

Abstract
YAP is a transcriptional coactivator that controls organ expansion and differentiation and is inhibited by the Hippo pathway in cells in interphase. Here, we demonstrated that, during mitosis, YAP localized to the midbody and spindle, subcellular structures that are involved in cytokinesis, the process by which contraction of the cytoskeleton produces two daughter cells. Furthermore, YAP was phosphorylated by CDK1, a kinase that promotes cell cycle progression. Knockdown of YAP by shRNA or expression of a nonphosphorylatable form of YAP delayed the separation of daughter cells (called abscission) and induced a cytokinesis phenotype associated with increased contractile force, membrane blebbing and bulges, and abnormal spindle orientation. Consequently, these defects led to an increased frequency of multinucleation, micronuclei, and aneuploidy. YAP was required for proper localization of proteins that regulate contraction during cytokinesis, including ECT2, MgcRacGap, Anillin, and RHOA. In addition, depletion of YAP increased the phosphorylation of myosin light chain, which would be expected to activate the contractile activity of myosin II, the molecular motor involved in cytokinesis. The polarity scaffold protein PATJ coprecipitated with YAP and colocalized with YAP at the cytokinesis midbody, and knockdown of PATJ phenocopied the cytokinetic defects and spindle orientation alterations induced by either YAP depletion or expression of a nonphosphorylatable YAP mutant. Together, these results reveal an unanticipated role for YAP in the proper organization of the cytokinesis machinery during mitosis through interaction with the polarity protein PATJ.
INTRODUCTION
The Yes-associated protein (YAP) was originally identified on the basis of its interaction with the Src family kinase Yes (1). YAP functions primarily as a co–transcription factor that interacts with various DNA binding proteins, including the transcription factors RUNX, TEAD/TEF, and p73, to regulate gene expression (2, 3). Genetic screens independently identified the Drosophila ortholog Yki as a gene that regulates cell proliferation and apoptosis (4). Both Yki and YAP are regulated by a conserved kinase cascade that includes Hippo (Hpo) and its mammalian orthologs MST1/2, which interact with Salvador (Sav) in flies or SAV1 (also known as WW45) in mammals. This kinase complex phosphorylates Warts (Wts) (LATS1/2 in mammals), which in turn phosphorylates Ser168 in Yki in flies and Ser127 in YAP. Yki/YAP phosphorylation tethers these proteins to 14-3-3 and sequesters them in the cytoplasm, inhibiting nuclear translocation and downstream transcriptional programs.
The YAP gene resides within a chromosomal region (11q22) that is amplified in various cancers, and several reports indicate that YAP can function as an oncogene. Indeed, YAP overexpression drives tumor formation in vivo (5, 6) and can transform cells in culture (7), and endogenous YAP is required for tumorigenicity induced by MST1/2 or NF2 loss of function in vivo (8–10). Increased YAP abundance is also found in various human cancers, including esophageal (11), gastric (11, 12), colon (13), lung (13), and ovarian carcinoma (13), non–small cell lung cancer (14), and hepatocellular carcinoma (15), for which increased YAP abundance is an adverse prognostic marker. However, YAP loss of heterozygosity and reduced protein abundance occur in some cancers, and YAP depletion can promote cell survival (16, 17), enhance migration and invasion, and promote tumor growth in immunocompromised mice (18), suggesting that YAP can also act as a tumor suppressor in some contexts.
In addition to the conserved functions in regulating tissue growth, the mammalian Hippo pathway orthologs have also been implicated in the regulation of mitosis. Loss-of-function alterations in LATS1 or LATS2 in Drosophila and mouse cells induce cytokinesis defects and multinucleation (19–23). Furthermore, MST2, WW45, and LATS, along with RASSF1A, a tumor suppressor protein, coassociate in a mitotic complex localized to centrosomes and the midbody (19, 24, 25). Like RASSF1A-deficient mouse embryonic fibroblasts (MEFs), LATS1-deficient MEFs fail cytokinesis (19, 24, 25). Deficiency in Drosophila Hpo leads to central spindle defects and the presence of lagging strands (26). Although these data demonstrate a mitotic role for members of the Hippo pathway, their precise functions in cell division and whether YAP, the key target of this pathway, plays a role in this process remain unknown.
Here, we identified a role for YAP in cytokinesis. We showed that YAP knockdown resulted in abnormal morphological alterations during cytokinesis as well as aberrant RHOA localization and myosin light chain (MLC) phosphorylation, spindle misorientation, and an increase in cytokinesis failure in HeLa and MCF-10A cells. In addition, we identified the polarity protein PATJ as a YAP-interacting partner that is also required for proper execution of cytokinesis. These data demonstrate that, in addition to its roles in transcription, YAP is a direct regulator of mitosis.
RESULTS
YAP localizes to the central spindle and midbody ring
Several mammalian Hippo pathway proteins localize to the mitotic machinery during cell division. To examine the localization of YAP during mitosis, we immunostained MCF-10A human mammary epithelial cells and HeLa cells with a panel of YAP antibodies. Using three different antibodies to YAP, we found that YAP, like MST and LATS proteins, localized to the mitotic machinery of dividing cells, at the central spindle during anaphase and at the midbody ring during telophase/cytokinesis (Fig. 1, A to G). We also found that YAP phosphorylated at Ser127, a product of LATS1-mediated phosphorylation, localized to the same mitotic structures (Fig. 1H). Moreover, exogenously expressed Flag-tagged YAP (Flag-YAP) or an enhanced green fluorescent protein–YAP (EGFP-YAP) fusion protein was also enriched at the midbody (Fig. 1, I and J). The validity of YAP midbody localization was supported by reduced YAP at this site in cells depleted of YAP by short hairpin RNA (shRNA) (fig. S1). Endogenous YAP colocalized with the midbody component Cep55 as well as upstream components of the Hippo pathway, such as MST and LATS (Fig. 1, K to M). These results showed that YAP associated with mitotic structures and raised the question of whether YAP regulates any aspect of mitosis.
Fig. 1. YAP localizes to the central spindle and to the midbody ring.

(A and B) Endogenous YAP (A) and LATS1 (B) localize to the central spindle of MCF-10A cells in anaphase. YAP (Cell Signaling Technology) and LATS1 (Bethyl Laboratories Inc.), green; tubulin, red; and 4′,6-diamidino-2-phenylindole (DAPI), blue. (C to F) YAP (Cell Signaling Technology) (C) and LATS1 (Bethyl Laboratories Inc.) (D) localize to the midbody ring of MCF-10A cells during cytokinesis (green); inset shows midbody ring. Other YAP antibodies [Abcam, (E); Sigma-Aldrich, (F)] show localization to the mid-body ring in MCF-10A cells (green). (G) YAP localizes to the midbody ring in HeLa cells (green). (H) YAP Ser127 (Cell Signaling Technology) localization in MCF-10A. (I to M) Overexpressed exogenous EGFP-YAP (I) or Flag-YAP (J) in HeLa cells localizes to the midbody ring. YAP (red) colocalizes with Cep55 (K), MST (L), and LATS1 (green) (M) at the midbody. Arrows indicate the central spindle or midbody region. Scale bars, 5 μm. (A) to (J) are representative of 95 images obtained from three independent experiments. (K) to (M) are representative of 35 images obtained from three independent experiments.
Knockdown of YAP disrupts cytokinesis
To examine if YAP plays a role in mitosis in MCF-10A or HeLa cells, we used five different shRNA interference sequences (shYAP) (fig. S2) or small interfering RNAs (siYAP) to knock down YAP. The effects of YAP knockdown on mitosis were examined by time-lapse microscopy in MCF-10A (Fig. 2A and movies S1 to S4) or HeLa cells (movies S5 and S6) expressing H2B-GFP (histone 2B–GFP) to label nuclei or GFP-tubulin to label mitotic spindles.
Fig. 2. Loss of YAP disrupts cytokinesis.

(A) Representative time-lapse images of mitosis of MCF-10A cells expressing GFP-tubulin (GFP-Tub) and infected with an empty lentiviral vector (control) or a vector encoding an shRNA targeting YAP (shYAP) during mitosis. Arrows indicate mitotic cells. shYAP cells show extensive blebbing and altered morphology during mitosis. Scale bar, 5 μm. Acq., acquisition. (B) Graph shows mean duration of mitosis (+SD) from three independent experiments for control (n = 127) and shYAP (n = 116) cells at the indicated division stages. The duration of anaphase was less than the time-lapse minimum interval for all cells. (C) Images show control and shYAP MCF-10A cells in cytokinesis. Graph shows percent of the cell divisions in control (n = 127) and shYAP-expressing (n = 116) cells with abnormal morphology, as represented in the images in (A). Scale bar, 5 μm. Bars are means + SEM from three independent experiments. (D) Top panels show representative images of cell nuclei (DAPI) in control and shYAP MCF-10A cells; dotted lines indicate multinucleated cells. Graphs show percent of multi-nucleate cells or cells with micronuclei pooled from two experiments for control (n = 676) and shYAP (n = 655) cells. Scale bar, 10 μm. (E) FISH analysis of control (n = 114) and siYAP-expressing (n = 129) MCF-10A cells. Graph depicts the number of centromeres per nucleus for the indicated cells pooled from two independent experiments. Statistical significance [***P < 001; **P < 0.01; *P < 0.05; NS (not significant), P > 0.05] was assessed by unpaired Student’s t test in (B) and (C) and by Fisher’s exact test in (D) and (E).
YAP knockdown elicited mitotic defects that involved an increased transit time from cytokinesis to abscission and a lengthening of mitosis by an average of 40 min over controls (Fig. 2B). Cells depleted of YAP also exhibited increased membrane blebbing during anaphase and telophase and a striking morphological disruption of cytokinesis involving “hyperdynamic” movements of the dividing cells and considerable blebbing and bulges (Fig. 2C and movies S1 to S6). In addition, YAP depletion caused misorientation of the spindle, which frequently resulted in one of the two daughter cells dividing perpendicular to the surface of the monolayer followed by abnormal suspension into the medium (fig. S3). Although most cells ultimately separated into two daughter cells, a small percentage failed to complete cytokinesis and became binucleated. YAP knockdown led to a sixfold increase in multinucleated cells and a threefold increase in the percentage of cells with micronuclei (Fig. 2D), which are a hallmark of disrupted cell division. Chromosome counting using fluorescently labeled centromeric FISH (fluorescence in situ hybridization) probes also revealed a 5% increase in the number of polyploid cells under conditions of YAP depletion (Fig. 2E). Together, these data demonstrate that YAP depletion causes a morphological and functional disruption of cytokinesis.
YAP is required for proper cleavage furrow contractility
RHOA is essential for actomyosin contraction during cytokinesis, and its precise localization and activation establish the cleavage furrow (27, 28). To determine if knockdown of YAP affects RHOA localization during mitosis, cells were monitored by time-lapse microscopy until late telophase and then immediately fixed in trichloroacetic acid (TCA) and stained to localize RHOA by immunofluorescence (29, 30). Z-stack reconstruction was performed (Fig. 3A and movie S7), and surface plot intensities were graphed to visualize RHOA. Control cells predominantly showed RHOA localization in a ring at the cleavage furrow with some small patches in the plasma membrane outside the cleavage furrow at the late stages of cytokinesis when the actin cortex reassembles in blebs generated during cytokinesis (31). In contrast, in YAP-depleted cells, RHOA not only localized to the midbody but also displayed considerable ectopic accumulation outside of the cleavage furrow region in 87.9% of the midbodies observed (Fig. 3A). Depletion of YAP resulted in additional ectopic RHOA foci but did not prevent RHOA localization to the midbody. Thus, the loss of YAP expression leads to the mislocalization of RHOA during late cell division.
Fig. 3. YAP is required for proper cleavage furrow contractility.

(A) Representative examples of RHOA immunostains at early (left panels) and late (right panels) cytokinesis in control (n = 27) and shYAP (n = 33) MCF-10A cells. Cells were monitored on gridded coverslips to identify cells undergoing cytokinesis and were immunostained for RHOA (red). Surface intensity plots are shown to the right of each image to visualize the positions of RHOA. White arrows indicate normal RHOA staining, and yellow arrows indicate abnormal RHO Aectopicfoci. Scale bar, 5 μm. Images were obtained from three independent experiments. (B) Representative images of ECT2 localization (green) during cytokinesis incontrol (n = 119) or shYAP (n = 127)MCF-10A cells. Corresponding surface plots of ECT2 staining intensity. Scale bar, 5 μm. Images were obtained from three independent experiments. (C) Colocalization (merge) of ECT2 (green) and RACGAP1 (red) for control (n = 46) or shYAP (n = 41) MCF10A cells. Scale bar, 5 μm. Images were obtained from three independent experiments.(D)Colocalization(merge) of ECT2 (green) and ANLN (red) for control(n =23) or shYAP (n = 27) MCF10A cells. Scale bar, 5 μm. Images were obtained from four independent experiments. (E) Quantification of the percent of cells with mislocalized foci for ECT2, RACGAP1, and ANLN and quantification of the percent of cells showing ectopic colocalization between RACGAP1 and ECT2 or ANLN and ECT2. RACGAP1 and ECT2 ectopic foci colocalized but ANLN and ECT2 foci did not. Statistical significance (***P<001;**P<0.01;*P<0.05;NS,P>0.05) was assessed by unpaired Student’s t test. Bars represent means + SEM from three independent experiments. (F) Lysates from HeLa cells expressing control plasmid or two different shYAP hairpins (#1 or #2) were immunoblotted for phosphorylated MLC (pMLC), YAP, and tubulin, at the indicated times after nocodazole release. Immunoblots are representative of two independent experiments. (G) Representative images from control (n = 40) and YAP knockdown (n = 26) cells were obtained from three independent experiments showing phosphorylated MLC immunostaining in cytokinesis. Surface intensity plots are shown to the right of each immunostaining image to visualize the intensity of phosphorylated MLC throughout the cell. Scale bar, 5 μm. (H) Representative time-lapse images of control or shYAP MCF-10A cells undergoing cell division. Analysis was obtained from YAP knockdown cells treated with no drug (n = 747), the Rho inhibitor C3 transferase (n = 973), or the ROCK inhibitor Y-27632 (n = 729) and LKO control cells (Control) treated with no drug (n = 983), C3 transferase Rho inhibitor (n = 514), or Y-27632 ROCK inhibitor (n=870). Arrows indicate mitotic cells. Scale bar, 10 μm.(I) Quantification of YAP-depleted mitotic cells that display the hyperdynamic cell division phenotype, which consists of considerable blebbing and bulging. C3 Trans, C3 transferase. (J) Quantification of failed division for the indicated treatments. Data in (I) and (J) were pooled from two independent experiments and assessed for statistical significance (***P < 001; **P < 0.01; *P < 0.05; NS, P > 0.05) with Fisher’s exact test.
RHOA activity is regulated during cytokinesis by the centralspindlin complex, which includes MgcRacGAP (RacGAP1) and MKLP and which recruits the RHOGEF ECT2. Anillin (ANLN) is a scaffold protein that links RHOA to actin and myosin components for furrow progression (32). Therefore, we sought to determine if these cleavage furrow components were mislocalized in cytokinesis-defective YAP-depleted cells. Immunofluorescence analysis was performed on cells fixed at the time of cytokinesis entry, as determined by time-lapse imaging, and surface plot intensities were generated to visualize ECT2 localization. In control cells, ECT2 was localized predominantly in a ring at the central spindle and at the midbody, whereas cells depleted of YAP showed considerable ectopic ECT2 accumulation outside of the central spindle region (Fig. 3, B to E). Similarly, RacGAP1 (Fig. 3, C to E) and ANLN (Fig. 3, D and E) also ectopically localized outside of the midbody region as well as the midbody. These results suggest that YAP affected RHOA activity and localization during cytokinesis by regulating the localization of the mitotic proteins ECT2, RacGAP1, and ANLN to the cleavage furrow.
To further investigate contractile activity in mitotic cells, we examined the phosphorylation of MLC, a downstream target of RHO. YAP knockdown induced an increase in the phosphorylation of MLC in mitotic HeLa cells generated by nocodazole treatment and release (Fig. 3F). Consistent with these results, immunofluorescence analysis revealed an elevation and mislocalization of phosphorylated MLC in 83% of YAP-depleted cells as compared to 5% of control cells (Fig. 3G).
To assess the importance of contractile activity in the YAP knockdown phenotype, we examined the effects of suppression of the RHO pathway using either C3 transferase, which inhibits RHO proteins by adenosine diphosphate ribosylation, or Y-27632, a pharmacological inhibitor of RHO kinase (ROCK), a crucial RHO effector that induces contractile activity. Inhibition of RHO in YAP-depleted cells reduced the morphological disruption and excessive cell contractions at division, although some small blebs were still apparent (Fig. 3, H and I, and movie S8). Treatment with C3 transferase blocked cell division because of the requirement for RHO activity for furrowing (33–36). Control cells (11.6%) failed division when treated with C3 transferase (Fig. 3J), an effect rescued by YAP depletion (2.5% of cells failed division), consistent with a role for YAP in RHO-regulated processes during division. In addition, treatment with the ROCK inhibitor Y-27632 reduced the percentage of morphologically disrupted mitoses by 91.2%, further supporting the hypothesis that the phenotype elicited by YAP knockdown was associated with inappropriate activation of the RHO pathway (Fig. 3, H and I, and movie S8). Together, these results suggest that YAP is important for proper contractile activity of cleavage furrow components during cytokinesis.
YAP is phosphorylated during mitosis
The YAP Western blots (Fig. 3F) revealed a substantial and transient mobility shift in nocodazole-released samples, suggesting that YAP was subjected to posttranslational modification during mitosis. The mobility of YAP was indeed retarded by nocodazole treatment of both HeLa and MCF-10A cells (Fig. 4A). Blotting of lysates from mitotic HeLa cells indicated that most of the YAP protein was modified under these conditions. Mitotic HeLa cells can be efficiently separated from interphase cells by mitotic “shake-off,” which was not effective in MCF-10A cells. Like endogenous YAP protein, exogenously expressed YAP displayed a similar shift in mobility during mitosis (Fig. 4B). Moreover, an alternative method of synchronizing cells, double thymidine blocking, also revealed a similar pattern of slower migrating YAP species during mitosis (Fig. 4C).
Fig. 4. YAP is differentially phosphorylated during mitosis.

(A) Immunoblot of endogenous YAP from MCF-10A (10A) or HeLa cells treated with nocodazole (Noc). Arrow indicates the phosphorylated form of YAP. Immunoblot is representative of two independent experiments. (B) YAP immunoblot of exogenously expressed pBabeFlagYAP (Flag) or pBabePuro control (Babe) in HeLa cells treated with nocodazole. Immunoblots are representative of two independent experiments. (C) Immunoblot of HeLa cells with double thymidine block shows that YAP was phosphorylated during mitotic phase of the cell cycle. Phosphohistone H3 S10 (PHH3 S10) is a mitotic marker of the cell cycle, and tubulin is used as a loading control. The number of hours after release from the double thymidine block is indicated. Immunoblots are representative of two independent experiments. (D) YAP and tubulin immunoblots of CIP- and PI-treated lysates from HeLa cells treated with nocodazole. Arrow indicates the phosphorylated form of YAP. Immunoblots are representative of two independent experiments. (E) Immunoblot of lysates of nocodazole-treated HeLa cells expressing either the Flag-YAP combination mutant for the five LATS phosphorylation sites YAP S61A, S109A, S127A, S164A, S381A (5SA) or wild-type (WT) Flag-YAP .Immunoblots are representative of two independent experiments. (F) Anti-YAP immunoblot of lysates of HeLa cells expressing shRNAs against YAP, LATS1 or LATS2 or LKO empty vector showing the YAP mobility shift with nocodazole treatment. Immunoblots are representative of two independent experiments. (G) Table indicating the single, double, or triple phosphorylation site mutants used to identify the mitotic sites. Nocodazole treatment was used to identify the combination of mutations that could reverse the mitotic shift in immunoblot assays. Highlighted in darker gray is the combination of mutations (YAP S138A, T143A, and S367A, referred to as Flag–YAP 3A) that reversed the mitotic shift. (H) Schematic map showing locations of our identified mitotic phosphorylation sites on YAP (red) relative to previously identified YAP phosphorylation sites (gray) and TEAD-, SH3-, WW1-, WW2-, and PDZ-binding domains. (I) Anti-Flag immunoblot of nocodazole analysis of the combination alanine mutant Flag–YAP 3A shows that loss of YAP sites Ser138, Thr143, and Ser367 reversed the mitotic shift. Immunoblot is representative of three independent experiments. (J) MS heat map of total spectral counts of WT YAP phosphosites from nonsynchronized (NS) or mitotic MCF-10A cells after release from nocodazole treatment (after 0, 30, 60, 90, 120, and 180 min). The right-hand labels indicate the phosphorylation site or sites identified. Data are representative of two independent experiments. (K) Immunoblot analysis of HeLa cells treated with CDK inhibitor Puravanol A or BMI1026. Arrow indicates the phosphorylated form of YAP. Immunoblots are representative of two independent experiments. (L) Quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis of cells expressing YAP WT, YAP 3A, YAP 3D, and empty vector control for the indicated TEAD-dependent transcriptional genes; representative of three biological replicates. Results were normalized to the housekeeping gene RPLP0 and are presented as mean fold change + SD. Statistical significance (***P < 001; **P < 0.01; *P < 0.05; NS, P > 0.05) was assessed by Student’s t test compared to control LKO. (M) Time-lapse analysis of the hyperdynamic phenotype of MCF-10A cells expressing the phosphomutants or shYAP cells reconstituted with the various phosphomutants. Quantification of the summary of three independent experiments from mitotic cells transfected with LKO (Control) and control pBABE (n = 542), YAP WT (n = 216), YAP 3A (n = 332), or YAP 3D (n = 229) or cells transfected with shYAP and control pBABE (n = 84), YAP WT (n = 117), YAP 3A (n = 119), or YAP 3D (n = 40). Data are represented as means + SEM, and statistical significance (***P < 001; **P < 0.01; *P < 0.05; NS, P > 0.05) was assessed by Student’s t test.
To examine if the retarded YAP mobility in mitotic cell lysates might be due to phosphorylation, we treated cell lysates with various combinations of calf intestinal phosphatase (CIP) and phosphatase inhibitor before SDS–polyacrylamide gel electrophoresis and Western blotting. CIP treatment of the mitotic cell lysates reversed the shift in YAP mobility; however, when phosphatase inhibitor was included in the presence of CIP treatment of mitotic YAP lysates, the YAP mobility shift was not affected, indicating that the retarded migration of this protein is due to phosphorylation (Fig. 4D). Because YAP is phosphorylated by kinases in the LATS family on any of five different sites (Ser61, Ser109, Ser127, Ser164, and Ser381), we examined the mobility of a YAP protein with alanine substitutions at each of these sites (YAP-5SA) from mitotic cell lysates. YAP-5SA mobility was shifted by nocodazole treatment to a similar extent as wild-type YAP, suggesting that the major mitotic YAP phosphorylation sites were distinct from LATS sites (Fig. 4E). Consistent with this result, depletion of LATS1 or LATS2 by shRNA did not alter the mitosis-induced mobility shift for YAP (Fig. 4F).
To identify the mitosis-specific YAP phosphorylation sites, we analyzed the mobility of exogenously expressed variants of YAP containing alanine substitutions for the eight amino acids reported to show enriched phosphorylation in mitotic HeLa cell lysates (S131A, S138A, T141A, T143A, T154A, S274A, S353A, and S367A) (37). First, we examined whether any of the single-site mutations prevented the phosphorylation shift induced by nocodazole treatment. Although four of the single mutants (YAP S138A, T143A, S274A, and S367A) displayed a slight reduction in the YAP band shift in mitotic cells, none prevented it entirely, suggesting that more than one phosphorylation site contributes to the retarded mobility. Therefore, we generated a set of double and triple YAP mutants (Fig. 4G), and we identified three sites in YAP—Ser138, Thr143, and Ser367—whose combined alanine substitution (YAP 3A) prevented the mitotic shift (Fig. 4, H and I, and fig. S4). Consistent with this result, increased phosphorylation of these sites was detected in mitotic cells by mass spectrometry (MS) (Fig. 4J).
The three mitosis-specific YAP phosphorylation sites matched the consensus site for cyclin-dependent kinase 1 (CDK1), a kinase that regulates many proteins that control mitosis. Moreover, the timing of YAP phosphorylation after release from nocodazole or double thymidine block is reminiscent of other CDK substrates, with greater phosphorylation at the entry of mitosis followed by dephosphorylation upon exit from mitosis (Figs. 3F and 4C). Indeed, short-term treatment of nocodazole-blocked cells with the CDK inhibitors Puravanol A or BMI1026 (38) prevented the YAP mobility shift (Fig. 4K). Although an indirect role for CDK1 cannot be ruled out, these findings suggest that CDK1 regulates the phosphorylation of YAP at Ser138, Thr143, and Ser367 during mitosis. Of note, another group has also provided evidence for CDK1-mediated phosphorylation of YAP during mitosis (39).
To address the functional relevance of YAP phosphorylation during mitosis, we generated phosphomimetic (YAP 3D) and nonphosphorylatable (YAP 3A) mutants of YAP containing either aspartate or alanine substitutions for the three mitotic phosphorylation sites, respectively. These substitutions did not affect the gain-of-function activities induced by YAP overexpression, including increasing TEAD-dependent transcription of CYR61, CTGF, and ITGFB2 (Fig. 4L). However, overexpression of YAP 3A, but not of YAP 3D, caused increased hyperdynamic activity and blebbing during cytokinesis to a similar extent as that observed in YAP-depleted cells (Fig. 4M and movie S9). Furthermore, reexpression of YAP 3A in cells depleted of endogenous YAP could not rescue the hyperdynamic cytokinesis observed in these cells. In contrast, reexpression of YAP 3D rescued the phenotype induced by YAP depletion to a similar extent as that of wild-type YAP. Additionally, we found that expression of YAP 3A induced an increase in the phosphorylation of MLC in mitotic HeLa cells (fig. S5), similar to the increase detected after knockdown of YAP (Fig. 3, F and G). These data demonstrate that the YAP phosphorylation sites Ser138, Thr143, and Ser367 are important for proper mitosis and cytokinesis.
PATJ coprecipitates with YAP and colocalizes with YAP to the midbody, and its knockdown disrupts cytokinesis
To investigate the mechanism by which phosphorylated YAP regulates cytokinesis, we analyzed the proteins that coimmunoprecipitate with YAP in both nonsynchronized MCF-10A cells and cells at different stages of mitosis (using nocodazole block and release). Exogenously expressed Flag-tagged wild-type YAP or YAP 3A or YAP 3D mutants were immunoprecipitated from MCF-10A cells, and the trypsinized immunoprecipitates were subjected to liquid chromatography–tandem MS (LC-MS/MS). To identify high-confidence candidate interacting proteins (HCIPs), we used Comparative Proteomics Analysis Software Suite (CompPASS) analysis, a software platform that uses unbiased metrics to assign confidence measurements to interactions from parallel nonreciprocal proteomic data sets (40, 41) (Fig. 5 and table S1).
Fig. 5. Proteins that coprecipitate with YAP.

Heat map of normalized total spectral counts of LC-MS/MS-identified and CompPASS-analyzed interacting proteins for YAP WT, YAP 3A, or YAP 3D from nonsynchronized or mitotic cells after release from nocodazole treatment at the indicated times. Data are representative of two independent experiments.
Multiple proteins previously identified as YAP interactors were detected in the YAP immunoprecipitates [TEAD1, TEAD3, PTPN14, AMOTL1, AMOTL2, LATS1, RASSF8, NF2, YWHAB, PALS (MPP5), and PATJ (INADL)], thus validating our methodology (42–51). No substantial differences were observed in the interaction of wild-type or mutant YAP proteins with TEAD1 or TEAD3, the major YAP-regulated transcription factors, providing additional evidence that YAP 3A and YAP 3D did not alter TEAD-dependent transcriptional activity (Fig. 5). Similarly, most of the interacting proteins displayed comparable amounts of coprecipitation with the wild-type and mutant YAP proteins, indicating that mutation of the mitotic phosphorylation sites did not grossly affect YAP protein folding or function.
PATJ was distinguished as the most abundantly detected protein in YAP immunoprecipitates by MS. To validate this interaction, we analyzed YAP or PATJ immunoprecipitates from untreated or nocodazole-treated MCF-10A cells. Indeed, endogenous YAP and PATJ coimmunoprecipitated, and the interaction could be detected using either anti-YAP or anti-PATJ antibody for immunoprecipitation (fig. S6). Although we did not consistently detect increased YAP-PATJ coimmunoprecipitation in mitotic cells, as observed in the MS data (Fig. 5), these results confirm that PATJ is a bona fide YAP-interacting protein.
To examine whether PATJ colocalizes with YAP during cytokinesis, we immunostained PATJ in nocodazole-treated MCF-10A cells. Indeed, PATJ was enriched at midbodies and colocalized with YAP on this structure (Fig. 6A). Knockdown of YAP resulted in a diffuse PATJ localization in the midbody region in 80% of cells (Fig. 6B), suggesting that YAP was required for the midbody localization of PATJ. In contrast, depletion of PATJ did not significantly affect YAP localization to the midbody ring (fig. S7). Moreover, PATJ knockdown induced a cytokinetic phenotype similar to that exhibited by YAP-depleted cells, with extensive protrusions, blebbing, and hyperdynamic behavior (Fig. 6C and movie S10), as well as misorientation of the spindle plane resulting in one of the two daughter cells dividing perpendicular to the surface of the monolayer (Fig. 6D). PATJ knockdown also caused an increase in the phosphorylation of MLC comparable to that observed in YAP-depleted cells (Fig. 6E). Together, our results showing YAP and PATJ colocalization at mitotic structures, combined with the comparable phenotypes elicited upon their knockdown, provide evidence that the YAP-PATJ interaction is important for proper cytokinesis.
Fig. 6. Interaction of PATJ and YAP in mitotic cells.

(A) Representative immunostaining images of the localization of endogenous PATJ (green) and YAP (red) to the midbody in mitotic cells. Zoom shows magnification of colocalized areas of the midzone and midbody. Loss of YAP results in diffuse immunostaining of PATJ in the cleavage and midbody region. Scale bars, 1 μm in zoomed images and 5 μm in all others. Representative images from 60 control cells and 50 shYAP cells. (B) Quantitation of the number of cells with diffuse immunostaining of PATJ in the cleavage and midbody region. (C and D) Quantification of cells with the hyperdynamic phenotype (D) or with the nonadherent daughter cell phenotype (D) in cells expressing shPATJ (n = 232), shYAP (n = 277), or control LKO (n = 2206). Bars in (B), (C), and (D) represent means + SEM of three experiments each performed in triplicate. Statistical significance (***P < 001; **P < 0.01; *P < 0.05; NS, P > 0.05) was assessed by unpaired Student’s t test compared to control LKO. (E) Contractile activity for these conditions was also analyzed by immunoblot of phosphorylated MLC activity. MLC and tubulin were used as internal loading controls. Immunoblots are representative of three independent experiments.
DISCUSSION
Execution of cytokinesis requires precise spatial and temporal activation of proteins that coordinate cell cycle control and cytoskeletal reorganization for the final separation of two daughter cells. The findings in this report indicate that YAP plays an important role in coordinating the organization of proteins that mediate this process. YAP localized to structures involved in mediating proper cytokinesis and was phosphorylated during mitosis by the mitotic regulatory kinase CDK1. Our studies showed that knockdown of YAP or mutation of the mitotic phosphorylation sites caused abnormal cortical activity and blebbing during cytokinesis. These results suggested that YAP coordinated with CDK1 to regulate mitotic exit. Although the precise mechanism whereby YAP regulates the coordination of cytokinesis remains to be established, our data implicate the polarity protein PATJ in this process. These findings reveal a previously unrecognized role for YAP distinct from its role in interphase, suggesting a specific function in the proper organization of the cytokinesis machinery through interactions of CDK1-phosphorylated YAP with PATJ (Fig. 7).
Fig. 7. Model for YAP regulation in cytokinesis.
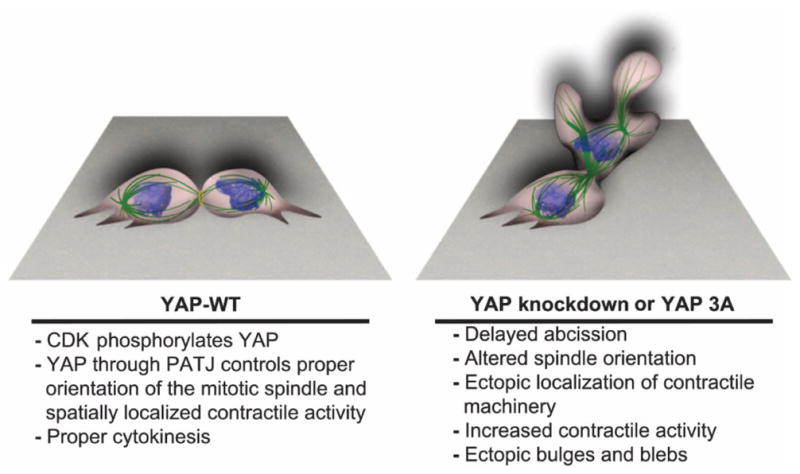
CDK-mediated phosphorylation promotes the interaction of YAP with the polarity protein PATJ to regulate proper spindle orientation and localized cellular contraction during cytokinesis.
Here, we showed that shRNA-mediated knockdown of YAP delayed abscission and also induced a cytokinesis phenotype associated with increased contractile force, membrane blebbing, and bulges. The specific nature of the defective cytokinesis phenotype elicited by YAP knockdown was similar to many other perturbations that affect the RHOA pathway. For example, overexpression of either active RHOA or unregulated catalytically active MLC kinase during cytokinesis impairs cortical activity and induces blebbing (52–54). Our data showed that cells depleted of YAP showed considerable ectopic RHOA accumulation outside of the cleavage furrow region, suggesting that YAP depletion caused mislocalization of RHOA.
Our data support a model in which YAP regulates the activation of contractile activity, possibly through regulation of the central spindle and furrow components during cytokinesis. Alterations in PLK1, ECT2, ANLN, RacGAP1, and p190RHOGAP, all of which affect RHOA activity, promote abnormal cortical activity, increased blebbing, and spindle mis-orientation, as well as impaired accumulation of RHOA at the equatorial cortex and ectopic localization at other membrane sites—phenotypes similar to those observed during cytokinesis in YAP-depleted cells (28, 32, 55–59). Because these proteins have been proposed to regulate not only the activation of RHO but also the organization of the contractile ring and linkage of RHO-dependent actomyosin contractility to the membrane at the equatorial plane (28), these phenotypes are believed to be caused by a failure to properly localize RHO-induced contractile activity. Consistent with YAP-mediated regulation of these proteins, depletion of YAP caused mislocalization of ECT2, ANLN, and RacGAP1, although ANLN foci were notably distinct from those containing colocalized RacGAP1 and ECT2 (Fig. 3E).
A model in which YAP regulates the coordination of contractile activity is also consistent with the substantial increase in the ROCK-mediated phosphorylation of MLC caused by the knockdown of YAP. ROCK is a downstream effector of RHOA, and its phosphorylation of MLC on Ser19 stimulates actomyosin contractility to promote furrow formation during cytokinesis (60–62). A role for YAP in the regulation of contractile activity is further supported by our finding that the hyperdynamic phenotype associated with YAP depletion could be rescued by inhibition of RHO or ROCK. It is feasible that the mislocalized excessive cortical activity during furrow ingression in YAP-depleted cells contributes to improper chromosome distribution during cytokinesis, leading to increased multinucleation, micronuclei, and abnormal chromosome numbers. Together, these data suggest that YAP is a component of the regulatory network that controls contractile activity during mitotic exit.
The identification of multiple amino acids that are specifically phosphorylated on YAP during mitosis provided valuable mechanistic insight relating to the role of YAP in cytokinesis. Our evidence suggested that these YAP sites (Ser138, Thr143, and Ser367) were CDK1 phosphorylation sites. Another group has reported that YAP is phosphorylated in vitro and in vivo by CDK1 (39) and identified Ser367 as one of the sites. However, we did not detect phosphorylation of the other two sites identified in that study (Thr119 and Ser289) by MS in MCF-10A cells (Fig. 4J), and phosphorylation of these sites was not enriched in mitotic HeLa cells (37). It is feasible that Taxol, the mitotic inhibitor used in the Yang et al. report, elicits distinct mitotic phosphorylation sites in the cell type(s) they examined.
CDK1 is responsible for synchronizing and timing the entry into various stages of cell division. CDK1 substrates are phosphorylated in early mitosis, when CDK1 activity is high and anaphase-promoting complex (APC) activity is low, thereby preventing cells from proceeding to mitotic exit. At metaphase, APC activity rises and begins to degrade the CDK1 activator cyclin B, which leads to a reduction of CDK1 activity and de-phosphorylation of CDK1 substrates, allowing cells to start exiting mitosis (63). Our evidence that the phosphorylated mitotic sites of YAP have a CDK consensus sequence (S/TPXK/R), that YAP displays a pattern of phosphorylation during mitosis similar to other CDK substrates, and that the mitotic phosphorylations are lost with CDK inhibition provides support that CDK phosphorylates these sites. The importance of these sites is highlighted by the similar phenotype of prolonged and abnormally contractile cytokinesis in cells overexpressing the phosphomutant YAP 3A and in cells in which YAP is depleted, as well as by the failure of YAP 3A to rescue these cytokinetic defects when reexpressed in cells lacking endogenous YAP. Thus, we propose that phosphorylation of YAP may contribute to the control of mitotic exit by CDK1. Of note, we also showed that the alterations in mitosis were not due to secondary effects on transcription because TEAD-dependent YAP target genes were unaffected by expression of YAP 3A or YAP 3D, and these YAP mutants interacted with TEAD family members to a similar extent as wild-type YAP on the basis of our coimmunoprecipitation and MS experiments (Fig. 5). Together, these data indicate that CDK1-mediated YAP phosphorylation early in mitosis is important for proper regulation of mitotic exit and cytokinesis.
Although we predicted that YAP would coimmunoprecipitate with cleavage furrow components, our analysis did not reveal any such interactions, possibly because the interactions are too transient to be detected by this method or because they may not be direct. PATJ was identified as a protein that coimmunoprecipitated with YAP and localized to the midbody during cytokinesis in a YAP-dependent fashion. Loss of PATJ also resulted in a hyperdynamic cytokinesis phenotype with altered spindle orientation similar to the phenotype induced by YAP depletion or expression of phosphomutant YAP 3A. Because PATJ knockdown did not disrupt the midbody localization of YAP, these data suggest that the interaction of PATJ with YAP is important for YAP’s regulation of cytokinesis.
PATJ is a scaffold protein that is part of the evolutionarily conserved Crumbs (crumbs-PALS1-PATJ) polarity complex that regulates tight junction formation, epithelial apicobasal polarity, and directional movement (64, 65). Along with other members of the Crumbs complex, PATJ has been implicated in the regulation of spindle orientation. We found that loss of PATJ or expression of the phosphomutant YAP 3A altered the spindle orientation of cells during mitosis and cytokinesis. Although little is known about the spatial cues that regulate spindle orientation in the z axis of symmetrically dividing cells, cadherin junctions and specific molecular cues that localize to the cortex through the Crumbs polarity complex are hypothesized to act by regulating astral microtubules or by exerting pulling or pushing forces on the spindle (66). Complexes associated with PATJ and YAP could influence spindle orientation through these spatial polarity cues. The localization of both YAP and PATJ to the midbody and central spindle raises the possibility that they could also regulate spatial cues that coordinate with checkpoint regulators to ensure the proper timing of mitosis, in a similar way that spatial cues regulate the timing of mitotic exit in yeast (67). The LATS and MST homologs DBF2 and CDC15 control the spatial and temporal aspects of mitosis in yeast; however, our data showing that the YAP-5SA mutant was still phosphorylated during mitosis suggested that the mitotic activities of YAP described in this report did not depend on LATS-mediated phosphorylation (68, 69).
These data are consistent with a model (Fig. 7) in which CDK1-phosphorylated YAP regulates cytokinesis through a mechanism involving interaction with the polarity protein PATJ, to spatially orient contractile activity and regulate the timing of mitotic exit. Alterations in YAP or PATJ abundance, or inhibition of CDK phosphorylation of YAP, cause distortion of the mitotic spindle and disrupt coordination of contractile processes and membrane dynamics. Further studies are required to define the nature of the interaction with PATJ and to elucidate the precise mechanism involved in PATJ/YAP-mediated coordination of cytokinesis.
Although YAP can act as an oncogene, YAP may also have a tumor suppressor–like function in some cancers (70). Consistent with this hypothesis, decreased YAP abundance correlates with reduced patient survival in a small fraction of aggressive and highly undifferentiated human colorectal carcinoma cases, and reexpression of YAP in colorectal carcinoma xenografts can restrict tumor growth (71). Moreover, YAP can enhance p73-mediated apoptosis (16–18, 46, 47). Loss of YAP or mutation of its mitotic phosphorylation sites resulted in cytokinesis failure that promoted micronuclei formation and aneuploidy, albeit in a small fraction of cells. It has been hypothesized that a failure in cell division leads to the formation of genetically unstable tetraploid cells with multiple centrosomes that can lead to aneuploidy and tumorigenesis (72, 73). A role for cytokinesis in tumorigenesis is supported by reports that mutations in certain tumor suppressors lead to increased rates of cytokinesis failure (74), and some components required for cytokinesis are either deregulated or encoded by genes located in chromosomal regions that are deleted or amplified in tumors and tumor-derived cell lines (75, 76). Tetraploidization, which frequently occurs in human cancers (77), often precedes aneuploidy and can induce tumorigenesis in mouse models (78, 79). Our results raise the possibility that loss of YAP may affect tumorigenic potential by contributing to genome instability events that occur as a result of failed cytokinesis.
MATERIALS AND METHODS
Cell culture and nocodazole treatment
MCF-10A cells were cultured as described in (80). HeLa cells were grown in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and penicillin and streptomycin (50 μg/ml each). The cells were then synchronized with nocodazole for 15 hours. Mitotic shake-off was performed for HeLa cells, and the cells were replated for nocodazole release. However, because MCF-10A cells could not shake-off, the entire population was released into normal growth media.
RNA interference
ON-TARGETplus SMARTpool siRNAs targeting YAP1 (Dharmacon) were transfected into HeLa cells, using FuGENE HD (Roche) according to the manufacturer’s protocol. Different shRNAs in human pLKO.1 lentiviral vectors (Open Biosystems) were used to knock down YAP1, LATS1, LATS2, or INADL (PATJ). The best two or three hairpins were used for subsequent experiments. The RNAi (RNA interference) Consortium (TRC) numbers used for the shRNAs are as follows: YAP: TRCN0000107265, TRCN0000107268, TRCN0000107269; LATS1: TRCN0000001776, TRCN0000001777; LATS2: TRCN0000000880, TRCN0000000884; INADL: TRCN0000158824, TRCN0000159109. The website for the TRC for human shRNA is www.broadinstitute.org/rnai/trc/lib. shRNA-expressing MCF-10A cell lines were selected with puromycin (2 μg/ml), and HeLa cells were also selected with puromycin (5 μg/ml).
Antibodies
Primary antibodies to the following proteins were used: YAP, phospho-YAP Ser127, PLK1 (208G4), MST2, phospho-MLC, and MLC (Cell Signaling Technology Inc.); LATS1 and LATS2 (Bethyl Laboratories Inc.); YAP, ECT2, tubulin, GFP, and PATJ (Abcam); YAP (H215), YAP (63.7), Anillin (H-300), RHOA (119), RHOA (26C4), ECT2 (H300), ECT2 (C-20), and Centrin-2 (N-17)-R (Santa Cruz Biotechnology); RacGAP1, YAP1, PLK1, Flag M2, and Flag M5 (Sigma-Aldrich); Cep55 (Abnova); and PATJ (Novus). Secondary antibodies used included Alexa Fluor goat anti-rabbit 488 and 568, goat anti-mouse 488 and 568, donkey anti-goat 488 and 568 (Invitrogen) or goat anti-mouse immunoglobulin G (IgG)–horseradish peroxidase (HRP), and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology). DAPI (Sigma-Aldrich) was used to stain DNA/nuclei.
Immunoblotting
Protein abundance was assessed by immunoblotting. Protein lysates for immunoblot analysis were prepared using radioimmunoprecipitation assay buffer [1% NP-40, 0.2% SDS, 0.5% sodium deoxycholate, 150 mM sodium chloride, 50 mM tris-HCl (pH 7.4), 10 mM sodium fluoride, 1 mM sodium orthovanadate, 10 mM β-glycerophosphate, apoprotinin (5 μg/ml), leupeptin (5 μg/ml), and 100 μM phenylmethylsulfonyl fluoride]. Lysates were run on 10% SDS-polyacrylamide gel or 4 to 20% tris-glycine gels (Invitrogen) and transferred onto polyvinylidene di-fluoride (PVDF) membranes (Millipore). Immunoblots were visualized with Pierce ECL Western Blotting Substrate (Thermo Scientific).
Immunofluorescence staining
Formaldehyde fixation was performed using 4% paraformaldehyde at room temperature for 15 min. Cells were rinsed three times with phosphate-buffered saline (PBS) containing 30 mM glycine and permeabilized with 0.2% Triton X-100/PBS for 5 min. Methanol/acetone (1:1) fixation was performed at −20°C for 15 min. For RHOA immunostaining, cells were fixed with 10% TCA on ice for 15 min (29, 30), rinsed three times in PBS/glycine, and permeabilized with 0.2% Triton X-100/PBS. All images were collected either with a Nikon C1si scanning confocal microscope on a Nikon TE2000U inverted microscope stand or with a Nikon A1R confocal microscope on a Nikon TiE inverted microscope stand. Both microscopes were equipped with Plan Apo 60× 1.4 NA (numerical aperture) oil objective lenses, and 405, 488, and 561 nm were used to excite DAPI, Alexa 488, and Alexa 568 fluorophores, respectively. Z-series optical sections were collected with a step size of 0.25 μm using a Nikon focus motor. Z-series were displayed as maximum z-projections. Gamma, brightness, and contrast were adjusted on displayed images (identically for compared image sets) using MetaMorph 7 software or NIS-Elements.
Time-lapse imaging
Cells were grown in multiwell glass bottom dishes (MatTek). MCF-10A or HeLa growth medium was used during image acquisition with a layer of mineral oil on top of the medium to prevent evaporation. All images were collected with a Nikon TE2000E or Nikon Ti motorized inverted microscope equipped with Perfect Focus System for continuous maintenance of focus. The microscopes were enclosed in incubators that provided both 37°C temperature control and 5% CO2. Both microscopes were equipped with 20× Plan Apo 0.75 NA or 60× Plan Apo 1.4 NA objective lenses. Histone H2B-GFP or GFP-tubulin fluorescence was excited with a mercury halide light source using a 480/40 excitation filter (Chroma) and collected with a 535/50 emission filter (Chroma). Images were acquired with a Hamamatsu ORCA-ER cooled charge-coupled device (CCD) camera controlled with MetaMorph 7 software or Hamamatsu ORCA-AG cooled CCD camera controlled with NIS-Elements image acquisition software. Time-lapse microscopy images were collected every 3 min, using an exposure time between 100 and 200 ms, with illumination light shuttered between acquisitions. Gamma, brightness, and contrast were adjusted on displayed images (identically for compared image sets) using MetaMorph 7 software or NIS-Elements.
Spindle misorientation
Spindle misorientation was determined by time-lapse differential interference contrast or phase microscopy in coordination with GFP expression. Cell divisions in which the division plane of one of the daughter cells was nonparallel to the plane of the plate were deemed to show spindle misorientation.
Treatment with C3 transferase or Y-27632
Cells were treated with CT04 Cell Permeable C3 Transferase (Cytoskeleton Inc.) at 0.5 μg/ml to inhibit RHOA activity or with Y-27632 (Calbiochem) at 10 μM (in water) to inhibit ROCK activity (33) for 5 hours at 37°C. No drug was added to the control, and a minimum of 100 mitotic events were observed in three individual experiments for each condition.
Site-directed mutagenesis
Hemagglutinin (HA) tag and mutations to generate non-phosphorylatable or phosphomimetic mutants were introduced by site-directed mutagenesis using the QuickChange Kit (Stratagene) on pBabe-Flag-human YAP (7). The YAP isoform 1 (NM_001130145) was used for all cloning. Additionally, for immunoprecipitations, complementary DNAs (cDNAs) encoding full-length human YAP were generated by RT-PCR from RNAs extracted from 293T or HeLa cells. Plasmids expressing N-Tap YAP proteins were made by cloning the cDNAs into a Gateway Flag or HAvectors. The peptide sequences for YAP isoform 1 sites that were mutated were as follows: YAP Ser138 (SLQLGAVSPGTLTPT), YAP Thr143 (AVSPGTLTPTGVVSG), and YAP Ser367 (GTQNPVSSPGMSQEL).
Immunoprecipitation and MS
Immunoprecipitation and sample preparation for MS analysis were performed as previously described (40, 41). MCF-10A or HeLa cells were infected at low multiplicities of infection with retroviruses expressing YAP wild type, YAP 3A, or YAP 3D tagged with Flag and HA tags at the N terminus (N-TapYAP). Because the expression of the transgenes is driven by the long terminal repeat of the virus protein, near-endogenous amounts are achieved. Mammalian Cell Lysis Buffer [50 mM tris (pH 7.5), 150 mM NaCl, and 0.5% NP-40] supplemented with protease (Roche, 11697498001) and phosphatase inhibitors (Roche, 04906837001) was used to lyse the cells. Lysates were collected from the untreated cells (without nocodazole) and designated the nonsynchronized population, and the nocodazole-treated cells were collected at 0, 30, 60, 90, 120, and 180 min after nocodazole release. Tryptic peptides were desalted before LC-MS/MS using STAGE (stop and go extraction) tips as previously described (40, 41) and resuspended in 10 ml of 5% acetonitrile, 5% formic acid. Technical replicates were loaded on the LC-MS/MS in succession analyzed using a top 10 method on an LTQ linear ion trap mass spectrometer (Thermo). Mass spectra were searched using the Sequest search algorithm (peptide false discovery rate <1.0% and protein false discovery rate <6.54%). Sequest identifications were loaded into the CompPASS informatics suite for analysis. CompPASS identifies HCIPs on the basis of the normalized weighted D score (NWD score), which incorporates the frequency with which they identified within the statistics table, the abundance (APSMs, average peptide spectral matches) when found, and the reproducibility of identification in technical replicates, and also determines a z score based on APSMs (40, 41). Proteins with NWD scores of ≥1.0 are considered HCIPs, although it is also likely that some bona fide interacting proteins may not reach the strict threshold set by an NWD score of ≥1.0. For our CompPASS analysis, we used a statistics table containing 52 bait proteins, composed of 39 unrelated proteins and 13 Hippo pathway components (excluding YAP) that were analyzed in an analogous manner. Individual experiments were analyzed using a statistics table derived from analogous AP-MS (affinity purification–MS) data for 52 (for MCF10A) or 171 (for HeLa) unrelated proteins to determine NWD scores and z scores based on spectral counts.
For the heat map analysis, total spectral counts were normalized to the total number of spectral counts of the bait protein found in the sample. To be included in the heat map, YAP-interacting proteins were required to have normalized total spectral counts greater than 3.0 in at least two time points for any YAP perturbation. YAP interaction heat maps were built with the heatmap.2 package in R version 3.1.1.
Phosphorylation sites were identified by searching mass spectra using a Sequest search database that incorporates all possible phosphorylated peptides. Phosphorylation sites were filtered using the A-score algorithm, which calculates the probability of correct phosphorylation site localization on the basis of the presence and intensity of site-determining ions in tandem mass spectra (81). Phosphorylation sites with A-scores >19 were considered to be correctly localized, and only these sites were considered in our analysis. YAP phosphorylation site heat maps were built with the heatmap.2 package in R version 3.1.1.
Coimmunoprecipitation of endogenous PATJ with YAP
MCF-10A cells were treated with nocodazole, and cell pellets were resuspended in Mammalian Cell Lysis Buffer supplemented with protease and phosphatase inhibitors (described above). Samples were centrifuged at 4°C, 14,000 rpm for 15 min. Lysates were precleared with 30 μl of beads of protein A/G agarose beads (Santa Cruz Biotechnology, sc-2003) per 500 μl of lysate. Sample buffer (6×) was added to 50 μg of the cleared lysate as 5% input, and input samples were heated to 100°C for 5 min. Protein lysate (1000 μg) was incubated with 60 μl of washed protein A/G agarose beads and either 2 μg of rabbit anti-PATJ (Abcam, ab102113), mouse anti-YAP (Santa Cruz Biotechnology, sc-101199), or IgG control [mouse (Santa Cruz Biotechnology, sc-2025) or rabbit (Santa Cruz Biotechnology, sc-2027)] for 4 hours at 4°C while rotating. Western blot analysis was performed using self-prepared 6 to 15% sucrose gradient acrylamide gels. Proteins were transferred onto methanol-activated PVDF membranes (EMD Millipore, IPFL00010) at 80 V. Primary antibodies were diluted 1:1000 in 5% bovine serum albumin (BSA), and the membranes were incubated with the primary antibodies for 1 hour at room temperature or overnight at 4°C (rabbit anti-PATJ, Abcam, ab102113; rabbit anti-PATJ, Novus, NBP2-19707; rabbit anti-YAP, Santa Cruz Biotechnology, sc-15407; mouse anti-YAP, Santa Cruz Biotechnology, sc-101199; and rabbit anti-tubulin, Abcam, ab6046). The membranes were incubated with the secondary antibodies anti-mouse IgG-HRP (GE Healthcare, NXA931) or anti-rabbit IgG-HRP (GE Healthcare, NA9340V), diluted 1:5000 in 5% BSA, for 1 hour at room temperature. Proteins were visualized using a film developer.
Supplementary Material
Mitosis of MCF-10A cells depleted of YAP with five distinct shRNA sequences.
Mitosis of PATJ-depleted MCF-10A cells.
Mitosis of YAP-depleted MCF-10A cells expressing GFP-tubulin.
Mitosis of MCF-10A cells treated with YAP siRNA.
Mitosis of YAP-depleted MCF-10A cells in a monolayer.
Mitosis of YAP-depleted HeLa cells.
Mitosis of YAP-depleted multiple HeLa cells.
RHOA localization in a mitotic YAP-depleted MCF-10A cell.
Mitosis of YAP-depleted MCF-10A cells treated with C3 transferase or Y-27632.
Mitosis of MCF-10A cells expressing wild-type YAP or YAP 3A or YAP 3D mutants.
Fig. S1. Immunofluorescence analysis of YAP-depleted cells.
Fig. S2. YAP hairpins.
Fig. S3. Spindle misorientation after YAP knockdown.
Fig. S4. YAP phospho-mutant combinations.
Fig. S5. Phosphorylation of MLC is increased by expression of the YAP 3A mutant.
Fig. S6. YAP and PATJ coimmunoprecipitation.
Fig. S7. Immunofluorescence of PATJ-depleted cells.
HCIPs for YAP wild type, YAP 3D, and YAP 3A.
Acknowledgments
We would like to thank T. Mitchison, D. Pellman, R. King, G. Danuser, G. Mouneimne, O. Florey, and T. Miki for helpful discussion and for the antibodies and reagents; S. Godinho for help with FISH; the Nikon Imaging Center at Harvard Medical School for help with light microscopy; M. A. Martinez-Gakidis for editing the manuscript; and G. Mouneimne for contributing artwork.
Funding: This project was supported by the Breast Cancer Research Foundation (J.S.B.), NIH grant AG011085 (J.W.H.), the National Research Service Award from the National Cancer Institute (1F32CA165738; A.E.W.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The MS data have been deposited at PeptideAtlas (www.peptideatlas.org) (identifier PASS00780).
Author contributions: D.A.B. carried out all of the experiments associated with the report except for the CDK inhibitor assay, the analysis of mutant variants of YAP that prevent mitotic phosphorylation, and the MS analysis. W.L. created point mutations, analyzed their gel shift, and performed CDK1 inhibitor assay. A.E.W. performed MS and used CompPASS to identify HCIPs and phosphosites under the direction of J.W.H. L.M.S. created heat maps for phosphorylation and interaction figures and provided statistical analysis support. R.C.J.S. provided support for the coimmunoprecipitation experiments. M.O. participated in the initial studies identifying the mitotic form of YAP and disruption of cytokinesis by its knockdown. D.A.B. and J.S.B. participated in the conception of experiments and their interpretation and drafted the manuscript. All authors reviewed and revised the manuscript.
REFERENCES AND NOTES
- 1.Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9:2145–2152. [PubMed] [Google Scholar]
- 2.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing Yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18:2551–2562. doi: 10.1093/emboj/18.9.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. 2006;103:12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc Natl Acad Sci USA. 2010;107:1431–1436. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam-Himlin DM, Daniels JA, Gayyed MF, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. The hippo pathway in human upper gastrointestinal dysplasia and carcinoma: A novel oncogenic pathway. Int J Gastrointest Cancer. 2006;37:103–109. doi: 10.1007/s12029-007-0010-8. [DOI] [PubMed] [Google Scholar]
- 12.Da CL, Xin Y, Zhao J, Luo XD. Significance and relationship between Yes-associated protein and survivin expression in gastric carcinoma and precancerous lesions. World J Gastroenterol. 2009;15:4055–4061. doi: 10.3748/wjg.15.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39:1582–1589. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010;101:1279–1285. doi: 10.1111/j.1349-7006.2010.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu MZ, Yao TJ, Lee NPY, Ng IOL, Chan YT, Zender L, Lowe SW, Poon RTP, Luk JM. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer. 2009;115:4576–4585. doi: 10.1002/cncr.24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276:15164–15173. doi: 10.1074/jbc.M010484200. [DOI] [PubMed] [Google Scholar]
- 17.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- 18.Yuan M, Tomlinson V, Lara R, Holliday D, Chelala C, Harada T, Gangeswaran R, Manson-Bishop C, Smith P, Danovi SA, Pardo O, Crook T, Mein CA, Lemoine NR, Jones LJ, Basu S. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008;15:1752–1759. doi: 10.1038/cdd.2008.108. [DOI] [PubMed] [Google Scholar]
- 19.Yabuta N, Okada N, Ito A, Hosomi T, Nishihara S, Sasayama Y, Fujimori A, Okuzaki D, Zhao H, Ikawa M, Okabe M, Nojima H. Lats2 is an essential mitotic regulator required for the coordination of cell division. J Biol Chem. 2007;282:19259–19271. doi: 10.1074/jbc.M608562200. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Yu K, Hao Y, Li D-m, Stewart R, Insogna KL, Xu T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6:609–617. doi: 10.1038/ncb1140. [DOI] [PubMed] [Google Scholar]
- 21.Yabuta N, Mukai S, Okamoto A, Okuzaki D, Suzuki H, Torigata K, Yoshida K, Okada N, Miura D, Ito A, Ikawa M, Okabe M, Nojima H. N-terminal truncation of Lats1 causes abnormal cell growth control and chromosomal instability. J Cell Sci. 2013;126:508–520. doi: 10.1242/jcs.113431. [DOI] [PubMed] [Google Scholar]
- 22.McPherson JP, Tamblyn L, Elia A, Migon E, Shehabeldin A, Matysiak-Zablocki E, Lemmers B, Salmena L, Hakem A, Fish J, Kassam F, Squire J, Bruneau BG, Hande MP, Hakem R. Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J. 2004;23:3677–3688. doi: 10.1038/sj.emboj.7600371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bothos J, Tuttle RL, Ottey M, Luca FC, Halazonetis TD. Human LATS1 is a mitotic exit network kinase. Cancer Res. 2005;65:6568–6575. doi: 10.1158/0008-5472.CAN-05-0862. [DOI] [PubMed] [Google Scholar]
- 24.Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 2005;65:92–98. [PubMed] [Google Scholar]
- 25.Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer GP. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr Biol. 2007;17:700–705. doi: 10.1016/j.cub.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 26.Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, Zafiropoulos PJ, Yamaguchi S, Winter S, Carthew RW, Cooper M, Jones D, Frenz L, Glover DM. Genome-wide survey of protein kinases required for cell cycle progression. Nature. 2004;432:980–987. doi: 10.1038/nature03160. [DOI] [PubMed] [Google Scholar]
- 27.Normand G, King RW. Understanding cytokinesis failure. Adv Exp Med Biol. 2010;676:27–55. doi: 10.1007/978-1-4419-6199-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bement WM, Miller AL, von Dassow G. Rho GTPase activity zones and transient contractile arrays. Bioessays. 2006;28:983–993. doi: 10.1002/bies.20477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi K, Yonemura S, Matsui T, Tsukita S. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J Cell Sci. 1999;112(Pt 8):1149–1158. doi: 10.1242/jcs.112.8.1149. [DOI] [PubMed] [Google Scholar]
- 30.Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localization in cells and tissues. Exp Cell Res. 2004;295:300–314. doi: 10.1016/j.yexcr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 31.Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175:477–490. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piekny AJ, Glotzer M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr Biol. 2008;18:30–36. doi: 10.1016/j.cub.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 33.Moorman JP, Bobak DA, Hahn CS. Inactivation of the small GTP binding protein Rho induces multinucleate cell formation and apoptosis in murine T lymphoma EL4. J Immunol. 1996;156:4146–4153. [PubMed] [Google Scholar]
- 34.Tsubakimoto K, Matsumoto K, Abe H, Ishii J, Amano M, Kaibuchi K, Endo T. Small GTPase RhoD suppresses cell migration and cytokinesis. Oncogene. 1999;18:2431–2440. doi: 10.1038/sj.onc.1202604. [DOI] [PubMed] [Google Scholar]
- 35.O’Connell CB, Wheatley SP, Ahmed S, Wang Y-l. The small GTP-binding protein rho regulates cortical activities in cultured cells during division. J Cell Biol. 1999;144:305–313. doi: 10.1083/jcb.144.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aepfelbacher M, Essler M, Luber De Quintana K, Weber PC. ADP-ribosylation of the GTP-binding protein RhoA blocks cytoplasmic division in human myelomonocytic cells. Biochem J. 1995;308(Pt 3):853–858. doi: 10.1042/bj3080853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dephoure N, Zhou C, Villén J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niiya F, Xie X, Lee KS, Inoue H, Miki T. Inhibition of cyclin-dependent kinase 1 induces cytokinesis without chromosome segregation in an ECT2 and MgcRacGAP-dependent manner. J Biol Chem. 2005;280:36502–36509. doi: 10.1074/jbc.M508007200. [DOI] [PubMed] [Google Scholar]
- 39.Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J. CDK1 phospho-rylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res. 2013;73:6722–6733. doi: 10.1158/0008-5472.CAN-13-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Couzens AL, Knight JDR, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras AC. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68:2789–2794. doi: 10.1158/0008-5472.CAN-07-6205. [DOI] [PubMed] [Google Scholar]
- 44.Vigneron AM, Ludwig RL, Vousden KH. Cytoplasmic ASPP1 inhibits apoptosis through the control of YAP. Genes Dev. 2010;24:2430–2439. doi: 10.1101/gad.1954310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283:5496–5509. doi: 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- 46.Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O’Neill E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell. 2007;27:962–975. doi: 10.1016/j.molcel.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oka T, Mazack V, Sudol M. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP) J Biol Chem. 2008;283:27534–27546. doi: 10.1074/jbc.M804380200. [DOI] [PubMed] [Google Scholar]
- 48.Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, Fu XD, Mills GB, Guan KL. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Huang J, Wang X, Yuan J, Li X, Feng L, Park JI, Chen J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012;26:1959–1971. doi: 10.1101/gad.192955.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paramasivam M, Sarkeshik A, Yates JR, III, Fernandes MJG, McCollum D. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell. 2011;22:3725–3733. doi: 10.1091/mbc.E11-04-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hauri S, Wepf A, van Drogen A, Varjosalo M, Tapon N, Aebersold R, Gstaiger M. Interaction proteome of human Hippo signaling: Modular control of the co-activator YAP1. Mol Syst Biol. 2013;9:713. doi: 10.1002/msb.201304750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JS, Kamijo K, Ohara N, Kitamura T, Miki T. MgcRacGAP regulates cortical activity through RhoA during cytokinesis. Exp Cell Res. 2004;293:275–282. doi: 10.1016/j.yexcr.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 53.Boyne JR, Yosuf HM, Bieganowski P, Brenner C, Price C. Yeast myosin light chain, Mlc1p, interacts with both IQGAP and class II myosin to effect cytokinesis. J Cell Sci. 2000;113(Pt 24):4533–4543. doi: 10.1242/jcs.113.24.4533. [DOI] [PubMed] [Google Scholar]
- 54.Fishkind DJ, Cao L-g, Wang Y-l. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells: Effects on mitosis and cytokinesis. J Cell Biol. 1991;114:967–975. doi: 10.1083/jcb.114.5.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol. 2004;164:233–241. doi: 10.1083/jcb.200309035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikawa M, Su L, Parsons SJ. Opposing roles of p190RhoGAP and Ect2 RhoGEF in regulating cytokinesis. Cell Cycle. 2008;7:2003–2012. doi: 10.4161/cc.7.13.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Somma MP, Fasulo B, Cenci G, Cundari E, Gatti M. Molecular dissection of cy-tokinesis by RNA interference in Drosophila cultured cells. Mol Biol Cell. 2002;13:2448–2460. doi: 10.1091/mbc.01-12-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Echard A, Hickson GRX, Foley E, O’Farrell PH. Terminal cytokinesis events uncovered after an RNAi screen. Curr Biol. 2004;14:1685–1693. doi: 10.1016/j.cub.2004.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- 61.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–1038. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. doi: 10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- 64.Shin K, Straight S, Margolis B. PATJ regulates tight junction formation and polarity in mammalian epithelial cells. J Cell Biol. 2005;168:705–711. doi: 10.1083/jcb.200408064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin K, Wang Q, Margolis B. PATJ regulates directional migration of mammalian epithelial cells. EMBO Rep. 2007;8:158–164. doi: 10.1038/sj.embor.7400890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schlüter MA, Pfarr CS, Pieczynski J, Whiteman EL, Hurd TW, Fan S, Liu CJ, Margolis B. Trafficking of Crumbs3 during cytokinesis is crucial for lumen formation. Mol Biol Cell. 2009;20:4652–4663. doi: 10.1091/mbc.E09-02-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan LY, Amon A. Spindle position is coordinated with cell-cycle progression through establishment of mitotic exit-activating and -inhibitory zones. Mol Cell. 2010;39:444–454. doi: 10.1016/j.molcel.2010.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiba S, Ikeda M, Katsunuma K, Ohashi K, Mizuno K. MST2- and Furry-mediated activation of NDR1 kinase is critical for precise alignment of mitotic chromosomes. Curr Biol. 2009;19:675–681. doi: 10.1016/j.cub.2009.02.054. [DOI] [PubMed] [Google Scholar]
- 69.Chiyoda T, Sugiyama N, Shimizu T, Naoe H, Kobayashi Y, Ishizawa J, Arima Y, Tsuda H, Ito M, Kaibuchi K, Aoki D, Ishihama Y, Saya H, Kuninaka S. LATS1/ WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J Cell Biol. 2012;197:625–641. doi: 10.1083/jcb.201110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bertini E, Oka T, Sudol M, Strano S, Blandino G. YAP: At the crossroad between transformation and tumor suppression. Cell Cycle. 2009;8:49–57. doi: 10.4161/cc.8.1.7259. [DOI] [PubMed] [Google Scholar]
- 71.Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, Kuo CJ, Camargo FD. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493:106–110. doi: 10.1038/nature11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(Suppl 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 73.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 74.Caldwell CM, Green RA, Kaplan KB. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 2007;178:1109–1120. doi: 10.1083/jcb.200703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corson TW, Gallie BL. KIF14 mRNA expression is a predictor of grade and outcome in breast cancer. Int J Cancer. 2006;119:1088–1094. doi: 10.1002/ijc.21954. [DOI] [PubMed] [Google Scholar]
- 76.Roversi G, Pfundt R, Moroni RF, Magnani I, van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers EFPM. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006;25:1571–1583. doi: 10.1038/sj.onc.1209177. [DOI] [PubMed] [Google Scholar]
- 77.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 79.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–115. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 81.Beausoleil SA, Villén J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mitosis of MCF-10A cells depleted of YAP with five distinct shRNA sequences.
Mitosis of PATJ-depleted MCF-10A cells.
Mitosis of YAP-depleted MCF-10A cells expressing GFP-tubulin.
Mitosis of MCF-10A cells treated with YAP siRNA.
Mitosis of YAP-depleted MCF-10A cells in a monolayer.
Mitosis of YAP-depleted HeLa cells.
Mitosis of YAP-depleted multiple HeLa cells.
RHOA localization in a mitotic YAP-depleted MCF-10A cell.
Mitosis of YAP-depleted MCF-10A cells treated with C3 transferase or Y-27632.
Mitosis of MCF-10A cells expressing wild-type YAP or YAP 3A or YAP 3D mutants.
Fig. S1. Immunofluorescence analysis of YAP-depleted cells.
Fig. S2. YAP hairpins.
Fig. S3. Spindle misorientation after YAP knockdown.
Fig. S4. YAP phospho-mutant combinations.
Fig. S5. Phosphorylation of MLC is increased by expression of the YAP 3A mutant.
Fig. S6. YAP and PATJ coimmunoprecipitation.
Fig. S7. Immunofluorescence of PATJ-depleted cells.
HCIPs for YAP wild type, YAP 3D, and YAP 3A.