

Abstract
Objective
The aim of this study was to investigate whether the extent and topography of cerebral demyelination correlates with and predicts disease progression in patients with juvenile metachromatic leukodystrophy (MLD).
Methods
A total of 137 MRIs of 46 patients with juvenile MLD were analyzed. Demyelination load and brain volume were quantified using the previously developed Software “clusterize.” Clinical data were collected within the German Leukodystrophy Network and included full scale intelligence quotient (FSIQ) and gross motor function data. Voxel‐based lesion‐symptom mapping (VLSM) across the whole brain was performed to investigate the spatial relationship of cerebral demyelination with motor or cognitive function. The prognostic value of the demyelination load at disease onset was assessed to determine the severity of disease progression.
Results
The demyelination load (corrected by the individual brain volume) correlated significantly with gross motor function (r = +0.55) and FSIQ (r = −0.55). Demyelination load at disease onset was associated with the severity of disease progression later on (P < 0.01). VLSM results associated frontal lobe demyelination with loss in FSIQ and more central region demyelination with decline of motor function. Especially progression of demyelination within the motor area was associated with severe disease progression.
Interpretation
We were able to show for the first time in a large cohort of patients with juvenile MLD that the demyelination load correlates with motor and cognitive symptoms. Moreover, demyelination load at disease onset, especially the involvement of the central region, predicts severity of disease progression. Thus, demyelination load seems a functionally relevant MRI parameter.
Introduction
Metachromatic leukodystrophy (MLD) is a rare inherited metabolic disease that belongs to the lysosomal storage disorders. The disease is caused by a deficiency of the enzyme arylsulfatase A (ASA), leading to accumulation of the metachromatic glycosphingolipid 3′‐O‐sulfogalactosylceramide, also called sulfatide.1 As a consequence, progressive demyelination in the central and peripheral nervous system occurs, leading to severe and progressive neurological symptoms.1, 2 It has been shown that especially the central demyelination is responsible for loss in gross motor function.3
Demyelination in the brain is sensitively reflected in brain magnetic resonance imaging (MRI) signal changes, especially as hyperintensity in T2‐weighted images.4 In MLD, these MRI changes can be quantified visually with the MRI severity score.5 More recently, the “demyelination load” was established and validated as an MR parameter in order to quantify the demyelinated white matter (WM),6, 7 which might detect subtle changes in the extent of demyelination more sensitively than the visual MR scoring system.8
In the early‐onset (late‐infantile) form of MLD, where neurological deterioration occurs rapidly with loss of gross motor function as the key symptom,9 it has already been shown that the “demyelination load” highly correlates with disease duration and loss of gross motor function.7 Disease onset in “juvenile patients” is not only more protracted, but first signs are more variable including cognitive and behavioral symptoms, in addition to gross and fine motor dysfunction.9, 10
Despite the heterogeneous clinical course of juvenile MLD, we found a positive correlation between disease progression and MRI severity score11; and more recently, FSIQ measures in eight juvenile patients were demonstrated to correlate with MRI severity score and the demyelination load.12 Furthermore, the topography of demyelination may be important for clinical characteristics of the patients. While late‐infantile children show a more homogeneous evolution of demyelination from parieto‐occipital to frontal and temporal WM, juvenile patients develop a more variable picture, partly with early frontal predominance.11, 13, 14
Several new therapeutic approaches are under investigation, for example, hematopoietic stem cell transplantation (HSCT), enzyme replacement therapy, and gene therapy.1, 15 Evaluation and monitoring of therapeutic studies require precise surrogate parameters and reference data. Therefore, it seems important to investigate whether the demyelination load is a clinically relevant parameter, not only to describe, but also to predict disease progression in juvenile MLD. Especially, as HSCT was proven to be a beneficial treatment when done at an early stage of the disease before the phase of rapid progression.16, 17, 18, 19 It seems essential to find predictors of disease progression already early during disease course in order to be able to judge on the “window of opportunity,” for example, whether a treatment has enough time to become effective.
The aim of this study was to evaluate the demyelination load as a clinical surrogate parameter in juvenile MLD. As symptoms in the juvenile form are variable, we correlated the demyelination load both with standardized gross motor and cognitive parameters. Moreover, we investigated the functional relevance of the topography of demyelination for the patients’ symptomatology, and then examined whether the extent and topography of the demyelination at time of diagnosis is predictive for the dynamic of the clinical course.
Methods
Patients
Clinical and MRI data of patients with MLD were collected within the German leukodystrophy network leukonet since 2006.8, 11 The study was approved by the local ethics committee (401/2005). Written informed consent was given by at least one caregiver.
The diagnosis of MLD was based on a deficiency of arylsulfatase A (ASA), increased sulfatide excretion in urine, and typical changes in MRI11 in the context of clinical symptoms. In 31 of the 46 patients, diagnosis was additionally supported by molecular genetic testing (mutations in ARSA gene). Juvenile MLD was defined as disease onset between the age of 30 months and 16 years.1, 20 In clinically presymptomatic patients, this was done on the basis of either first MRI changes and/or decreased nerve conduction velocity. In addition, a juvenile disease course in a sibling was considered confirmatory and genotype had to be compatible with a juvenile course.
A total of 137 MRIs of 46 children with juvenile MLD (24 female) were included. Age at onset was between 2.5 and 13.75 years (mean: 6.7 ± 3.3 years) and patients’ age at MRI was between 0.92 and 35.5 years (mean: 9.8 ± 6.8 years). Fifty‐one MRIs of 11 patients were performed after they received HSCT. All other MRIs were performed during the natural disease course. Data under treatment were included in this study only for correlation of MRI changes with the patients’ motor and cognitive function. For the prognostic analysis, data under treatment were excluded.
Clinical parameter
The deterioration of gross motor function is a key clinical feature in MLD. We applied the standardized and validated Gross Motor Function Classification in MLD (GMFC‐MLD score 21).
Results from standardized neuropsychological testing were used to assess cognitive abilities. The full scale intelligence quotient (FSIQ) was tested with the Wechsler Intelligence Scale for Children (WISC; n = 26), Wechsler Adult Intelligence Scale (n = 15), and Kaufman Assessment Battery for Children (K‐ABC; n = 4). In some patients (n = 12), numerical neuropsychological test results were given in the medical records, however, without indication of the testing procedure (whether WISC or K‐ABC).
In order to assess the predictive value of both the demyelination load and its spatial extent at the time of diagnosis for the dynamic of the disease course, we retrospectively categorized the disease course of patients with a clinical follow‐up of at least 2 years into “slowly progressive” and “rapidly progressive.” This was done using the time interval between disease onset, that is, first clinical signs (whether motor or cognitive/behavioral), and entering GMFC‐MLD level 2 (=loss of independent walking). As we had shown before that GMFC‐MLD 2 marked the onset of rapid regression in the natural history of juvenile MLD patients, we used the median (=27 months) characterizing the time between GMFC‐MLD levels 1 and 2, as previously published.11 Therefore, patients with less than or equal to 27 months between levels 1 and 2 were categorized as having a “rapidly progressive” disease course, and patients with more than 27 months between levels 1 and 2 or more than 27 months since onset of the disease (without deteriorating to level 1, i.e., only cognitive symptoms) were defined as “slowly progressive.”
MRI parameter
MRI data were acquired on different clinical scanners. For this study, axial T2‐weigthed images (with a high mean in‐plane resolution of 0.56 × 0.56 mm2 ± 0.19/0.19 mm) and a mean slice thickness of 4.77 mm (±1.3 mm) were selected. Furthermore, high‐resolution 3D T1‐weighted images (mean 0.97 × 0.76 × 0.84 mm3 ± 0.31/0.24/0.22 mm3) or axial FLAIR‐weighted images (mean slice thickness 4.73 mm ± 1.4 mm) were included.
To quantify the volume of demyelinated WM, the “demyelination load” was measured semiautomatically as described before using the “clusterize”6 toolbox (www.medizin.uni-tuebingen.de/kinder/en/research/neuroimaging/software). To account for the variability of different brain volumes and the brain atrophy at late stages of MLD,11 we corrected the demyelination load by the children's individual brain volume as done before using the clusterize toolbox.6
Voxel‐based lesion‐symptom mapping
To identify the effect of the local distribution of demyelination, voxel‐based lesion‐symptom mapping (VLSM) was applied. This method analyzes the relation of tissue lesions with symptoms on a voxel‐by‐voxel basis.22 In order to detect joint areas of demyelination causing certain symptoms, images have to be brought into a standard space; this was achieved by spatially normalizing all demyelination load masks using the spatial deformation field generated by spatially normalizing high‐resolution T1 3D datasets to MNI space, applying segmentation functionality available within the statistical parametric mapping (SPM12) software (http://www.fil.ion.ucl.ac.uk/spm/software/spm12/).23
Statistical analysis
In order to investigate the association of the amount of demyelination with clinical parameters, the demyelination load was correlated with the GMFC‐MLD score using Spearman's rank correlation (categorical variable) coefficient and with the FSIQ using Pearson's correlation coefficient (continuous variable).
In addition, using the VLSM approach, we analyzed the whole brain WM for symptom‐causing areas of demyelination correlating with clinical parameters. Here, the MRIcron‐tool “nonparametric mapping” was used to generate the demyelination symptom maps (www.mccauslandcenter.sc.edu/crnl/chris-rordens-neuropsychology-lab). Every analysis had eight threads and only voxels damaged in at least 10% of the patients were included. GMFC‐MLD and FSIQ were tested applying the nonparametric Brunner–Munzel test.24 For multiple comparison correction, a permutation correction was used at a 5% level, using 1000 permutations, as recommended for this kind of analysis.25
In order to assess the predictive value of the demyelination load at disease onset for later disease progression, the first MRI after onset of symptoms was evaluated. The amount of the demyelination load was compared between the “slowly” and “rapidly progressing” patients, as defined above using the Mann–Whitney U test. For investigation of spatial extent of the demyelination load between the two groups, the VLSM approach was used and a Liebermeister test was applied for binomial data, again using permutation correction for multiple comparison correction.25
Results
Demyelination load and gross motor function
A total of 137 MRIs and clinical data of all 46 patients were available. When assessing the interrelation of demyelination load and GMFC‐MLD scores, there was a significant positive correlation (ρ = 0.55, P < 0.0001; Fig. 1A), indicating that patients with a higher demyelination load also had higher GMFC‐MLD score (i.e., more motor disability). When only investigating cross‐sectional data, that is, their last available MRI (n = 46, mean age: 12.2 ± 8.5 years), this correlation remained strong (ρ = 0.60, P < 0.0001).
Figure 1.
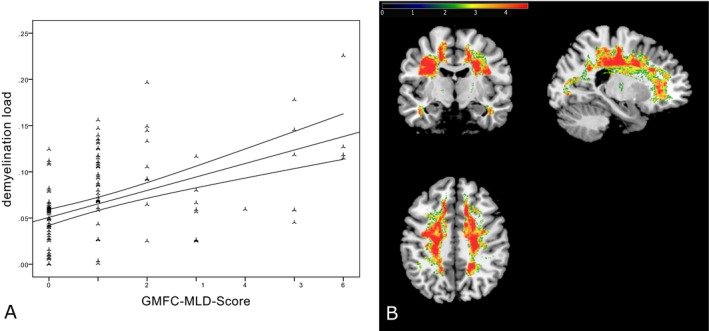
Correlation between GMFC‐MLD score and demyelination load. (A) The positive correlation between the corrected demyelination load and gross motor function loss, indicated by higher GMFC‐MLD scores (ρ = 0.55, P < 0.0001). (B) The spatial association between demyelination of white matter (WM) in the central region and gross motor deterioration (higher GMFC‐MLD scores). For illustration, significance thresholds of 5% false discovery rate correction24, 34 are displayed. The color bar represents z scores.
For the VLSM analysis, 125 demyelination load masks of 44 patients were available after visual inspection of successful spatial normalization. Figure 1B shows WM areas where the demyelination correlated with deterioration of gross motor function (as assessed by the GMFC‐MLD). The WM underlying the primary cortical motor region was particularly associated with gross motor deterioration and to some degree also prefrontal and parieto‐occipital areas (Fig. 1B).
Demyelination load and IQ
Fifty‐seven MRIs and FSIQ results of 25 patients were available (mean age at MRI: 14.2 ± 6.0 years). When assessing the interrelation of corrected demyelination load and the FSIQ scores, there was a strongly significant negative correlation (r = −0.55, P < 0.0001), indicating that patients with a higher demyelination load had a lower FSIQ score (i.e., more cognitive impairment). This relation is illustrated in Figure 2A. Again, this correlation was also found in cross‐sectional data (n = 25, r = −0.54, P < 0.01, mean age: 12.4 ± 6.6 years).
Figure 2.
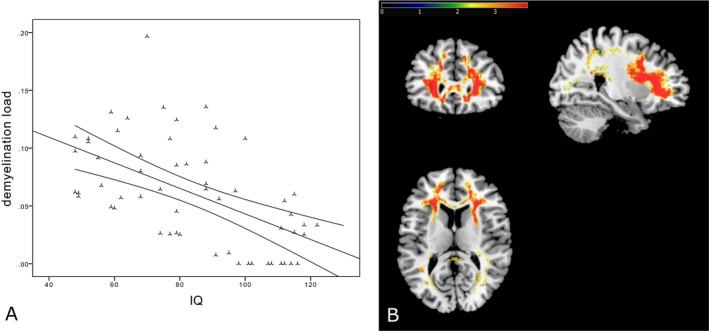
Correlation between IQ and demyelination load. (A) The negative correlation between the corrected demyelination load and cognitive function (correlation coefficient = −0.55, P < 0.0001). (B) The spatial relation between loss of cognitive function (decreasing IQ) and demyelination in prefrontal regions. For illustration, significance thresholds of 5% false discovery rate correction24, 34 are displayed. The color bar represents z scores.
For the VLSM analysis, 50 demyelination load masks of 24 patients were available (mean age at MRI: 13.8 ± 5.9 years). Figure 2B shows significant areas of negative correlation between FSIQ and areas of demyelination. Especially, demyelination load in frontal regions revealed a significant correlation with lower FSIQ. In contrast, central WM regions were spared and only few temporal and parietal regions were involved (Fig. 2B).
Prediction of disease progression
From 15 patients, a long clinical follow‐up was available which allowed long‐term evaluation of disease course. Of these, seven patients showed a rapidly progressive and eight patients a slowly progressive course as defined above. Slowly progressive patients had significantly lower demyelination load at their first MRI than rapidly progressing patients as shown in Figure 3A (P = 0.003), although rapidly progressing patients had their first MRI even earlier after disease onset (mean: 1.2 ± 0.98 years after onset), compared to slowly progressing patients (mean: 4.1 ± 3.5 years after onset, P = 0.03, Mann–Whitney U test). Age at first MRI of these 15 patients was between 4.5 and 18.2 years (mean: 9.6 ± 4.3 years). It is important to note that age at onset, which was between 2.5 and 13.5 years (mean: 6.9 ± 3.2 years), did not differ between the groups (P > 0.1), and thus, did not predict disease progression in this cohort. However, there was a significantly negative correlation of the age of onset with the demyelination load (ρ = 0.57, P = 0.024), indicating that patients with an early disease onset have on average a higher demyelination load at their first MRI after onset. This is also illustrated in Figure 3, where results were color coded by the age of onset.
Figure 3.
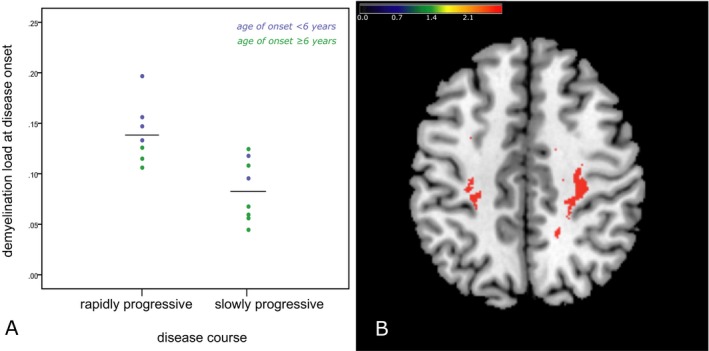
Predictive value of the demyelination load at diagnosis. (A) Comparison of demyelination load at presentation between the patients with slow and rapid disease progression (P = 0.003). Mean values are indicated as horizontal black lines. Blue indicates disease onset below 6 years of age, green 6 years and above. The spatial lesion map on the right (B) suggests the early demyelination of subcortical white matter (WM) (U‐fibers) in the central region is related to rapid progression of the following disease course. For illustration, significance thresholds of 5% false discovery rate correction24, 34 are displayed. The color bar represents z scores.
The VLSM revealed that particularly demyelination involving areas in the central region (and especially toward the U‐fibers of the WM) at time of diagnosis was associated with a rapid progression of their disease course (Fig. 3B).
Discussion
Metachromatic leukodystrophy is a rare, but invariably progressive and ultimately fatal neurodegenerative disorder. New therapeutic approaches are currently under investigation.1, 15 There is an increasing body of evidence suggesting that successful therapy is related to early treatment, that is, presymptomatic and early symptomatic,16, 17, 18, 19 and to a slower disease progression, that is, treatments such as HSCT being more effective in the more slowly progressive juvenile form than in the rapidly progressive late‐infantile form.17, 18 Thus, one of the clinical challenges when to recommend HSCT in juvenile MLD is to judge on the dynamics of the disease course. And before the loss of independent walking announcing rapid regression,9 this is not possible on clinical parameters alone. One might expect that age at onset, for example, early juvenile versus late juvenile, might be such a parameter, but there is no clear evidence supporting this notion. This was also seen in our study where age at onset did not significantly differ between rapid and slow disease progression. However, we found that age of onset did correlate negatively with the demyelination load in the MRI done at onset. Therefore, it can be concluded that age of onset might have an additional influence, but alone (without incorporating the demyelination load) did not predict disease progression in our cohort. It therefore seems particularly important to evaluate potential biomarkers which are not only able to characterize but also to predict different aspects of disease progression.
In a large cohort of patients with the juvenile form of MLD we were not only able to show that the demyelination load correlated with regression of motor and cognitive function, but we could especially demonstrate that the demyelination load in the first MRI after onset of symptoms was predictive for disease progression, that is, a higher demyelination load indicates a more severe and rapid clinical course. In addition, we were able to show that the topography of areas showing demyelination is functionally relevant.
It has already been shown that the demyelination load is a functionally relevant parameter for patients with late‐infantile MLD.7 In the current study, we were able to show that the demyelination load correlates with motor and cognitive function also in the juvenile form, where the MRI changes were expected to be more variable.11 We could corroborate findings from a recent correlation of demyelination load and IQ in a mixed (late‐infantile, juvenile, adult form) MLD cohort, where a similar degree of correlation was found.12
In addition, we could show that the topography of demyelination within the cerebral WM is functionally relevant, which is clearly a new finding. In particular, regression of cognitive function was associated with more pronounced involvement of frontal WM areas, sparing the central (motor) parts of the WM. It seems noteworthy that frontal demyelination, causing cognitive symptoms, can appear independently without impairment of the central region. Clinically, an isolated longer lasting cognitive manifestation has been reported especially in MLD of later juvenile or adult onset.10, 26 The frontal lobe is generally considered to be responsible for multiple cognitive domains (especially related to executive functions, e.g., working memory, episodic memory, problem solving, and perception).27 Its impairment therefore explains the typical cognitive symptoms in juvenile MLD, which include decreasing school performance, deficiency in concentration and attention, and psychiatric symptoms.1, 10 The deterioration of gross motor function was associated with more widespread WM involvement, but particularly including the WM underlying the central motor area (and thus, presumably, the corticospinal tract). Further affected regions included prefrontal and parieto‐occipital WM regions. The latter most likely explains the clinical finding typical for both the late‐infantile and juvenile forms of MLD that severe motor regression (once independent walking is lost) occurs often in parallel with loss of cognitive function and a generally severe clinical deterioration.9, 10 Our findings are therefore well in line with the clinical impression that, if widespread central and prefrontal demyelination is present and independent walking is lost, a rapid loss of cognitive functions can also be expected.
These findings support the functional relevance of the demyelination load and show that it is a useful surrogate parameter to evaluate disease progression and indicate its usefulness also for evaluation of therapy effects. It has to be kept in mind in this context that MRI changes usually precede neurological symptoms in juvenile MLD,11 thus, first signs of demyelination do not immediately lead to a functional deficit. But the further progression of demyelination, and especially the involvement of specific WM areas, seems to correlate well with specific aspects of clinical deterioration. In the evaluation of therapeutic effects and cerebral changes on follow‐up, the demyelination load was already demonstrated to be more sensitive than the MRI scoring system.8, 28 Still, the sensitivity and specificity of new (more myelin‐specific) MRI parameters might be important to evaluate further in detecting functionally relevant WM changes in the brain as suggested recently, for example, diffusion‐weighted imaging, MR spectroscopy, magnetization transfer imaging, myelin water imaging, cortical thickness determination, and GM/WM volumetry.12, 29, 30, 31
The probably most intriguing and clinically relevant finding of this study was the prognostic value of the demyelination load at the time of diagnosis for the severity of the to‐be‐expected disease course. Here, patients with slowly progressing disease showed a significantly lower demyelination load. This finding seems all the more relevant as the mean time from disease onset to the first MRI was even significantly longer in the group with slower progression than in the group with rapid progression. Hence, even though the initial MRI was obtained later, there was less demyelination. Given the variability in the clinical course, this is not only important for patient counseling, but also for treatment decision as discussed above. It has been shown that the treatment effect of HSCT, currently the only clinically available treatment option for juvenile MLD, might take some time to have an effect.8, 32, 33 Therefore, it seems essential to have additional parameters in order to predict whether the individual patient will deteriorate rapidly or might still have a wide enough window of opportunity for HSCT. For this reason, it is an important finding that patients with rapid disease progression have a significantly higher demyelination load already at time of diagnosis. Especially, our findings that demyelinated U‐fibers in the central region of the WM might predict rapid disease progression seem of importance in this respect.
In conclusion, we were able to assess, quantify, and localize the interrelation between demyelination load and gross motor as well as cognitive function in patients with juvenile MLD. Most importantly, our results provide evidence that rapidly progressing juvenile patients have a higher demyelination load already at time of diagnosis, compared to more slowly progressing children. Especially, the central WM U‐fibers seem to be of critical importance for predicting the severity of the clinical disease course. Our data confirm the demyelination load to be a clinically relevant MR parameter to describe functional parameters of disease progression in juvenile MLD; it may therefore also be an important surrogate marker when it comes to evaluating different treatment regimes in this otherwise fatal neurodegenerative disease.
Conflict of Interest
M. Strölin, I. Krägeloh‐Mann, C. Kehrer, and M. Wilke have nothing to disclose. S. Groeschel reports an institutional research grant from Shire plc, outside of the submitted work.
Acknowledgments
We thank all participating centers of the German Leukonet, coordinator Volkmar Gieselmann, as well as patients and their families. We are also grateful for the technical advice given by Dr Bianca de Haan, Division of Neuropsychology, University of Tübingen, Germany. Furthermore, we thank Dr Judith Böhringer, Neurometabolic Lab, Children's Hospital of Tübingen, Germany, for providing biochemical and genetic diagnostic information of the patients.
References
- 1. Gieselmann V, Krägeloh‐Mann I. Metachromatic Leukodystrophy In: D Valle, AL Beaudet, B Vogelstein, KW Kinzler, SE Antonarakis, A Ballabio, KM Gibson. G Mitchell. The online metabolic and molecular bases of inherited disease. New York: McGraw‐Hill; 2014. p. chapter 148. [Google Scholar]
- 2. Gieselmann V, Krägeloh‐Mann I. Metachromatic leukodystrophy–an update. Neuropediatrics 2010;41:1–6. [DOI] [PubMed] [Google Scholar]
- 3. Dali CÍ, Barton NW, Farah MH, et al. Sulfatide levels correlate with severity of neuropathy in metachromatic leukodystrophy. Ann Clin Transl Neurol 2015;2:518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eichler F, Grodd W, Grant E, et al. Metachromatic Leukodystrophy: a Scoring System for Brain MR Observations. AJNR AmJNeuroradiol 2009;30:1893–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van der Voorn JP, Pouwels PJ, Kamphorst W, et al. Histopathologic correlates of radial stripes on MR images in lysosomal storage disorders. AJNR AmJNeuroradiol 2005;26:442–446. [PMC free article] [PubMed] [Google Scholar]
- 6. Clas P, Groeschel S, Wilke M. A semi‐automatic algorithm for determining the demyelination load in metachromatic leukodystrophy. Acad Radiol 2012;19:26–34. [DOI] [PubMed] [Google Scholar]
- 7. Groeschel S, i Dali C, Clas P, et al. Cerebral gray and white matter changes and clinical course in metachromatic leukodystrophy. Neurology 2012;79:1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krägeloh‐Mann I, Groeschel S, Kehrer C, et al. Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non‐transplanted cohort. Bone Marrow Transplant 2013;48:369–375. [DOI] [PubMed] [Google Scholar]
- 9. Kehrer C, Blumenstock G, Gieselmann V, Krageloh‐Mann I. The natural course of gross motor deterioration in Metachromatic Leukodystrophy. DevMedChild Neurol 2011;53:850–855. [DOI] [PubMed] [Google Scholar]
- 10. Kehrer C, Groeschel S, Kustermann‐Kuhn B, et al. Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet J Rare Dis 2014;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Groeschel S, Kehrer C, Engel C, et al. Metachromatic Leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. JInheritMetab Dis 2011;34:1095–1102. [DOI] [PubMed] [Google Scholar]
- 12. Tillema J‐M, Derks MG, Pouwels PJW, et al. Volumetric MRI data correlate to disease severity in metachromatic leukodystrophy. Ann Clin Transl Neurol 2015;2:932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schiffmann R, van der Knaap MS. Invited article: an MRI‐based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van der Knaap MS, Valk J. Magnetic resonance in myelin, myelination, and myelin disorders. Berlin Heidelberg: Springer, 2005. [Google Scholar]
- 15. Krägeloh‐Mann I, Groeschel S. Therapies of lysosomal storage disorders targeting the brain. Lancet Lond Engl 2016;388:440–442. [DOI] [PubMed] [Google Scholar]
- 16. Groeschel S, Kühl J‐S, Bley AE, et al. Long‐term Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in Patients With Juvenile Metachromatic Leukodystrophy Compared With Nontransplanted Control Patients. JAMA Neurol 2016;73:1133–1140. [DOI] [PubMed] [Google Scholar]
- 17. Boucher AA, Miller W, Shanley R, et al. Long‐term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single‐institution cohort report. Orphanet J Rare Dis 2015;10:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin HR, Poe MD, Provenzale JM, et al. Neurodevelopmental Outcomes of Umbilical Cord Blood Transplantation in Metachromatic Leukodystrophy. Biol. Blood Marrow Transplant. J Am Soc Blood Marrow Transplant 2013;19:616–624. [DOI] [PubMed] [Google Scholar]
- 19. van Rappard DF, Boelens JJ, van Egmond ME, et al. Efficacy of hematopoietic cell transplantation in metachromatic leukodystrophy: the Dutch experience. Blood 2016;127:3098–3101. [DOI] [PubMed] [Google Scholar]
- 20. Moser H, Lees M. Sulfatide lipidosis: metachromatic leukodystrophy In: JB Stanbury, JB Wyngaarden, DS Fredrickson. The metabolic basis of inherited disease. New York: McGraw‐Hill; 1965. p. 539–559. [Google Scholar]
- 21. Kehrer C, Blumenstock G, Raabe C, Krageloh‐Mann I. Development and reliability of a classification system for gross motor function in children with metachromatic leucodystrophy. DevMedChild Neurol 2011;53:156–160. [DOI] [PubMed] [Google Scholar]
- 22. Bates E, Wilson SM, Saygin AP, et al. Voxel‐based lesion‐symptom mapping. Nat Neurosci 2003;6:448–450. [DOI] [PubMed] [Google Scholar]
- 23. Malone IB, Leung KK, Clegg S, et al. Accurate automatic estimation of total intracranial volume: a nuisance variable with less nuisance. NeuroImage 2015;104:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rorden C, Karnath HO, Bonilha L. Improving lesion‐symptom mapping. JCogn Neurosci 2007;19:1081–1088. [DOI] [PubMed] [Google Scholar]
- 25. Medina J, Kimberg DY, Chatterjee A, Coslett HB. Inappropriate usage of the Brunner‐Munzel test in recent voxel‐based lesion‐symptom mapping studies. Neuropsychologia 2010;48:341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colsch B, Afonso C, Turpin J‐C, et al. Sulfogalactosylceramides in motor and psycho‐cognitive adult metachromatic leukodystrophy: relations between clinical, biochemical analysis and molecular aspects. Biochim Biophys Acta 2008;1780:434–440. [DOI] [PubMed] [Google Scholar]
- 27. Duncan J, Owen AM. Common regions of the human frontal lobe recruited by diverse cognitive demands. Trends Neurosci 2000;23:475–483. [DOI] [PubMed] [Google Scholar]
- 28. Groeschel S, Kehrer C, Krägeloh‐Mann I. Standardized description of rare diseases ‐ the natural course and treatment of metachromatic leukodystrophy. Rev Neurol 2014;48:C84–C93. [Google Scholar]
- 29. Groeschel S, Hagberg GE, Schultz T, et al. Assessing White Matter Microstructure in Brain Regions with Different Myelin Architecture Using MRI. PLoS ONE 2016;11:e0167274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pouwels PJW, Vanderver A, Bernard G, et al. Hypomyelinating leukodystrophies: translational research progress and prospects. Ann Neurol 2014;76:5–19. [DOI] [PubMed] [Google Scholar]
- 31. i Dali C, Hanson LG, Barton NW, et al. Brain N‐acetylaspartate levels correlate with motor function in metachromatic leukodystrophy. Neurology 2010;75:1896–1903. [DOI] [PubMed] [Google Scholar]
- 32. Stillman AE, Krivit W, Shapiro E, et al. Serial MR after bone marrow transplantation in two patients with metachromatic leukodystrophy. AJNR Am J Neuroradiol 1994;15:1929–1932. [PMC free article] [PubMed] [Google Scholar]
- 33. van Egmond ME, Pouwels PJW, Boelens J‐J, et al. Improvement of white matter changes on neuroimaging modalities after stem cell transplant in metachromatic leukodystrophy. JAMA Neurol 2013;70:779–782. [DOI] [PubMed] [Google Scholar]
- 34. Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. NeuroImage 2002;15:870–878. [DOI] [PubMed] [Google Scholar]