

Key Points
FVL platelet-poor and platelet-rich plasma have a reduced threshold for the activation of blood coagulation, which is modulated by TFPIα.
Prothrombinase assembled with FVL is less susceptible to inhibition by TFPIα than is prothrombinase assembled with FV.
Abstract
Activated factor V (FVa) and factor X (FXa) form prothrombinase, which converts prothrombin to thrombin. The α isoform of tissue factor pathway inhibitor (TFPI) dampens early procoagulant events, partly by interacting with FV. FV Leiden (FVL) is the most common genetic thrombophilia in whites. Thrombosis risk is particularly elevated in women with FVL taking oral contraceptives, which produce acquired TFPIα deficiency. In mice, FVL combined with a 50% reduction in TFPI causes severe thrombosis and perinatal lethality. However, a possible interaction between FVL and TFPIα has not been defined in humans. Here, we examined this interaction using samples from patients with FVL in thrombin generation and fibrin formation assays. In dilute TF- or FXa-initiated reactions, these studies exposed a TFPI-dependent activation threshold for coagulation initiation that was greatly reduced by FVL. The reduced threshold was progressively overcome with higher concentrations of TF or FXa. Plasma assays using anti-TFPI antibodies or a TFPI peptide that binds and inhibits FVa, demonstrated that the decreased activation threshold resulted from reduced TFPIα inhibition of prothrombinase. In assays using purified proteins, TFPIα was a 1.7-fold weaker inhibitor of prothrombinase assembled with FVL than with FV. Thus, FVL reduces the threshold for initiating coagulation, and this threshold is further reduced in situations of low TFPIα concentration. Individuals with FVL are likely prone to thrombosis in response to weak procoagulant stimuli that would not initiate blood clot formation in individuals with FV.
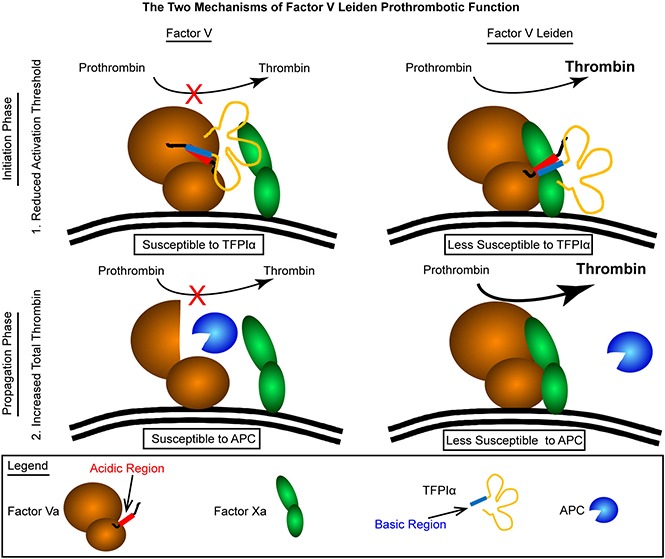
Visual Abstract
Introduction
The initiation phase of hemostasis commences when the complex of tissue factor (TF) and factor VIIa (FVIIa) generates the serine protease factor Xa (FXa).1 FXa activates a small amount of the cofactor factor Va (FVa),2,3 and the two combine to form the thrombin-generating prothrombinase complex.4-6 This slow initiation phase is followed by a rapid burst in thrombin generation, termed the “propagation” phase, which is mediated by a series of amplification reactions, including further activation of FV by thrombin.2 The α and β isoforms of the TF pathway inhibitor (TFPI) modulate the initiation phase by inhibiting the TF-FVIIa complex.7-9 TFPIα, but not TFPIβ, also modulates the initiation phase by inhibiting early forms of prothrombinase.10 TFPIα contains 3 Kunitz-type protease inhibitor domains (K1, K2, and K3), the second of which inhibits FXa.11 An evolutionarily conserved basic region in the TFPIα C terminus binds with high affinity to a similarly conserved FV B-domain acidic region in FXa-activated and platelet-released forms of FVa.10,12 This acidic region is rapidly removed by thrombin.2,13 The TFPIα basic region does not bind any other acidic region in thrombin-activated FVa.10 As a result, TFPIα does not inhibit prothrombinase activity during the propagation phase.14,15 TFPIα does inhibit prothrombinase containing partially proteolysed forms of FVa that retain the B-domain acidic region during the initiation phase. Instead, the propagation phase is downregulated by a number of endogenous anticoagulants, including activated protein C (APC).16 APC proteolytically inactivates FVa, through initial cleavage at Arg506, and subsequent cleavages at Arg306 and Arg679.17
The factor V Leiden (FVL) mutation is the substitution of Arg506 with Gln.18-20 This prevents APC from cleaving at residue 506, and slows subsequent cleavages at Arg306 and Arg679.21 It is the most common hereditary thrombophilia in white populations, with 5% heterozygous for this mutation.22 Heterozygous individuals have a three- to eightfold increased risk of venous thromboembolism, whereas homozygous individuals have an 80-fold increased risk.22
Although the thrombotic risk has long been attributed to the resistance of FVL to proteolytic inactivation by APC, several lines of evidence suggest that the APC and TFPI anticoagulant systems function cooperatively, and that TFPI contributes to FVL pathogenesis. In vitro coagulation reactions using purified proteins at mean plasma concentrations found that, although TFPIα inhibition of coagulation initiation is not altered by APC, the ability of APC to inhibit the propagation phase of TF-initiated thrombin generation is enhanced by the addition of 2.5 nM TFPIα.23 When the same assays were performed substituting FVL for FV, 2.5 nM TFPIα was required for APC to inhibit the propagation phase, and a 50% reduction in TFPIα (to 1.25 nM) resulted in almost complete loss of APC anticoagulant function.24 A striking procoagulant effect of FVL combined with a 50% reduction in TFPI has also been observed in vivo; mice with FVL homozygosity and TFPI haploinsufficiency die of severe perinatal intravascular thrombosis.25 A similar synergistic procoagulant relationship between FVL and TFPI is possibly present in humans, where estrogen-containing oral contraceptives decrease plasma TFPI concentration by 25% to 30%26 and further increase the risk of thrombosis in women with FVL fivefold.27-29
Here, we investigated the effect of FVL on prothrombinase inhibition by TFPIα. Because TFPIα only inhibits prothrombinase during the initiation phase, these studies focused on the effect of FVL and TFPIα on the lag time of thrombin generation. We found that FVL decreases the ability of TFPIα to inhibit the initiation phase of coagulation. These data provide a potential mechanism to explain the synergistic effect of combined FVL and TFPI deficiency.
Materials and methods
Materials
Human prothrombin, thrombin, and FXa were from Enzyme Research Laboratories. Recombinant human APC was a gift from John Griffin (Scripps Research Institute, La Jolla, CA), and the inhibitory anti-APC antibody HAPC1555 was a gift from Patricia Liaw (McMaster University, Hamilton, Ontario, Canada). TF (Innovin) was from Siemens, and bovine type I collagen was from Invitrogen. Antibodies against the K2 and K3C domains of TFPI were provided by Novo Nordisk, and the antibody against K1 was a gift from George Broze Jr (Washington University, St. Louis, MO). FV810 and FVL810 were gifts from Rodney Camire (University of Pennsylvania, Philadelphia, PA). Recombinant human TFPIα was as described.30 Dansylarginine-N-(3-ethyl-1,5-pentanediyl)amine (DAPA) and corn trypsin inhibitor (CTI) were from Haematologic Technologies. The peptide Leu-Ile-Lys-Thr-Lys-Arg-Lys-Arg-Lys-Lys (LIKTKRKRKK) was synthesized by the Peptide Core Facility (BloodCenter of Wisconsin, Milwaukee, WI), and Gly-Pro-Arg-Pro was purchased from GenScript. Spectrozyme TH was from Sekisui Diagnostics. Phosphatidylcholine, phosphatidylserine, and phosphatidylethanolamine were from Avanti Polar Lipids (Alabaster, AL). Phospholipid vesicles containing 40% phosphatidylcholine, 40% phosphatidylethanolamine, and 20% phosphatidylserine (PC:PE:PS) were prepared by sonication, as described by Morrissey.31 FV-deficient plasma was purchased from either George King Bio-Medical or HRF Inc.
Platelet-poor plasma (plasma) and platelet-rich plasma (PRP)
All studies using human subjects were approved by the Institutional Review Board of the Medical College of Wisconsin (Milwaukee, WI). Written informed consent was obtained prior to phlebotomy. Participants were either referred to the study from the Benign Hematology Clinic (Froedtert Hospital, Milwaukee, WI) or identified based upon FVL test results using the i2b2 Cohort Discovery Tool (Clinical and Translational Science Institute of Southeast Wisconsin, Milwaukee, WI). Prospective participants taking a direct thrombin inhibitor were excluded from the study. Ten participants were recruited in each genotype group (homozygous FVL, heterozygous FVL, and control). Two tubes of blood were collected. The first was discarded to limit TF contamination. The second was collected into 3.2% citrate and 50 μg/mL CTI, and centrifuged (150 × g, 15 minutes) to prepare PRP. PRP was subsequently centrifuged (700 × g, 10 minutes) to prepare plasma. PRP was diluted with plasma to a platelet concentration of 1.5 × 108/mL.
Calibrated automated thrombography
PRP or plasma was diluted 1:1 with FV-deficient plasma. For plasma experiments, plasma (40 μL) was incubated (10 minutes at 37°C) with an activation mixture (10 μL) containing FXa or TF (varying concentrations) and PC:PE:PS (4 μM final) in 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 150 mM NaCl, 0.1% bovine serum albumin, pH 7.4 (HBSA). For PRP experiments, the activation mixture contained FXa (varying concentrations) and collagen (20 μg/mL final) in HBSA. For some experiments, anti-K1 (50 nM), anti-K2 (50 nM), anti-K3C (50 nM), LIKTKRKRKK (1 μM), TFPIα (1 nM), APC (5 nM), and/or HAPC1555 (50 nM) was included in the incubation. Thrombin generation was initiated with a mixture of calcium and thrombin fluorogenic substrate (FluCa; Diagnostica Stago) (10 μL). Fluorescence was monitored on a Fluoroskan Ascent microplate reader (Thermo Scientific), and thrombin calculated with Thrombinoscope version 5 software (Thrombinoscope BV). All samples were measured in triplicate.
Prothrombinase activity assays
FV810 or FVL810 (0.5 nM) was incubated with PC:PE:PS (20 μM), DAPA (3 μM), and the indicated concentrations of TFPIα in HBS containing 5 mM CaCl2 and 0.1% PEG-8000. Reactions were initiated by the addition of prothrombin (1.4 μM) and FXa (5 nM). Timed aliquots were quenched by the addition of EDTA (33.3 μM), and thrombin concentrations were determined by monitoring cleavage of a thrombin chromogenic substrate (Spectrozyme TH, 0.32 mM) at 405 nm using a SpectraMax 384 Plus microplate reader (Molecular Devices), and comparison with a thrombin standard curve. Fifty percent inhibitory concentration (IC50) were determined as described.10
Fibrin formation assays
Fibrin formation assays were adapted from Campbell et al.32 Briefly, FXa (indicated concentrations) or thrombin (1 nM) was incubated with PC:PE:PS (4 μM) in HBSA containing 5 mM CaCl2 (80 μL total volume). Reactions were initiated by the addition of plasma (20 μL; diluted 1:1 with FV-deficient plasma), and fibrin formation was monitored at 405 nm for 1 hour. In control reactions, the addition of phospholipid vesicles alone, without any initiator, did not produce a measurable change in absorbance, the addition of thrombin (1 nM) increased absorbance, and this increase was blocked by the peptide Gly-Pro-Arg-Pro (2 mM), which prevents fibrin polymerization,33 confirming that the change in absorbance was due to fibrin clot formation (supplemental Figure 1). All samples were measured in triplicate.
Total TFPI and TFPIα antigen
Anti-K2 was conjugated to AlphaLISA acceptor beads (PerkinElmer). Anti-K1, for measurement of total TFPI, and anti-K3C, for measurement of TFPIα, were conjugated with biotin. Plasma (20 μL, diluted in HBSA containing 0.1% Tween-20) and 20 μL of a mixture containing the conjugated acceptor bead (12.5 μg/mL) and biotinylated anti-K1 (2.5 nM) or anti-K3C (5 nM) was added to the well of a 96-well plate and incubated (1 hour at 37°C). Streptavidin-conjugated donor beads (PerkinElmer) were added (25 μL, 20 μg/mL) and incubated (1 hour at room temperature). The plate was read on an EnSpire microplate reader (PerkinElmer) and the results compared with a TFPIα standard curve.
Statistical analyses
The thrombin generation parameters were not normally distributed. Therefore, nonparametric Kruskal-Wallis tests were performed, followed by correction with Dunn’s multiple comparisons tests using GraphPad Prism version 6.07 (GraphPad Software).
Results
Study participants
Plasma and PRP were collected from 10 heterozygous FVL, 10 homozygous FVL, and 10 normal FV individuals (Table 1). All study participants were white, because FVL is predominantly a white mutation.22 The control and homozygous FVL donor populations were of a similar average age (37.8 vs 40.9 years, respectively; P = .60), whereas the heterozygous FVL donors were older (58.4 years; P = .01, compared with controls).
Table 1.
Study participant information
| Control (n = 10) | Heterozygous FVL (n = 10) | Homozygous FVL (n = 10) | |
|---|---|---|---|
| M/F | 7/3 | 5/5 | 3/7 |
| Age | 37.8 (25.5-59.0)* | 58.4 (42.9-76.3) | 40.9 (18.8-63.8) |
F, female; M, male.
*Mean age (age range).
FVL reduces the TFPI-dependent activation threshold for TF-initiated thrombin generation in plasma
The anticoagulant potential in blood establishes an activation threshold that must be overcome for the initiation of coagulation. These studies evaluated the effect of the FVL gene mutation on the activation threshold. All samples were mixed 1:1 with FV-deficient plasma prior to analysis, thereby reducing the interdonor variability in other plasma parameters, such as coagulation protein concentrations or lipid profiles. TF-initiated thrombin generation was first measured in FV and homozygous FVL plasmas. The lag time for thrombin generation was the same for FV and FVL plasma in reactions initiated with 1 pM TF, although the peak thrombin was greater in FVL plasma (Figure 1A). Because the TF concentration was progressively reduced to 0.05 pM, the differences between FVL and FV plasma became progressively more pronounced, with faster and greater thrombin production observed in FVL plasma. The differences in lag time were normalized at each TF concentration by the addition of a monoclonal antibody against the TFPI K2 domain (anti-K2), whereas the FVL plasmas continued to have greater peak thrombin in the presence of anti-K2.
Figure 1.

The threshold for TF- and FXa-initiated thrombin generation is lower in FVL plasma than in FV plasma and is dependent on TFPI. (A) A total of 0.05 to 1 pM TF or (B) 5 to 100 pM FXa initiated thrombin generation in plasma anticoagulated with citrate and CTI containing FV (black) or FVL (gray). Reactions were started by the addition of CaCl2 to plasma containing 4 μM PC:PE:PS in the presence (dashed line) or absence (solid line) of anti-K2 antibody (50 nM). At 0.05 pM TF or 5 pM FXa, no thrombin generation occurred unless TFPI activity was blocked. TF experiments were performed using 2 each of FV and FVL donors. FXa experiments were performed with 10 donors of each genotype. Representative thrombin generation curves from a single FV donor and a single FVL donor are shown (A-B).
FVL reduces the activation threshold for FXa-initiated thrombin generation in plasma
In TF-initiated thrombin generation assays, TFPI can exert anticoagulant effects by inhibiting TF-FVIIa or prothrombinase. The influence of the FV genotype in the TF-initiated thrombin generation assays (Figure 1A) suggested that prothrombinase inhibition is a major component of the TFPI anticoagulant activity observed. Therefore, FXa-initiated thrombin generation experiments were performed using FV and FVL plasmas to study the effect of FVL on prothrombinase inhibition by TFPI without the confounding factor of TFPI-mediated TF-FVIIa inhibition.10 Similar to that observed in the TF-initiated reactions, the impact of TFPI on thrombin generation increased because the initiating FXa concentration was progressively reduced to the point where 5 pM FXa produced no thrombin generation unless TFPI activity was blocked with anti-K2 (Figure 1B). The inhibitory anti-APC antibody HAPC155534 did not alter thrombin generation in FV or FVL plasmas, but did reverse the effects of exogenous APC added to either plasma (supplemental Figure 2). Therefore, the difference between FVL and normal FV plasmas was not caused by APC contamination.
Collectively, these experiments identified an FV genotype-dependent difference in the concentration of FXa required to initiate coagulation. All 10 FV plasmas generated thrombin in response to 20 pM FXa, 3 of 10 in response to 10 pM FXa, and none in response to 5 pM FXa (data not shown). By contrast, the threshold for thrombin generation was lower in FVL plasmas; 9 of 10 homozygous FVL plasmas generated thrombin in response to 10 pM FXa, and 6 of 10 in response to 5 pM FXa (data not shown). Heterozygous FVL plasmas had an intermediate and highly variable response (supplemental Figure 3). These differences in the sensitivity of the plasmas to a procoagulant trigger reveal an activation threshold that varies with FV genotype and is TFPI dependent.
The difference in lag time between FV and FVL plasmas is TFPI dependent
In reactions initiated with 100 pM FXa, the average lag time was longer in plasmas containing FV (13.01 ± 1.12 minutes) compared with heterozygous FVL (12.16 ± 2.57 minutes) or homozygous FVL (11.73 ± 1.08 minutes). Although these differences were not statistically significant, they suggested an FVL dose response for initiation of thrombin generation. The role of TFPIα in this process was evaluated in 2 ways. First, the addition of recombinant TFPIα prolonged the lag time more in FV plasma than in homozygous FVL plasma (61.4% ± 14.3% vs 39.4% ± 5.8%, respectively; P < .01), with an intermediate effect in heterozygous FVL plasma (46.5% ± 20.7%; P < .05 compared with FV plasma) (Figure 2A). The resulting lag times were significantly different between FV and homozygous FVL plasmas (21.03 ± 2.95 minutes vs 16.37 ± 1.87 minutes, respectively; P < .05), whereas the average lag time for heterozygous FVL plasmas was intermediate (17.99 ± 5.21 minutes) and not statistically different from FVL or FV plasmas. Second, anti-K2 decreased the lag time more in FV plasma than homozygous FVL plasma (30.5% ± 4.0% vs 25.3% ± 3.0%, respectively), whereas an intermediate response was observed in heterozygous FVL plasma (29.1% ± 4.1%). However, these decreases were not statistically different (Figure 2B). The effects of anti-K2 were more apparent in reactions initiated with 20 pM FXa (Figure 1). In the absence of antibody, the average lag time in FV plasma initiated with 20 pM FXa (55.77 ± 32.22 minutes) was significantly longer than in either heterozygous FVL plasma (30.64 ± 25.19 minutes; P < .05) or homozygous FVL plasma (26.62 ± 8.61 minutes; P < .05). Anti-K2 decreased the average lag time in FV plasma by 61.0% ± 14.1%, in heterozygous FVL plasma by 46.6% ± 17.8% (P < .05), and in homozygous FVL plasma by 39.2% ± 14.3% (P < .01) (Figure 2C). This corresponded to average lag times, in the presence of anti-K2, of 17.79 ± 3.24 minutes, 12.69 ± 3.90 minutes (P < .01), and 15.34 ± 3.44 minutes (P = .12), respectively.
Figure 2.

Inhibition of plasma TFPI reduces the lag time for initiation of thrombin generation to a greater extent in FV plasma than in FVL plasma. Plasma thrombin generation assays were initiated with either (A-B) 100 pM, (C) 20 pM, or (D-F) also 100 pM FXa. Assays were performed in the absence or presence of (A) 1 nM TFPIα, (B-C) 50 nM anti-K2, (D) 50 nM anti-K3C, (E) 50 nM anti-K1, or (F) 1 μM LIKTKRKRKK. Shown are the changes in lag time, relative to the untreated plasma control (n = 10 for each genotype). The boxes represent the 25th to 75th percentile; the whiskers represent minimum and maximum values; and the lines represent the median values. Het, heterozygous.
Altered prothrombinase inhibition by TFPIα produces the differences in lag time for thrombin generation
The impact of FV genotype on thrombin generation suggested that TFPIα is a weaker inhibitor of prothrombinase assembled with FVL, than prothrombinase assembled with FV. Prothrombinase inhibition was further probed in 2 ways. First, an anti-K3C antibody, which blocks prothrombinase inhibition by TFPIα,10 shortened the lag time in 100 pM FXa-initiated thrombin generation assays more in normal FV plasma than in homozygous FVL plasma (20.7% ± 4.1% vs 16.8% ± 1.3%) with an intermediate effect in heterozygous FVL plasma (19.3% ± 2.5%), although the differences were not statistically significant (Figure 2D). Similar to anti-K2, anti-K3C normalized the lag times between the genotypes (10.29 ± 0.56 minutes, 9.78 ± 1.83 minutes, and 9.78 ± 1.02 minutes). As a control, an anti-K1 antibody, which blocks TF-FVIIa inhibition by TFPIα, had little effect on thrombin generation in any of the plasmas, consistent with the lack of TF-FVIIa activity in this assay (Figure 2E). Second, the peptide LIKTKRKRKK, which mimics a portion of the TFPIα C-terminal basic region and inhibits prothrombinase by tightly binding to the FVa acidic region,10 prolonged the lag time in FV plasma by 27.8% ± 5.4%, in heterozygous FVL plasma by 18.6% ± 7.7% (P < .05), and in homozygous FVL plasma by 8.0% ± 9.2% (P < .001) (Figure 2F). The resistance of FVL plasma to the effects of this peptide was very strong, such that in 3 of 10 homozygous FVL plasmas, it had no impact on thrombin generation, an observation not made in any of the FV or heterozygous FVL plasmas. The average lag time in the presence of LIKTKRKRKK was greater in FV plasmas than in homozygous FVL plasmas (16.62 ± 1.65 minutes vs 12.74 ± 2.09 minutes; P < .01), whereas heterozygous FVL plasmas had an intermediate response to the peptide (14.47 ± 3.44 minutes; P < .05).
FVL reduces the TFPI-dependent activation threshold for FXa-initiated thrombin generation in PRP
Platelets contain pools of TFPIα and FV/FVa, along with a physiologic surface for prothrombinase assembly.5,10,35 Therefore, the impact of FVL on platelet-mediated thrombin generation was examined in collagen-treated PRP from donors with FV or homozygous FVL (Figure 3; supplemental Figure 4). The PRP was treated with collagen to activate platelets. Similar to that observed in plasma, FXa titrations demonstrated an approximate twofold difference in the threshold for initiation of thrombin generation: 20 pM FXa in FV PRP (5/5 donors) vs 10 pM FXa in FVL PRP (4/5 donors) (supplemental Figure 4). No PRP sample generated thrombin in response to 5 pM FXa, unless anti-K2 was added (Figure 3). The interdonor variability was increased at lower FXa concentrations. When initiating reactions with 10 pM FXa, the average lag time in homozygous FVL PRP was 86.3 ± 19.7 minutes, compared with >120 minutes for FV PRP, because none of the FV PRP samples generated any detectable thrombin (P < .01). This difference was normalized by the anti-K2 antibody, which decreased the lag times to 29.90 ± 6.39 minutes and 35.20 ± 7.96 minutes (P = .46) for FVL and FV PRP, respectively.
Figure 3.

FVL reduces the TFPI-dependent activation threshold for FXa-initiated thrombin generation in PRP. Thrombin generation was initiated in PRP, containing FV (black) or FVL (gray), with 5 to 100 pM FXa and 20 μg/mL collagen, in the absence (lines) or presence (dashes) of 50 nM anti-K2. Experiments were performed using 5 each of FV and FVL donors. Shown are representative thrombin generation curves from a single FV donor and a single FVL donor.
FVL reduces the TFPI-dependent activation threshold for FXa-initiated fibrin formation
The results of the thrombin generation assays were validated in fibrin formation assays, an independent assay system that is sensitive to sub-nanomolar concentrations of thrombin.36 Fibrin formation was measured in FV and homozygous FVL plasmas. In this assay, all plasmas generated fibrin in response to 5 pM FXa, with a shorter lag time in FVL plasma than in FV plasma (Figure 4A). The maximum rate of fibrin formation (estimated by the slope between the onset of fibrin formation and the plateau) was similar between the 2 groups. The difference between FV and FVL plasma was more pronounced when the concentration of FXa used to initiate was reduced. In reactions initiated with 0.5 pM FXa, only 1 of 8 FV plasmas generated a stable fibrin clot, as indicated by the absorbance reaching a plateau within 60 minutes. By comparison, 7 of 8 homozygous FVL plasmas produced stable clots under these conditions (Figure 4B). The differences between FV and FVL plasmas were normalized by the addition of anti-K2, which blocks all TFPI functions, or anti-K3C, which specifically blocks the TFPIα/FV interaction (Figure 4B).
Figure 4.

FVL reduces the TFPI-dependent activation threshold for FXa-initiated fibrin formation. Fibrin formation was initiated in FV plasma (red) or FVL plasma (blue) by the addition of (A) 5 pM or (B) 0.5 pM FXa, 4 μM PC:PE:PS, and 5 mM CaCl2, and monitored at 405 nm. Experiments were performed in the absence (solid lines) or presence of anti-K2 (long dashed lines) or anti-K3C (short dashed line). Experiments were performed using 8 each of FV and FVL donors. Shown are representative fibrin formation curves from a single FV donor and a single FVL donor. OD, optical density.
Prothrombinase assembled with FVL is less susceptible to TFPIα inhibition
The results from the thrombin generation and fibrin formation assays indicated that TFPIα had decreased prothrombinase inhibitory activity in the presence of FVL. We tested whether these results could be explained by decreased TFPIα protein in individuals with FVL. However, the total TFPI concentration was similar in FVL and FV plasmas (1.22 ± 0.24 nM vs 1.07 ± 0.09 nM, respectively), as was the TFPIα concentration (0.39 ± 0.05 nM vs 0.37 ± 0.01 nM). The FV-deficient plasma had reduced total TFPI and reduced TFPIα (0.55 nM and 0.16 nM, respectively).
Therefore, we quantified the inhibitory function of TFPIα in a purified prothrombinase assay using FV810, a recombinant form of FV containing the acidic region that binds the basic region of TFPIα,37,38 as well as FV810 containing the Leiden mutation (FVL810). The baseline rates of thrombin generation in these assays were similar between FV and FVL prothrombinase (average of 1.55 vs 1.46 nM per second). Prothrombinase containing FV810 was inhibited by TFPIα in a dose-dependent manner (IC50 = 4.7 nM; 95% confidence interval [CI] = 4.3-5.1 nM) (Figure 5A). TFPIα was a poorer inhibitor of prothrombinase assembled with FVL810. The IC50 increased 1.7-fold to 8.1 nM (95% CI = 7.4-8.9 nM). Similarly, the IC50 for LIKTKRKRKK inhibition of prothrombinase assembled with FVL810 was 2.1-fold higher than observed for prothrombinase assembled with FV810 (2.8 μM vs 6.0 μM; 95% CI = 2.4-3.2 μM and 4.6-7.8 μM, respectively) (Figure 5B).
Figure 5.

Prothrombinase containing FVL is less susceptible to inhibition by TFPIα. Various amounts of (A) TFPIα or (B) LIKTKRKRKK were incubated with 0.5 nM FV810 (□) or FVL810 (●), 20 μM PC:PE:PS, and 3 μM DAPA. Reactions were initiated with mixtures of 1.4 μM prothrombin and 5 nM FXa. The prothrombinase activity ± SD is reported as percent of control from 3 sets of experiments.
Discussion
Resistance to the anticoagulant activity of APC in plasma clotting assays was found to be prevalent and heritable among those with a history of venous thrombosis in the early 1990s.18 Shortly thereafter, FVL was identified as the most common genetic cause of this resistance.19,20,39 Mutation of Arg506 is thought to increase thrombotic risk through two APC-dependent mechanisms. First, the resistance to inactivation of FVL by APC prolongs the time prothrombinase is active in plasma clotting assays.21 Second, APC cleavage of FV at Arg506 produces an anticoagulant FV molecule,40 which acts as a cofactor for APC-mediated inactivation of FVIIIa41,42 and FVa.43 The data presented here uncover diminished inhibition of prothrombinase by TFPIα during the initiation of coagulation as a third, and APC-independent, biochemical mechanism contributing to the procoagulant phenotype associated with FVL. This mechanism is primarily manifest by lowering the activation threshold for initiation of the procoagulant response. The role of TFPI as a regulator of the activation threshold for coagulation has previously been proposed by Dahm et al,44 based on their studies of deep vein thrombosis risk, which was identified among individuals with plasma TFPI in the lowest 10th percentile of the normal range.
The APC-independent procoagulant tendency of FVL was apparent in thrombin generation assays, where differences between FV and FVL in plasma or PRP were normalized by blocking TFPI function with the anti-K2 antibody. These results, along with the lack of APC activity in the assay system, implicated a reduced ability of TFPIα to inhibit prothrombinase assembled with FVL as the procoagulant mechanism. This hypothesis was independently confirmed in: 1) thrombin generation experiments using the peptide LIKTKRKRKK, which showed reduced anticoagulant activity in FVL plasma; 2) in thrombin generation experiments using the anti-K3C antibody that blocks prothrombinase inhibition by TFPIα and had reduced procoagulant activity in FVL plasma; and 3) in turbidity experiments measuring FXa-initiated fibrin formation.
The plasma TFPI concentration varies with the plasma FV concentration.45-47 FV-deficient patients also have TFPIα deficiency, and immunodepletion of FV from plasma also removes TFPIα.46 Moreover, two FV mutants have been identified that bind to TFPIα and cause its concentration to increase 10- to 20-fold.45,47 These two mutations suggest the importance of prothrombinase inhibition by TFPIα, because they are associated with a moderately severe bleeding phenotype. Importantly, however, the FVL patients had normal total TFPI and TFPIα plasma concentrations, indicating the reduced anticoagulant activity of TFPI in FVL plasma was not caused by reduced TFPI concentration. Therefore, the decreased ability of TFPIα to inhibit prothrombinase assembled with FVL is likely responsible for the reduced anticoagulant activity observed. Although TFPIα was only a 1.7-fold weaker inhibitor of prothrombinase assembled with FVL than with FV, this relatively small difference in activity may be physiologically important under conditions where coagulation is initiated by weak stimuli. Moreover, its impact will be amplified when the TFPIα concentration is reduced. This is illustrated in Figure 6. The IC50 for inhibition of prothrombinase containing plasma FV is 0.9 nM, which is approximately the plasma TFPIα concentration.10 The average plasma TFPIα concentration would inhibit ∼30% of prothrombinase activity following the 1.7-fold increase in IC50 associated with FVL. The combination of a 50% reduction of the TFPIα concentration and the 1.7-fold increase in the IC50 results in inhibition of only ∼10% of prothrombinase activity. This greatly reduces the threshold for initiation of procoagulant activity, as was observed in the in vitro experiments presented here and the in vivo experiments of Eitzman et al, who found that the combination of FVL and reduced TFPI is substantially more procoagulant than either condition alone, as was observed in mice with combined FVL and TFPI haploinsuffiency.25
Figure 6.

A small change in inhibitory kinetics may explain the thrombosis associated with combined FVL and TFPIα haploinsufficiency. TFPIα inhibition of prothrombinase containing FXa-FVa (red) is reproduced from Wood et al.10 TFPIα inhibition of prothrombinase containing FXa-activated FVL (blue) is extrapolated, assuming a 1.7-fold increase in IC50. With FV, 50% inhibition is achieved by 0.9 nM TFPIα (i; green dashed line). This same concentration of TFPIα would be expected to inhibit ∼30% of prothrombinase containing FVL (ii; purple dashed line), and a 50% reduction in TFPIα would result in ∼10% prothrombinase inhibition (iii; orange dashed line).
In humans, plasma TFPI is decreased 25% to 30% by the use of either oral contraceptives26 or hormone replacement therapy,48 and women with FVL using oral contraceptives have a striking increased risk of venous thrombosis.27-29 The data presented here suggest the increased thrombotic risk in these women may be at least partially attributable to the combination of FVL and reduced TFPIα. Following a weak activation stimulus, a small amount of FVa may become available to assemble prothrombinase, either via release from activated platelets or activation by FXa. Under normal circumstances, this FVa would be rapidly bound by TFPIα. However, this interaction is less effective in the presence of FVL. The interaction of TFPIα with FV or FVL may be particularly relevant at the surface of activated platelets, which release TFPIα35,49 and forms of FVa that bind TFPIα,10 and provide a surface for prothrombinase assembly.5 The reduced TFPI inhibitory activity observed in PRP supports this supposition, demonstrating that even the high local concentration of platelet-released TFPIα is not capable of inhibiting FVL prothrombinase to the same extent as FV prothrombinase.
The molecular mechanism by which the Leiden mutation modifies the ability of TFPIα to inhibit prothrombinase is not yet clear. The interaction between the TFPIα basic region (LIKTKRKRKK) and the FVa acidic region is tight-binding and occurs in a charge-dependent manner.10,50 However, broad evolutionary conservation of the Leu, Ile, and Thr residues in FV and TFPIα51 strongly suggests that inhibition of FVa cofactor activity also requires a non–charge-based interaction between the two proteins. Several lines of experimental evidence support this notion. First, polylysine does not inhibit FV810 cofactor activity.52 Second, cleavage of FV810 at Arg1545 produces a form of FVa retaining the acidic region that is totally resistant to inhibition by a peptide mimicking the FV B-domain basic region (residues 963-1008; FV-BR)52 and by TFPIα.10 Third, FV-BR competes with FXa for binding to FV810, even though the B-domain is not needed for prothrombinase assembly.52 Taken together, these data suggest that LIKTKRKRKK may interact with a region on the FVa heavy chain near the FXa binding site, and the Leiden mutation weakens this interaction. Much work is needed to further define the interactive sites between TFPIα and FVa.
In conclusion, the FVL mutation appears to be prothrombotic through biochemical mechanisms acting during separate phases of coagulation. Prothrombinase assembled with FVL is less susceptible to inhibition by TFPIα during the initiation phase of coagulation. This reduces the activation threshold for initiation of blood clot formation. Second, the well-documented resistance of FVL to degradation by APC results in an enhancement of the propagation phase of coagulation and amplifies the extent of blood clot development. Therefore, it appears that patients with FVL are susceptible to venous thrombosis because weak prothrombotic stimuli, which would be rapidly dampened by TFPIα in individuals with normal FV, initiate the formation of blood clots in a process that is then further amplified by the resistance of FVL to degradation by APC.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Hartmut Weiler, Walter Bialkowski, and the phlebotomists of the BloodCenter of Wisconsin for their assistance with this study.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute (HL068835 [A.E.M.] and HL129193 [J.P.W.]), and by the National Center for Advancing Translational Sciences (UL1TR001436).
The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: J.P.W. and P.E.R.E. designed and performed experiments, analyzed data, and wrote the manuscript; L.M.B.K. recruited study participants and wrote the manuscript; S.A.M. designed experiments and wrote the manuscript; and A.E.M. designed the research, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: A.E.M. receives grant support from Novo Nordisk. The remaining authors declare no competing financial interests.
Correspondence: Alan E. Mast, Blood Research Institute, BloodCenter of Wisconsin, PO Box 2178, Milwaukee, WI 53201; e-mail: alan.mast@bcw.edu.
References
- 1.Komiyama Y, Pedersen AH, Kisiel W. Proteolytic activation of human factors IX and X by recombinant human factor VIIa: effects of calcium, phospholipids, and tissue factor. Biochemistry. 1990;29(40):9418-9425. [DOI] [PubMed] [Google Scholar]
- 2.Monković DD, Tracy PB. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265(28):17132-17140. [PubMed] [Google Scholar]
- 3.Schuijt TJ, Bakhtiari K, Daffre S, et al. . Factor Xa activation of factor V is of paramount importance in initiating the coagulation system: lessons from a tick salivary protein. Circulation. 2013;128(3):254-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton PG, Jackson CM, Hanahan DJ. Relationship between factor V and activated factor X in the generation of prothrombinase. Nature. 1967;214(5091):923-924. [DOI] [PubMed] [Google Scholar]
- 5.Miletich JP, Kane WH, Hofmann SL, Stanford N, Majerus PW. Deficiency of factor Xa-factor Va binding sites on the platelets of a patient with a bleeding disorder. Blood. 1979;54(5):1015-1022. [PubMed] [Google Scholar]
- 6.Nesheim ME, Taswell JB, Mann KG. The contribution of bovine factor V and factor Va to the activity of prothrombinase. J Biol Chem. 1979;254(21):10952-10962. [PubMed] [Google Scholar]
- 7.Baugh RJ, Broze GJ Jr, Krishnaswamy S. Regulation of extrinsic pathway factor Xa formation by tissue factor pathway inhibitor. J Biol Chem. 1998;273(8):4378-4386. [DOI] [PubMed] [Google Scholar]
- 8.Girard TJ, Warren LA, Novotny WF, et al. . Functional significance of the Kunitz-type inhibitory domains of lipoprotein-associated coagulation inhibitor. Nature. 1989;338(6215):518-520. [DOI] [PubMed] [Google Scholar]
- 9.Maroney SA, Ellery PE, Wood JP, Ferrel JP, Martinez ND, Mast AE. Comparison of the inhibitory activities of human tissue factor pathway inhibitor (TFPI)α and TFPIβ. J Thromb Haemost. 2013;11(5):911-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor-alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci USA. 2013;110(44):17838-17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wun TC, Kretzmer KK, Girard TJ, Miletich JP, Broze GJ Jr. Cloning and characterization of a cDNA coding for the lipoprotein-associated coagulation inhibitor shows that it consists of three tandem Kunitz-type inhibitory domains. J Biol Chem. 1988;263(13):6001-6004. [PubMed] [Google Scholar]
- 12.Ndonwi M, Girard TJ, Broze GJ Jr. The C-terminus of tissue factor pathway inhibitor α is required for its interaction with factors V and Va. J Thromb Haemost. 2012;10(9):1944-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monkovic DD, Tracy PB. Activation of human factor V by factor Xa and thrombin. Biochemistry. 1990;29(5):1118-1128. [DOI] [PubMed] [Google Scholar]
- 14.Franssen J, Salemink I, Willems GM, Wun TC, Hemker HC, Lindhout T. Prothrombinase is protected from inactivation by tissue factor pathway inhibitor: competition between prothrombin and inhibitor. Biochem J. 1997;323(pt 1):33-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mast AE, Broze GJ Jr. Physiological concentrations of tissue factor pathway inhibitor do not inhibit prothrombinase. Blood. 1996;87(5):1845-1850. [PubMed] [Google Scholar]
- 16.Walker FJ, Sexton PW, Esmon CT. The inhibition of blood coagulation by activated protein C through the selective inactivation of activated factor V. Biochim Biophys Acta. 1979;571(2):333-342. [DOI] [PubMed] [Google Scholar]
- 17.Kalafatis M, Rand MD, Mann KG. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J Biol Chem. 1994;269(50):31869-31880. [PubMed] [Google Scholar]
- 18.Svensson PJ, Dahlbäck B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med. 1994;330(8):517-522. [DOI] [PubMed] [Google Scholar]
- 19.Zöller B, Dahlbäck B. Linkage between inherited resistance to activated protein C and factor V gene mutation in venous thrombosis. Lancet. 1994;343(8912):1536-1538. [DOI] [PubMed] [Google Scholar]
- 20.Bertina RM, Koeleman BP, Koster T, et al. . Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369(6475):64-67. [DOI] [PubMed] [Google Scholar]
- 21.Kalafatis M, Bertina RM, Rand MD, Mann KG. Characterization of the molecular defect in factor VR506Q. J Biol Chem. 1995;270(8):4053-4057. [DOI] [PubMed] [Google Scholar]
- 22.Kujovich JL. Factor V Leiden thrombophilia. Genet Med. 2011;13(1):1-16. [DOI] [PubMed] [Google Scholar]
- 23.van ’t Veer C, Golden NJ, Kalafatis M, Mann KG. Inhibitory mechanism of the protein C pathway on tissue factor-induced thrombin generation. Synergistic effect in combination with tissue factor pathway inhibitor. J Biol Chem. 1997;272(12):7983-7994. [DOI] [PubMed] [Google Scholar]
- 24.van ’t Veer C, Kalafatis M, Bertina RM, Simioni P, Mann KG. Increased tissue factor-initiated prothrombin activation as a result of the Arg506--> Gln mutation in factor VLEIDEN. J Biol Chem. 1997;272(33):20721-20729. [DOI] [PubMed] [Google Scholar]
- 25.Eitzman DT, Westrick RJ, Bi X, et al. . Lethal perinatal thrombosis in mice resulting from the interaction of tissue factor pathway inhibitor deficiency and factor V Leiden. Circulation. 2002;105(18):2139-2142. [DOI] [PubMed] [Google Scholar]
- 26.Harris GM, Stendt CL, Vollenhoven BJ, Gan TE, Tipping PG. Decreased plasma tissue factor pathway inhibitor in women taking combined oral contraceptives. Am J Hematol. 1999;60(3):175-180. [DOI] [PubMed] [Google Scholar]
- 27.Bloemenkamp KW, Rosendaal FR, Helmerhorst FM, Büller HR, Vandenbroucke JP. Enhancement by factor V Leiden mutation of risk of deep-vein thrombosis associated with oral contraceptives containing a third-generation progestagen. Lancet. 1995;346(8990):1593-1596. [DOI] [PubMed] [Google Scholar]
- 28.Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood. 1995;85(6):1504-1508. [PubMed] [Google Scholar]
- 29.Vandenbroucke JP, Koster T, Briët E, Reitsma PH, Bertina RM, Rosendaal FR. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet. 1994;344(8935):1453-1457. [DOI] [PubMed] [Google Scholar]
- 30.Lockett JM, Mast AE. Contribution of regions distal to glycine-160 to the anticoagulant activity of tissue factor pathway inhibitor. Biochemistry. 2002;41(15):4989-4997. [DOI] [PubMed] [Google Scholar]
- 31.Morrissey JH. Morrissey laboratory protocol for preparing phospholipid vesicles (SUV) by sonication. 2001. Available at: http://tf7.org/suv.pdf. Accessed May 2012.
- 32.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood. 2009;114(23):4886-4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laudano AP, Doolittle RF. Synthetic peptide derivatives that bind to fibrinogen and prevent the polymerization of fibrin monomers. Proc Natl Acad Sci USA. 1978;75(7):3085-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liaw PC, Ferrell G, Esmon CT. A monoclonal antibody against activated protein C allows rapid detection of activated protein C in plasma and reveals a calcium ion dependent epitope involved in factor Va inactivation. J Thromb Haemost. 2003;1(4):662-670. [DOI] [PubMed] [Google Scholar]
- 35.Maroney SA, Haberichter SL, Friese P, et al. . Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007;109(5):1931-1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apheresis Sci. 2008;38(1):15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bos MH, Camire RM. A bipartite autoinhibitory region within the B-domain suppresses function in factor V. J Biol Chem. 2012;287(31):26342-26351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu H, Toso R, Camire RM. Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J Biol Chem. 2007;282(20):15033-15039. [DOI] [PubMed] [Google Scholar]
- 39.Zöller B, Svensson PJ, He X, Dahlbäck B. Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest. 1994;94(6):2521-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahlbäck B, Hildebrand B. Inherited resistance to activated protein C is corrected by anticoagulant cofactor activity found to be a property of factor V. Proc Natl Acad Sci USA. 1994;91(4):1396-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen L, Dahlbäck B. Factor V and protein S as synergistic cofactors to activated protein C in degradation of factor VIIIa. J Biol Chem. 1994;269(29):18735-18738. [PubMed] [Google Scholar]
- 42.Thorelli E, Kaufman RJ, Dahlbäck B. Cleavage of factor V at Arg 506 by activated protein C and the expression of anticoagulant activity of factor V. Blood. 1999;93(8):2552-2558. [PubMed] [Google Scholar]
- 43.Cramer TJ, Griffin JH, Gale AJ. Factor V is an anticoagulant cofactor for activated protein C during inactivation of factor Va. Pathophysiol Haemost Thromb. 2010;37(1):17-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dahm A, Van Hylckama Vlieg A, Bendz B, Rosendaal F, Bertina RM, Sandset PM. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood. 2003;101(11):4387-4392. [DOI] [PubMed] [Google Scholar]
- 45.Cunha ML, Bakhtiari K, Peter J, Marquart JA, Meijers JC, Middeldorp S. A novel mutation in the F5 gene (factor V Amsterdam) associated with bleeding independent of factor V procoagulant function. Blood. 2015;125(11):1822-1825. [DOI] [PubMed] [Google Scholar]
- 46.Duckers C, Simioni P, Spiezia L, et al. . Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood. 2008;112(9):3615-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vincent LM, Tran S, Livaja R, Bensend TA, Milewicz DM, Dahlbäck B. Coagulation factor V(A2440G) causes east Texas bleeding disorder via TFPIα. J Clin Invest. 2013;123(9):3777-3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Høibraaten E, Qvigstad E, Andersen TO, Mowinckel MC, Sandset PM. The effects of hormone replacement therapy (HRT) on hemostatic variables in women with previous venous thromboembolism--results from a randomized, double-blind, clinical trial. Thromb Haemost. 2001;85(5):775-781. [PubMed] [Google Scholar]
- 49.Novotny WF, Girard TJ, Miletich JP, Broze GJ Jr. Platelets secrete a coagulation inhibitor functionally and antigenically similar to the lipoprotein associated coagulation inhibitor. Blood. 1988;72(6):2020-2025. [PubMed] [Google Scholar]
- 50.Wood JP, Baumann Kreuziger LM, Desai UR, Mast AE. Blocking inhibition of prothrombinase by tissue factor pathway inhibitor alpha: a procoagulant property of heparins. Br J Haematol. 2016;175(1):123-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123(19):2934-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bunce MW, Bos MH, Krishnaswamy S, Camire RM. Restoring the procofactor state of factor Va-like variants by complementation with B-domain peptides. J Biol Chem. 2013;288(42):30151-30160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.