05 main effect of CHOP siRNA; n = 7 to 11 experiments.
In the presence of CHOP knockdown under basal conditions, mitochondrially encoded COX-I increased by 1.2-fold, while nuclear-encoded proteins, COX-IV, decreased by 1.4-fold, and Tfam remained unaffected (P < 0.05; Fig. 7, B–E). In response to CCA, the increase in COX-I was further augmented by ~1.5-fold as expected, whereas remarkably, the COX-IV decrease observed basally in knockdown cells was compensated by three-fold to levels comparable with scrambled-treated cells (Fig. 7, C and D). Additionally, Tfam was elevated by 1.3-fold with CCA (P < 0.05; Fig. 7E). Altogether, the findings suggest that CHOP influences mitochondrial composition under basal conditions where mitochondrial biogenesis may be potentially impaired by orphaned electron transport chain subunits, but this can be rescued with CCA. Moreover, the contractile activity-induced mitochondrial biogenesis does not rely on CHOP induction of either the ER or mitochondrial UPR.
Of note, general UPR signaling of both the mitochondria and ER remained unaffected by CHOP silencing, as represented by the lack of changes observed at basal levels (Fig. 8, A–F). UPRER proteins BiP and ATF4 (P < 0.05; Fig. 8, B and C), as well as UPRMT markers mtHSP70, mtHSP60, and Sirt3, were all elevated with CCA, indicative of persistent stress induction and UPR activation associated with muscle contractile activity (P < 0.05; Fig. 8, D–F). Contrary to TUDCA-treated cells undergoing CCA, CPN10 levels did not increase, but remained unaltered (Fig. 8A).
Fig. 8.

CHOP knockdown in skeletal muscle cells had no impact on general UPR signaling. C2C12 cells were transfected with either 30 nM of scrambled (Scr) or CHOP siRNA (si-CHOP) on days 4 and 7 of differentiation. Chronic contractile activity (CCA) began on day 5 vs. control (CON) nonstimulated cells. A: representative blots of UPRMT/ER markers. Graphical representations of UPRER markers ATF4 (B), BiP (C), and UPRMT markers mtHSP70 (D), mtHSP60 (E), and Sirt3 (F). Data are expressed as means ± SE and are measured in arbitrary units (AU). *P < 0.05 main effect of CCA; φP < 0.05 main effect of CHOP siRNA; n = 4 to 7 experiments.
DISCUSSION
Mitochondria provide energy in the form of ATP to maintain cellular metabolic homeostasis (38). However, mitochondria also have alternative roles in regulating other pathways, such as autophagy (12, 18, 30), apoptosis (20, 25, 36, 41), and calcium homeostasis (10, 13, 22), all of which are vital to skeletal muscle health. During both muscle development and exercise, PGC-1α is activated, resulting in an increase in mitochondrial content (2, 8, 21, 23, 39). Mitochondrial biogenesis involves the synthesis of many nuclear and mitochondrially encoded proteins, and the increase in protein synthesis that occurs can potentially perturb cellular homeostasis by exceeding the protein-folding capacity of the cell. Thus, to prevent this accumulation of unfolded proteins, the unfolded protein response (UPR) occurs in both the endoplasmic reticulum (UPRER) and the mitochondrion (UPRMT). This involves the activation of a series of transcription factors to promote the transcription of genes encoding chaperones designed to assist in protein folding. Additionally, in an attempt to regain proteostasis, global protein translation is reduced. In this study, we hypothesized that this breadth of UPR signaling activation would impact mitochondrial adaptations induced either during skeletal muscle differentiation, or as a result of chronic contractile activity (CCA).
Similar to earlier work from our laboratory, C2C12 muscle cell differentiation led to the upregulation of mitochondrial markers as a result of the induced biogenesis (8). Along with this mitochondrial biogenesis triggered during muscle development, our data depict the selective activation of UPR components, which corroborates previous findings (26, 27). To investigate the role of ER stress, in particular, the terminal UPRER branch was partially inhibited using TUDCA. TUDCA functions as a chaperone mimetic to ameliorate ER stress by assisting in protein folding (9, 40). As a result, proapoptotic factors CHOP and caspase-12 induction are reduced with TUDCA treatment in response to cellular stress (23, 40, 45). The partial inhibition of UPRER signaling before the onset of differentiation was verified in our model by the decrease in myotube differentiation. Reduced differentiation is characteristic of ER stress attenuation, due to less myoblast fusion and diminished stress-induced myoblast apoptosis (26, 27, 43). Additionally, the ER stress marker BiP protein was decreased at day 4 of differentiation. This is consistent with another study in which the attenuated protein expression of BiP in human liver cells was observed in response to UPR inhibition (45).
Surprisingly, despite the reduced myotube differentiation, augmented mitochondrial biogenesis was observed, indicated by increased mitochondrial markers in the presence of TUDCA. These changes in specific mitochondrial markers occurred with the accompaniment of large increases in ATF4, no changes in CHOP, and a decrease in BiP protein level. This suggests that the attenuation of ER stress with TUDCA facilitated the synthesis of mitochondria and that ATF4 may be important in directing this process. This occurred in the absence of increases in UPRMT proteins mtHSP70, mtHSP60, and CPN10, suggesting that this induction is not central for differentiation-induced mitochondrial biogenesis.
We have previously shown that our cell culture model of CCA results in marked increases in mitochondrial biogenesis, as indicated by changes in MitoTracker green staining, increases in cellular respiration, as well as large changes in mitochondrial markers (24, 39). The current study confirmed those data and further suggests that the increases in mitochondrial content are accompanied by only modest changes in UPRMT markers, but large increases in BiP, as well as transcription factors ATF4 and CHOP. The inhibition of ER stress with TUDCA led to a further increase in the expression of mitochondrial markers, including PGC-1α, which had an additive CCA effect. This occurred despite the fact that the levels of an important transcription factor of the UPR, CHOP, were reduced or unchanged in the presence of TUDCA, while the increases in ATF4 and BiP were unaffected. These suggest that increases in mitochondrial content produced by CCA occur independently of CHOP, but that ATF4 and BiP may be important for the adaptation observed. Importantly also, components of the UPRMT (mtHSP70, mtHsp60, and CPN10) tended to be further elevated during CCA in the presence of TUDCA, suggesting an improvement in intraorganelle protein trafficking and folding capacity as a result of the combined treatments of contractile activity and ER stress attenuation.
Our results indicate that the dampening of ER stress with TUDCA improves both differentiation- and CCA-induced mitochondrial biogenesis, while selectively enhancing the UPRMT response during CCA. We were surprised by this result because our previous in vivo findings indicated that mitochondrial adaptations during CCA were largely independent of the presence of TUDCA and partial UPRER inhibition (23). Indeed, the UPRER inhibition with TUDCA in vivo did not amplify CCA-induced mitochondrial markers, such as PGC-1α protein and COX-IV (23), as we found in the current study. This may be the result of a differential magnitude of the TUDCA effect in vitro vs. in vivo, and this requires further investigation.
Wu et al. (44) have previously shown that the absence of CHOP had the potential to lead to an improvement in exercise tolerance. Since favorable changes in endurance are most often attributed to alterations in mitochondrial content, we wanted to verify whether the specific knockdown of CHOP would enhance mitochondrial content in the presence of CCA. Thus, we employed siRNA techniques, as we have done previously (17, 39), to partially reduce CHOP levels. siRNA treatment of myotubes effectively decreased CHOP levels by 43% under basal conditions and prevented the typical large increase in CHOP, which is normally induced by CCA. Importantly, the increases in other important components of the UPRER pathway, including BiP and ATF4, were unaffected by CHOP knockdown, and were increased by CCA, as expected. The reduction of CHOP under basal conditions led to parallel decreases in nuclear encoded COX-IV levels, no change in Tfam protein, and reciprocal levels of COX-I subunits, suggesting that CHOP would normally impact the transcription of COX-IV and mtDNA in an opposite fashion. The effect of CCA served to rescue the decline in COX-IV brought about by the absence of CHOP and had an additive effect on the level of the mtDNA-encoded subunit, COX-I. The expression of the UPRMT components mtHSP70, mtHSP60, and CPN10 was unaffected by CHOP knockdown under basal conditions, or as a result of CCA, as a normal increase was observed. This occurred despite the fact the UPRMT genes are activated through a CHOP-dependent pathway (1, 46). In the nucleus, CHOP forms a heterodimer with C/EBPβ (CCAAT enhancer-binding protein) creating an active transcription factor to upregulate mitochondria-quality control genes (1, 16, 46). However, an overexpression study of CHOP in monkey kidney cells revealed that this protein is not sufficient for the induction of the UPRMT proteins in the presence of stress. Thus, other transcription factors are likely involved (1), certainly in muscle. Alternatively, it may be that the degree of CHOP knockdown was insufficient to exert its effects upon its UPRMT downstream targets. What is evident from our study is that any mitochondrial protein expression imbalance created by the absence of CHOP can be fully rescued by CCA, indicating that CCA triggers alternative signaling pathways to maintain mitochondrial content and composition.
In both TUDCA-treated cells, as well as in the presence of CHOP siRNA, CHOP-induction was reduced under cellular stress. However, in contrast to the results obtained in TUDCA-treated cells, siRNA-induced CHOP knockdown did not further augment mitochondrial biogenesis, under either basal or CCA conditions. As CHOP knockdown did not reproduce similar results to TUDCA-treated cells in enhancing mitochondrial content, the observed increase in mitochondrial content with the drug treatment could be attributed to its pleiotropic effects in activating other mitochondrial biogenesis pathways. An alternative explanation may be that TUDCA has an overall attenuating effect upon cellular stress via facilitation of protein folding in the ER lumen (4, 9, 28, 40). Smiles and Camera (34) have suggested that this may result in a reduced ATP consumption by the UPRER pathway, potentially increasing the energy availability for mitochondrial biogenesis. For example, a potential increase in the activity of the energy-dependent protein import pathway into the mitochondria may further activate the UPRMT to ameliorate the mitochondrial stress associated with increased biosynthesis, as observed with our results.
Altogether, it is clear that the UPR is involved to some extent, in exercise-induced remodeling. In Wu et al.’s (44) study, the adaptation of ER stress markers to repeated bouts of treadmill running occurred before increases in mitochondrial cytochrome c, a surrogate marker of mitochondrial content, suggesting that initial improvements to ER proteostasis precede that of the synthesis of mitochondrial proteins (35). Additionally, ATF6 has been implicated during the recovery phase following exercise through a physical interaction with PGC-1α to trigger UPR signaling (44). Thus, a PGC-1α-mediated response suggests that the triggered UPR may be interlinked with other training adaptive responses that are similarly regulated by PGC-1α (35). Moreover, it is likely that different components of the UPR engage in crosstalk with other signaling pathways that are integral to skeletal muscle health (5). Thus, to better delineate what function the UPR serves in mitochondrial adaptations, UPR components other than CHOP, such as ATF6 or ATF5, should be eliminated to tease out whether distinct mechanisms are utilized by different arms of the UPR, as they may differentially be engaged in crosstalk with other pathways.
In summary, our findings suggest that mitochondrial biogenesis activated during muscle development from the myoblast to the myotube stage is CHOP-independent, and it may rely on other components of the UPRER to facilitate mitochondrial synthesis. Under basal, steady-state conditions, CHOP can influence mitochondrial composition by altering the correct stoichiometry of nuclear and mitochondrially encoded proteins of the electron transport chain. This can be salvaged by contractile activity in which other compensatory and redundant pathways of mitochondrial biogenesis are triggered. Indeed, CCA-induced mitochondrial adaptations occur, irrespective of CHOP induction and may be augmented via amelioration of ER stress to increase mitochondrial content further. Our study sheds light upon the necessity of UPR signaling for mitochondrial biogenesis during muscle phenotypic adaptations.
GRANTS
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada to D. A. Hood. D. A. Hood is also the holder of a Canada Research Chair in Cell Physiology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Z.S.M.M. and D.A.H. conceived and designed the research; Z.S.M.M. performed experiments; Z.S.M.M. analyzed data; Z.S.M.M. and D.A.H. interpreted results of experiments; Z.S.M.M. prepared figures; Z.S.M.M. drafted manuscript; Z.S.M.M. and D.A.H. edited and revised manuscript; Z.S.M.M. and D.A.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge the expert technical assistance of Claudia Tran.
REFERENCES
- 1.Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2: e874, 2007. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886, 2002. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 3.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci 36: 254–261, 2011. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Berger E, Haller D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun 409: 610–615, 2011. doi: 10.1016/j.bbrc.2011.05.043. [DOI] [PubMed] [Google Scholar]
- 5.Bohnert KR, Gallot YS, Sato S, Xiong G, Hindi SM, Kumar A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J 30: 3053–3068, 2016. doi: 10.1096/fj.201600250RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradshaw RA, Dennis EA (Editors). Regulation of Organelle and Cell Compartment Signaling. London: Elsevier Academic Press, 2011, p. 374, 413. [Google Scholar]
- 7.Carter HN, Hood DA. Contractile activity-induced mitochondrial biogenesis and mTORC1. Am J Physiol Cell Physiol 303: C540–C547, 2012. doi: 10.1152/ajpcell.00156.2012. [DOI] [PubMed] [Google Scholar]
- 8.Collu-Marchese M, Shuen M, Pauly M, Saleem A, Hood DA. The regulation of mitochondrial transcription factor A (Tfam) expression during skeletal muscle cell differentiation. Biosci Rep 35: e00221, 2015. doi: 10.1042/BSR20150073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gani AR, Uppala JK, Ramaiah KVA. Tauroursodeoxycholic acid prevents stress-induced aggregation of proteins in vitro and promotes PERK activation in HepG2 cells. Arch Biochem Biophys 568: 8–15, 2015. doi: 10.1016/j.abb.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Poletti F, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 12: 77–85, 2012. doi: 10.1016/j.mito.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gollnick PD, King DW. Effect of exercise and training on mitochondria of rat skeletal muscle. Am J Physiol 216: 1502–1509, 1969. [DOI] [PubMed] [Google Scholar]
- 12.Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signaling pathways. EMBO J 30: 2101–2114, 2011. doi: 10.1038/emboj.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrington JL, Murphy E. The mitochondrial calcium uniporter: mice can live and die without it. J Mol Cell Cardiol 78: 46–53, 2015. doi: 10.1016/j.yjmcc.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242: 2278–2282, 1967. [PubMed] [Google Scholar]
- 15.Hoppeler H, Lüthi P, Claassen H, Weibel ER, Howald H. The ultrastructure of the normal human skeletal muscle. A morphometric analysis on untrained men, women and well-trained orienteers. Pflugers Arch 344: 217–232, 1973. doi: 10.1007/BF00588462. [DOI] [PubMed] [Google Scholar]
- 16.Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2: e835, 2007. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iqbal S, Ostojic O, Singh K, Joseph A-M, Hood DA. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 48: 963–970, 2013. doi: 10.1002/mus.23838. [DOI] [PubMed] [Google Scholar]
- 18.Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D’Agati VD, Duffield JS. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26: 1040–1052, 2015. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HJ, Jamart C, Deldicque L, An GL, Lee YH, Kim CK, Raymackers JM, Francaux M. Endoplasmic reticulum stress markers and ubiquitin–proteasome pathway activity in response to a 200-km run. Med Sci Sports Exerc 43: 18–25, 2011. doi: 10.1249/MSS.0b013e3181e4c5d1. [DOI] [PubMed] [Google Scholar]
- 20.Kluck RM. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 275: 1132–1136, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Kraft CS, LeMoine CMR, Lyons CN, Michaud D, Mueller CR, Moyes CD. Control of mitochondrial biogenesis during myogenesis. Am J Physiol Cell Physiol 290: C1119–C1127, 2006. doi: 10.1152/ajpcell.00463.2005. [DOI] [PubMed] [Google Scholar]
- 22.Lim J-A, Li L, Kakhlon O, Myerowitz R, Raben N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 11: 385–402, 2015. doi: 10.1080/15548627.2015.1009779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Memme JM, Oliveira AN, Hood DA. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am J Physiol Cell Physiol 310: C1024–C1036, 2016. doi: 10.1152/ajpcell.00009.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menzies KJ, Singh K, Saleem A, Hood DA. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288: 6968–6979, 2013. doi: 10.1074/jbc.M112.431155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohamad N, Gutiérrez A, Núñez M, Cocca C, Martín G, Cricco G, Medina V, Rivera E, Bergoc R. Mitochondrial apoptotic pathways. Biocell 29: 149–161, 2005. [PubMed] [Google Scholar]
- 26.Nakanishi K, Dohmae N, Morishima N. Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB J 21: 2994–3003, 2007. doi: 10.1096/fj.06-6408com. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol 169: 555–560, 2005. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313: 1137–1140, 2006. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 1833: 410–416, 2013. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rambold AS, Lippincott-Schwartz J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 10: 4032–4038, 2011. doi: 10.4161/cc.10.23.18384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529, 2007. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 32.Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813: 1269–1278, 2011. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789, 2005. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 34.Smiles WJ, Camera DM. More than mitochondrial biogenesis: alternative roles of PGC-1α in exercise adaptation. J Physiol 593: 2115–2117, 2015. doi: 10.1113/JP270177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smiles WJ, Hawley JA, Camera DM. Effects of skeletal muscle energy availability on protein turnover responses to exercise. J Exp Biol 219: 214–225, 2016. doi: 10.1242/jeb.125104. [DOI] [PubMed] [Google Scholar]
- 36.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446, 1999. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 37.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13: 184–190, 2011. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tait SWG, Green DR. Mitochondria and cell signaling. J Cell Sci 125: 807–815, 2012. doi: 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uguccioni G, Hood DA. The importance of PGC-1α in contractile activity-induced mitochondrial adaptations. Am J Physiol Endocrinol Metab 300: E361–E371, 2011. doi: 10.1152/ajpendo.00292.2010. [DOI] [PubMed] [Google Scholar]
- 40.Vang S, Longley K, Steer CJ, Low WC. The unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob Adv Health Med 3: 58–69, 2014. doi: 10.7453/gahmj.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet 43: 95–118, 2009. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol 197: 857–867, 2012. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiles B, Miao M, Coyne E, Larose L, Cybulsky AV, Wing SS. USP19 deubiquitinating enzyme inhibits muscle cell differentiation by suppressing unfolded-protein response signaling. Mol Biol Cell 26: 913–923, 2015. doi: 10.1091/mbc.E14-06-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Boström P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab 13: 160–169, 2011. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, Yoffe B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 36: 592–601, 2002. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]