

Abstract
Aortopathies pose a significant healthcare burden due to excess early mortality, increasing incidence, and underdiagnosis. Understanding the underlying genetic causes, early diagnosis, timely surveillance, prophylactic repair, and family screening are keys to addressing these diseases. Next-generation sequencing continues to expand our understanding of the genetic causes of heritable aortopathies, rapidly clarifying their underlying molecular pathophysiology and suggesting new potential therapeutic targets. This review will summarize the pathogenetic mechanisms and management of heritable genetic aortopathies with attention to specific forms of both syndromic and nonsyndromic disorders, including Marfan syndrome, Loeys-Dietz syndrome, vascular Ehlers-Danlos syndrome, and familial thoracic aortic aneurysm and dissection.
Keywords: Thoracic aortic aneurysm, genetic aortopathy, Marfan syndrome, Loeys-Dietz syndrome, vascular Ehlers-Danlos syndrome, familial thoracic aortic aneurysm and dissection
Introduction
Aortic diseases, or aortopathies, include thoracic abdominal aortic aneurysm (TAA) and abdominal aortic aneurysm (AAA). They are the 18th most common cause of death, responsible for 1% to 2% of all deaths in the industrialized world.1,2 An aneurysm is a localized arterial dilation to a diameter 50% above normal.3 By the Laplace law, wall tension equates to lumen radius multiplied by the pressure such that arterial dilation begets further dilation due to a progressive rise in wall tension.3 As such, the natural history of aneurysmal dilation is progressive expansion, occasionally culminating in catastrophic consequences such as dissection or rupture. Prior to a potentially lethal event, only 5% of the patients are forewarned by symptoms, whereas death is the first symptom for the remaining majority.1 Aortic aneurysms convey a significant mortality burden that is likely underestimated due to their silent nature and increasing incidence.1 The reported increasing incidence is likely from a combination of the major growth in diagnostic imaging as well as an aging population.4,5 The largely silent, unpredictable, and deadly features of aneurysmal disease all necessitate a better understanding by clinicians of the relevant risk factors, pathophysiology, screening modalities, and available treatments.
Aortic aneurysms include both TAA and AAA. Abdominal aortic aneurysms are common and are typically degenerative disorders associated with traditional atherosclerotic risk factors such as advanced age, cigarette smoking, hypertension, and hypercholesterolemia.2,6 These aneurysms likely arise from complicated interactions of multiple predisposing genes and environmental risk factors.2 In contrast, thoracic aortic aneurysms occur in all age groups and are more likely to be associated with a genetic background, presenting as either part of a syndromic disorder or an isolated aberration.2,7 In addition, TAAs may occur as a sporadic phenomenon or as a familial disorder; the latter may follow classic Mendelian inheritance or nonclassic inheritance of a complex trait.2,8 Elefteriades et al1 classified aortic aneurysms by their anatomic relationship to the ligamentum arteriosum. Aneurysms proximal to the ligament are more likely to be nonarteriosclerotic, whereas those distal to the ligamentum are primarily of arteriosclerotic origin. Familial and some syndromic TAAs often have faster growth rates than sporadic TAAs and therefore may present earlier.5 As such, thoracic aortic aneurysm dissection (TAAD) is an important cause of premature death in young adults. In fact, Puranik et al9 found that aortic dissection caused 5.4% of sudden cardiac deaths in an autopsy series of 427 individuals with mean age of 26.8 years.
Genetic aortopathies are an underappreciated group of disorders which pose a significant healthcare burden. The goal of this review is to familiarize the clinician with some of the key inherited TAA entities as well as the current recommended guidelines for surveillance and management. Areas requiring further investigation are also discussed.
Genetic Thoracic Aortic Aneurysms
Thoracic aortic aneurysms and dissections, respectively, have a strong genetic basis. Examples of syndromic TAA disorders include Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS), and vascular Ehlers-Danlos syndrome (vEDS). Familial TAAD (FTAAD) denotes a group of nonsyndromic disorders which generally present with isolated TAAs, without associated characteristic systemic features. Advances in next-generation sequencing are rapidly uncovering novel genes and/or loci associated with hereditary TAAs.7 Most genetic causes of heritable TAA disorders are inherited as monogenic defects with an autosomal dominant pattern of inheritance with high penetrance.7 Altogether, genetic triggers account for about 20% of thoracic aortic disease; however, this is likely an underestimate due to underdiagnosis of silent TAAs in family members of probands and infrequent use of genetic testing in the clinical setting.1,8,10 This review is not a comprehensive catalogue of all known genetic TAA disorders but rather offers an overview of 4 important entities: MFS, LDS, vEDS, and FTAAD (Table 1). Awareness of the more common genetic TAA disorders is paramount due to variable natural progression, different surveillance and management, and excessive early mortality.
Table 1.
Thoracic aortic aneurysm disorders.

Genetically mediated thoracic aortic disorders share histopathologic features of medial degeneration, characterized by destructive matrix remodeling with elastin fragmentation, impaired proliferation of vascular smooth muscle cells, and proteoglycan deposition.2,3 There is increased matrix metalloproteinase (MMP)-2 and MMP-9 activity which lyse elastic fibers and break down extracellular matrix (ECM).3 Because genes mutated in MFS and vEDS both encode ECM proteins (fibrillin-1 and type III procollagen, respectively), the original hypothesis was that altered ECM contents led to structural weakness in the aortic wall, thereby leading to progressive aneurysmal dilation.2 However, this purely mechanical hypothesis was later found to be too simplistic. An important hint to the contrary came from studying fibrillin-1–deficient mice which were noted to have abnormal septation of distal alveoli; these mice displayed increased levels of free transforming growth factor β (TGF-β) and increased activation of its downstream effectors (pSMAD2/3).20 Transforming growth factor β inhibition in this MFS mouse model restored distal alveolar septation, implicating increased TGF-β signaling as a key abnormality in MFS.20 Fibrillin proteins bear significant homology to latent TGF-β–binding proteins which sequester TGF-β, thereby downregulating the bioavailability and activity of TGF-β; fibrillin-1 deficiency therefore leads to increased TGF-β levels and its downstream effectors, including MMPs (Figure 1).2
Figure 1.

TGF-β regulation and signaling. Increased TGF-β signaling is associated with TAAs. In Marfan syndrome, mutations in fibrillin-1 result in increased TGF-β bioavailability and therefore increased activity of both the canonical (blue) and noncanonical (green) TGF-β signaling pathways, with overexpression of target genes including matrix metalloproteinases. Loeys-Dietz syndrome results from loss of function mutations in the TGF-β ligands, receptors, or R-Smad effectors which paradoxically result in increased TGF-β signaling. TAAs indicate thoracic aortic aneurysms; TGF-β, transforming growth factor β. Reproduced with permission from Verstraeten et al.21
Transforming growth factor β is a regulatory cytokine expressed by vascular smooth muscle that regulates MMP activity and is involved in numerous pathways related to cell growth, differentiation, and oncogenesis.3,7 There is mounting evidence that dysregulation of TGF-β signaling may be a primary etiologic factor underlying numerous TAA disorders that includes the following: (1) serum TGF-β levels correlate with rate of aneurysmal growth in mouse MFS models,22 (2) serum TGF-β levels are markedly elevated in patients with acute aortic dissection,23,24 (3) in patients with MFS, TGF-β levels correlate with both size and rate of growth of aortic root aneurysms,25 (4) TGF-β levels predict aortic dissection and elective aortic root surgery,25 and (5) high TGF-β levels have been noted in other TAA disorders ranging from FTAAD to bicuspid aortic valve–associated TAA.2 Notably, TGF-β levels may serve as a prognostic and therapeutic marker in patients with TAA, although this remains theoretical.2,22,23,25
Marfan syndrome
Marfan syndrome is an autosomal dominant syndromic disorder with variable expression and pleiotropic features affecting the skeletal, ocular, and cardiovascular systems, with the latter being the major source of morbidity and mortality.21,26 Marfan syndrome affects approximately 1 in every 5000 persons and accounts for 5% of all aortic dissections, without a sex, racial, or ethnic bias. 11,27,28 French pediatrician Antoine-Bernard Marfan classically described the skeletal features of the syndrome in 1896, including arachnodactyly, pectus deformity, and disproportionately long extremities, among others.29 McKusick26 described the cardiovascular manifestations of MFS in a case series of 105 patients and their families in 1955, calling attention to the occurrence of aortic aneurysms and dissections.
Aortic dilation in MFS typically occurs at the sinuses of Valsalva and the tubular portion of the ascending aorta forming a “pear-shaped” annuloaortic ectasia (Figure 2), and these aneurysms typically grow approximately 0.1 cm/year.5,11 Without surgical intervention, the lifetime risk for aortic dissection is an alarming 50% with rare survival past the 1940s.11,28 Indeed, progressive aortic root enlargement resulting ultimately in dissection is the main cause of premature mortality in patients with MFS.27 Treatment with β-blockers and improved surgical technique has prolonged median survival from 48 years in the 1972 to 72 years in 1993.30 Nevertheless, despite adequate medical management, 90% of patients with MFS will have aortic surgery or suffer an aortic dissection in their lifetime.7
Figure 2.
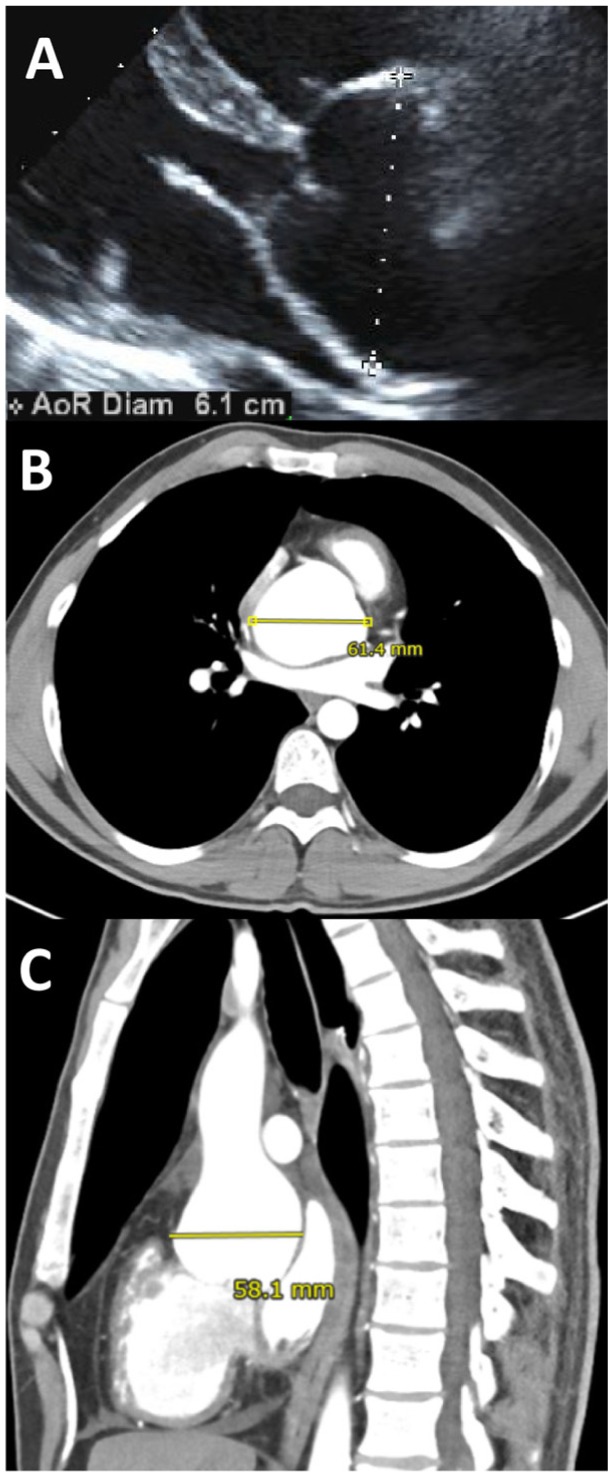
Aortic root dilation in Marfan syndrome. These images show aortic root dilation in a 27-year-old male with Marfan syndrome. Note the dilation of the sinuses of Valsalva, forming a pear-shaped appearance. Importantly, different imaging modalities, cross sections, and planes give different measurements of the same aortic aneurysm. Follow-up imaging should therefore use the same imaging modality and measurements should be made perpendicular to the axis of the aorta for reliable longitudinal comparisons.
(A) Transthoracic echocardiography in the parasternal long-axis view. (B) and (C) Contrast enhanced computed tomographic (CT) scan in the transverse (B) and sagittal (C) planes.
Despite the description of Marfan syndrome in 1896, it was not until 1991 that the causative gene for MFS was discovered to be FBN-1 encoding fibrillin-1, which is an ECM glycoprotein abundantly present in the suspensory ligament of the lens, the periosteum of bone, and the aortic media.31–33 As noted above, this mutation leads to fibrillin-1 deficiency, which then results in an excess of TGF-β (Figure 1). More than 800 distinct FBN-1 mutations have been described.34 Individual families may have their own private mutation, but family members sharing the same mutation often display a heterogeneous phenotype.11 Notably, 25% of MFS arises de novo from a sporadic mutation.21
Diagnostic criteria for MFS have undergone several revisions. Revision by Loeys et al35 in 2010 placed greater weight on the 2 cardinal manifestations of MFS: ectopia lentis and aortic root dilation. With a family history of MFS, the diagnosis can be made if the patient has at least one of the following: ectopia lentis, aortic root dilation, or a systemic score ≥7 based on systemic correlates. In the absence of family history, the diagnosis requires a combination of the cardinal features, systemic score, and FBN-1 gene mutation (Figure 3 and Table 2). Notably, FBN-1 gene mutation is neither necessary nor sufficient for the diagnosis because testing is not yet broadly available in the clinical setting and a given mutation may be either silent or result in phenotypes other than MFS.7
Figure 3.

Revised Ghent criteria for diagnosis of Marfan syndrome.35 *Caveat: without discriminating features of Shprintzen-Goldberg syndrome, Loeys-Dietz syndrome, or vascular Ehlers-Danlos syndrome (as defined elsewhere) and after TGFBR1/2, collagen biochemistry, and COL3A1 testing if indicated; other conditions/genes may emerge with time. In the absence of a family history of Marfan syndrome, ectopia lentis and fibrillin-1 mutation indicate either (a) Marfan syndrome in the presence of aortic dilation or (b) ectopia lentis syndrome in the absence of aortic dilation. See Table 2 for the systemic score. FBN1 indicates fibrillin-1.
Table 2.
Systemic score for Marfan syndrome.

Loeys-Dietz syndrome
First described in 2005, LDS is an autosomal dominant syndromic aortopathy with variable expression characterized by aggressive TAAs.36 Loeys-Dietz syndrome type 1 is associated with craniofacial defects (bifid uvula, cleft palate, orbital hypertelorism, and craniosynostosis), and LDS type 2 has vascular EDS-type features (easy bruising, joint laxity, visceral rupture, and thin, translucent skin), both caused by TGF-β receptor (TGFBR) mutations (TGFBR1 and TGFBR2).12 Loeys-Dietz syndrome types 3, 4, and 5 were also subsequently described and arise, respectively, from mutations of the TGF-β downstream effector SMAD3 gene, the TGF-β 2 ligand (TGFB2) gene, and the TGF-β 3 ligand (TGFB3) gene (Figure 1).21,37 MacCarrick et al37 proposed a revised nosology in 2014 to reduce emphasis on systemic dysmorphic features because the primary concern with all the LDS-associated mutations is the underlying vascular disease. Accordingly, they proposed that the presence of an aneurysm or dissection and any of the associated mutations is sufficient to make the diagnosis of LDS irrespective of other systemic features.37 There is also significant clinical variability between family members with the same mutation.12
The vascular-related morbidity and mortality of LDS is alarming. Thoracic aortic aneurysms in LDS can grow faster than 1.0 cm/year, 10 times faster than the average MFS TAA.5 In a series of 90 patients with LDS, Loeys et al12 found that the mean age at death was 26 years (range: 0.5–47 years), where TAADs claimed 67%, abdominal aortic dissections claimed 22%, and cerebral bleeding claimed 7%. A total of 32% had a vascular event (dissection, surgery, or death from dissection or rupture) before 19 years of age. Arterial involvement was widespread beyond the aorta, involving both the head and neck as well as abdominal arterial branches.
Loeys-Dietz syndrome and MFS likely share aberrant TGF-β signaling as part of their pathogenesis (Figure 1). As described above, fibrillin-1 deficiency in MFS results in increased TGF-β bioavailability and activity such that fibrillin-1–deficient mice have increased free TGF-β levels and downstream effectors such as pSMAD2/3.2 In LDS, despite loss-of-function mutations in TGF-β receptors, the aortic walls of patients with heterozygous mutations indicate increased TGF-β activity: they have increased collagen, nuclear phosphorylated SMAD2, and expression of connective tissue growth factor which is a TGF-β–responsive gene.36,38 This paradox is poorly understood. Therefore, both MFS and LDS share enhanced TGF-β signaling as a core abnormality at the molecular level.
There is phenotypic overlap between LDS and both MFS and vEDS. However, genetic testing to distinguish among these disorders is paramount given differences in prognosis and management. Indeed, the median survival for LDS is only 37 years, lower than with both vEDS (48 years) and treated MFS (70 years).12,30,39 Although there are no established diagnostic criteria for LDS, mutation in any of the 5 LDS genes (TFBR1, TGFBR2, SMAD3, TGFB2, and TGFB3) in combination with arterial aneurysm, dissection, or family history of LDS is sufficient to make the diagnosis.37
Vascular Ehlers-Danlos syndrome
Vascular Ehlers-Danlos syndrome, or Ehlers-Danlos syndrome type IV, is an autosomal dominant disorder characterized by risk for spontaneous intestinal, uterine, and arterial rupture as well as joint and cutaneous manifestations.39 The prevalence of vEDS is about 1 in 90 000.40 In a study of 419 patients with vEDS and affected relatives, Pepin et al39 described a median survival of 48 years with the most (79%) dying of arterial rupture and the rest mostly of either organ (uterus, left ventricle, liver, spleen) or gastrointestinal rupture. Of all arterial complications identified, approximately half involved the thoracic or abdominal arteries, and the rest were split between the head and neck, and limb arteries. Mean age at first complication was 30.6 years.40
The culprit gene (COL3A1) encodes for type III procollagen, a component of skin, vessel wall, and hollow organs.7,13 The defect results in friable aortic tissue with tears along the aorta and its branches, leading to rupture and dissection often without prerequisite aneurysm and high surgical mortality.7,39
The clinical diagnosis of vEDS, as per revised nosology of Villefranche, requires the presence of 2 of the following: (1) thin, translucent skin; (2) arterial, intestinal, or uterine rupture; (3) easy bruising; and (4) characteristic facial appearances.41 Diagnosis is confirmed by either finding of structurally abnormal type III procollagen in a culture of dermal fibroblasts or COL3A1 gene mutation.13 Due to its rarity, the diagnosis of vEDS is often not made until arterial or organ rupture.40
Familial thoracic aortic aneurysm
Familial thoracic aortic aneurysms and dissections are a heterogeneous group of nonsyndromic TAA disorders which are heritable with autosomal dominance, variable expression, and incomplete penetrance.7 Isolated TAAs are discovered in 11% to 19% of first-degree relatives of patients with TAAs, although the true rate may be higher due to undiscovered aneurysms.38 Although by definition, “isolated” TAAD occurs in the absence of other manifestations, many affected families do have identifiable extra-aortic features.7 Causative mutations in the following genes have been associated with FTAAD: smooth muscle–specific α-actin (ACTA2), vascular smooth muscle contractile protein β-MHC (MYH11), TGFBR2, myosin light chain kinase (MYLK), and a type 1 cyclic guanosine monophosphate–dependent protein kinase that controls smooth muscle relaxation (PRKG1).3,42
Mutations in ACTA2 are the most commonly identified, accounting for 10% to 14% of FTAAD.14 Patients present with TAAs, livedo reticularis, iris flocculi, a patent ductus arteriosus, and nonthoracic aneurysms.38 These patients are also at higher risk for premature coronary artery disease, ischemic stroke, and moyamoya.15 Mutations in MYH11, which encodes a major contractile protein in vascular smooth muscle, are found in FTAAD associated with patent ductus arteriosus, possibly due to impaired smooth muscle proliferation, migration, and contraction.16,38 As with LDS, FTAAD has also been described in association with TGFBR2 mutations, specifically involving Arg460 in the intracellular domain associated with paradoxically increased TGF-β activity.17,38 In addition to TAA, mutations of the TGFBR2 Arg460 residue are also associated with abdominal, cerebral, and coronary aneurysms.17 Mutations in MYLK are associated with acute aortic dissection with little or no aortic enlargement.18 A gain of function mutation of PRKG1 is associated with dissections occurring at a young age—as early at 17 years—as well as coronary aneurysm and dissection.19 Identifying other genetic links to FTAAD is a subject of ongoing research.
The paucity of data on specific FTAAD conditions limits understanding of the natural history of its various forms. Diagnosis relies on a typical family history and exclusion of known genetic syndromic disorders.
The clustering of mutations involving matrix components, TGF-β signaling, and smooth muscle contractile elements among the genetic TAA entities is beginning to clarify the pathogenetic underpinnings of thoracic aneurysmal disease, opening avenues for rational medical therapy.
Management of Thoracic Aortic Aneurysms
In caring for patients with TAAs, the virtues of early diagnosis, family screening, timely surveillance, medical management, and early elective aortic root replacement to avert a lethal catastrophe cannot be overstated. The bedrock of any clinical diagnosis remains obtaining a complete medical history and a detailed family history and performing a comprehensive physical exam. However, this bedrock is undermined by the variable expression, incomplete penetrance, phenotypic overlap, and clinically silent albeit life-threatening features of the genetic aortopathies.7,8 Only 5% of TAAs are symptomatic.1 For the remainder, the diagnosis is made either incidentally on imaging performed for other indications or following the potentially deadly complications of dissection and/or rupture. Noninvasive imaging and genetic testing are key to the diagnosis of genetic aortopathies. Once the diagnosis is made, treatment includes medical therapy with the goal of reducing dilation rate to prevent aortic catastrophe, periodic surveillance to monitor rate of dilation, and prophylactic surgical correction in high-risk persons.
Medical therapy
The goal of medical management of patients with TAAs is to retard the rate of aneurysmal expansion to mitigate the risk of a potentially catastrophic dissection or rupture. The therapeutic arsenal includes aggressive risk factor management and medication. Risk factor reduction includes the following: (1) treating hypertension to the lowest tolerated blood pressure, (2) treating dyslipidemia, (3) promoting smoking cessation, (4) avoidance of strenuous isometric exercise, (5) discouraging cocaine and other adrenergic agents, (6) stress management, and (7) close multidisciplinary monitoring during pregnancy.3,43 Figure 4 illustrates the pathogenetic rationale for currently available medical therapies.
Figure 4.

Pathogenetic rationale for medical therapies against thoracic aneurysms. Angiotensin II both promotes aneurysm formation by stimulating AT1 receptors by potentiating transforming growth factor β (TGF-β) signaling and attenuates aneurysmal dilation by stimulating AT2 receptors via extracellular signal–regulated kinase antagonism. Angiotensin receptor blockers (ARBs) preferentially inhibit the AT1 receptor, whereas angiotensin-converting enzyme inhibitors (ACEIs) decrease both AT1 and AT2 receptor activation. Increased AT1 receptor activity results in increased reactive oxygen species through the NADH/NADPH system, in turn increasing matrix metalloproteinase (MMP) levels and inflammation which result in medial degeneration and aneurysm formation. Statins inhibit the NADH/NADPH system and doxycycline inhibits MMPs. β-blockers reduce shear stress by reducing dP/dT. Reproduced with permission from Danyi et al.44
β-blocker therapy
In 1982, Boucek et al45 demonstrated that propranolol reduced dissections in a turkey model of aortic aneurysms, postulating this effect to be due to reduced hypertension, decreased pulsatile wall stress, and by directly affecting tissue elasticity. More than just blood pressure reduction, the specific reduction in change in pressure with respect to time (dP/dT) by β-blockers reduces sheer stress and likely contributes to their efficacy in TAA treatment (Figure 4).46 In 1994, Shores et al47 published an open-label, randomized trial of propranolol in patients with MFS demonstrating a significant reduction in rate of aortic dilation in the treatment group (0.023 vs 0.084 cm/year) despite a larger baseline aortic diameter (34.6 vs 30.2 cm). Ladouceur et al48 published a retrospective study in 2007 comparing 77 MFS children treated with β-blockers (atenolol in >70%, nadolol in 17%, and propranolol in 6%) with 78 untreated MFS children; the β-blocker–treated children had a statistically significant decrease in rate of dilation at the sinuses of Valsalva by 0.16 mm/year. β-blockers thus became the mainstay of medical therapy for patients with TAAs.
The use of β-blockers for patients with TAAs is not without controversy. A meta-analysis of β-blocker treatment in 802 patients with MFS from 6 studies found no difference in rates of aortic dissection, rupture, surgery, or death.49 The studies showing benefit with β-blockers are limited to patients with MFS; there is currently no evidence of benefit in the other syndromic TAAs or FTAAD. β-blockers may further reduce the elasticity of the aortic wall, possibly compounding medial degeneration already present in TAAs.1 Furthermore, lifetime β-blocker therapy in young patients with TAA is not without side effects.
Although β-blockers have become the mainstay of medical treatment for those with TAAs, their role in the prevention of TAA complications remains equivocal.
Renin-angiotensin-aldosterone inhibition
As previously discussed, increased TGF-β activity may be a key aberration underlying multiple genetic aortopathies.2 Angiotensin II receptor blockers (ARBs) inhibit TGF-β activity via selective AT1 receptor blockade without affecting the AT2 receptor (Figure 4).44 Treating fibrillin-1–deficient mice with TGF-β–neutralizing antibodies corrects the arterial and alveolar abnormalities associated with TGF-β hyperactivity.20,50 Habashi et al50 found that treatment with the ARB losartan similarly corrected these abnormalities. The group also tested fibrillin-1–deficient mice with documented aortic root aneurysms with placebo, propranolol, or losartan; in contrast with the other 2 groups, the losartan-treated mice lacked elastic fiber fragmentation, had reduced nuclear pSMAD2, and had slower aortic root growth rates indistinguishable from that of wild-type mice (Figure 5).50 Aortic root growth rate in the propranolol-treated MFS mice was slower than in the placebo-treated MFS mice but still faster than in the wild-type mice. Two years later, Brooke et al27 published a case series of 18 patients with MFS in whom an ARB was added to β-blocker therapy at the Johns Hopkins Hospital; they found that the rate of aortic root dilation decreased significantly from 3.54 ± 2.87 to 0.46 ± 0.62 mm/year after adding an ARB (irbesartan in 1 and losartan in 17 patients). Interestingly, there was no blood pressure change, indicating an alternate mechanism of benefit. This laid the groundwork for further inquiry into the role of ARBs in the medical treatment of TAAs.
Figure 5.
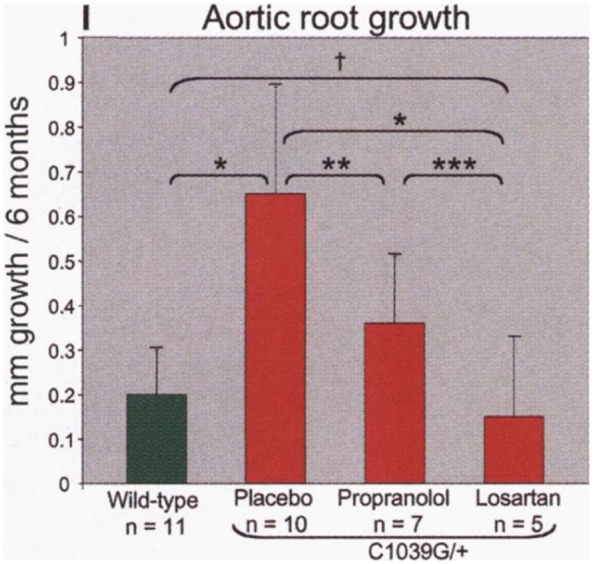
Aortic root growth rate in MFS mice (Fbn1C1039G/+) treated with placebo, propranolol, and losartan. This graph compares the average aortic root aneurysm growth rate during 6 months of treatment in (from right to left): (1) wild-type mice, (2) MFS mice treated with placebo, (3) MFS mice treated with a β-blocker (propranolol), and (4) MFS mice treated with an angiotensin receptor blocker (losartan). Growth rate is greatest for the placebo-treated MFS mice. The growth rate in losartan-treated MFS mice is indistinguishable from that of wild-type mice. Propranolol treatment results in a growth rate intermediate between placebo treatment and the wild-type state. Reproduced with permission from Habashi et al.50
Further suggesting the efficacy of ARBs was the COMPARE study, a multicenter, open-label, randomized controlled trial which randomized 233 patients with MFS to either losartan (n = 116) or no additional treatment (n = 117).51 Over 3 years of follow-up, the rate of aortic root dilation was significantly lower in the losartan arm than in the control arm (0.77 ± 1.36 vs 1.35 ± 1.55 mm). Again, there was no apparent correlation between the degree of blood pressure lowering and aortic root dilation rate. However, despite mechanistic plausibility and early positive clinical studies, the benefit of ARBs in TAAs has been questioned in subsequent studies. The Pediatric Heart Network published a randomized controlled trial of 608 patients with MFS who received either atenolol or losartan.52 Over a 3-year period, there was no significant difference in the rate of aortic root change. Notably, both groups experienced a decrease in aortic root size, and younger age was associated with a greater decrease in aortic root size. The lack of a placebo group was a significant shortcoming. The Marfan Sartan study, a double-blind, randomized controlled trial of 303 patients with MFS given either losartan or placebo, further weakened the case for ARBs.53 Over a median follow-up of 3.5 years, there was no difference in the change in aortic root diameter. The authors concluded that losartan should not be considered standard therapy for patients with MFS.
Angiotensin-converting enzyme inhibitors (ACEIs) block the conversion of angiotensin I to angiotensin II, a peptide hormone which acts on both AT1 and AT2 receptors. Although ARBs specifically block the AT1 receptor, ACEIs reduce both AT1 and AT2 receptor signaling (Figure 4). AT1 receptor stimulation promotes aneurysmal dilation by potentiating TGF-β signaling, whereas AT2 receptor stimulation attenuates dilation by inhibiting TGF-β–mediated activation of extracellular signal–regulated kinase.54 Because of this, ACEIs, which decrease both AT1 and AT2 receptor signaling, would logically be of lesser benefit compared with ARBs, which selectively inhibit AT1 receptors. Indeed, the ACEI enalapril was less effective in attenuating aortic root growth than the ARB losartan in a mouse model of MFS.54
As with β-blockers, the benefit of ARBs and ACEIs in attenuating progression of TAAs and preventing complications remains unproven. In addition, the weight of evidence is specific to MFS and cannot be extrapolated to treatment of TAAs of other causes.
Other pharmacologic interventions
Other promising, though unproven, medical options for TAAs include statin drugs, doxycycline, immunosuppressants (rapamycin), and anti-inflammatory agents (COX inhibitors).3,7,38 In a retrospective review of 1560 patients with TAAs, those receiving statin therapy (n = 369, 24%) had lower rates of adverse events (death, dissection, or rupture) and of requiring surgery for those with ascending, arch, and descending TAAs; no benefit was seen in those with root TAAs.55 Doxycycline inhibits MMPs which contribute to aortic aneurysm by lysing elastic fibers and breaking down the ECM (Figure 4).3,38 In a mouse model of MFS, doxycycline treatment lowered MMP-2 and MMP-9 levels, reduced elastic fiber degradation, and prolonged life (132 vs 79 days).56 Another study compared the effects of doxycycline versus atenolol in MFS mice; in contrast to the atenolol-treated mice, the doxycycline-treated mice displayed decreased MMP-2 and MMP-9 activation, decreased TGF-β expression, and normalized aortic wall stiffness.57 Most importantly, the doxycycline-treated mice did not develop TAAs, whereas the atenolol-treated mice did.
The past decade has seen incredible advances in our understanding of the molecular mechanisms underlying thoracic aortic aneurysms. This understanding has provided the theoretical grounds for exciting new medical interventions. However, clinical data are either lacking or conflicting. High-quality trials are necessary to clarify the role of medical therapy in managing thoracic aortic aneurysms.
Surveillance and Timing of Repair
Individuals with known TAAs should undergo periodic surveillance of aneurysm diameter with noninvasive imaging. Prophylactic surgery is recommended once a critical diameter is reached to avert emergent thoracic aortic syndromes given the high mortality in the acute setting. In a series of 675 patients with MFS who underwent aortic root replacement, 30-day mortality rate was 1.5% for elective repair versus 11.7% for emergent repair.58 In the same series, nearly half of the aortic dissections occurred with an aortic diameter of 6.5 cm or less, prompting the authors to recommend prophylactic repair when the diameter is “well below” this size. The excess mortality of realized acute aortic syndromes compared with elective repair underscores the critical role of active surveillance with prophylactic surgery.
Elefteriades et al identified “hinge points” of 6 cm for ascending TAAs and 7 cm for descending TAAs, above which the risk or dissection or rupture increases by 32.1% and 43% points, respectively, far above the risk of death associated with elective surgery (Figure 6).1,28,59 Prophylactic repair is therefore generally recommended once the ascending aorta reaches 5.5 cm for asymptomatic nonsyndromic TAAs or with aneurysm growth rate greater than 0.5 cm/year.3,42,43 Guidelines for surveillance and repair of TAAs differ depending on the underlying predisposition.
Figure 6.

Risk of aortic complications as a function of aortic size. The y-axis depicts the probability of aortic complication (dissection or rupture) as a function of aortic size, demonstrating the “hinge points” at which the lifetime risk jumps substantially: 6 cm for the ascending aorta (top) and 7 cm for the descending aorta (bottom). Reproduced with permission from Coady et al.59
Patients with MFS should undergo imaging on diagnosis, at 6 months, and then annually if stable.3,43 Transthoracic echocardiography should evaluate the proximal aorta including the diameter of the root at its largest dimension, the sinotubular junction, and the ascending aorta; when technical difficulty limits echocardiographic views, computed tomographic (CT) or magnetic resonance (MR) angiography may be used (Figure 2).60 The recommended threshold for surgical repair in patients with MFS is 5.0 cm, smaller than for nonsyndromic TAAs due to a greater tendency to rupture at smaller diameters.42,43,61 Repair at 4.5 cm may be appropriate in those with high-risk features including those with a family history of dissection at a diameter less than 5.0 cm, dilation rate >3 to 5 mm/year, severe aortic regurgitation, or desired pregnancy.42,43,60 Following prophylactic repair of the ascending aorta, surveillance imaging of the aortic arch and descending aorta should be performed as these are sites of later-onset aneurysms and dissections.43
Patients with LDS should undergo CT or MR angiography from head to pelvis at baseline, at 6 months, and then at 1 to 2-year intervals due to widespread vascular involvement beyond the aorta.37,43 Although aortic dissection risk increases with MFS at or above 5.0 cm aortic root dimension, dissections have been reported in patients with LDS at 3.9 to 4.0 cm.12,37 In addition, vascular surgical mortality in LDS is only 1.7% versus about 45% in vEDS.12,39 Prophylactic surgical aortic root repair in patients with LDS with TAA is therefore recommended once the maximal root diameter reaches 4.2 cm internal diameter by transesophageal echocardiography or 4.4 to 4.6 cm external diameter by CT or MR angiography.37,43
In patients with vEDS, imaging surveillance and prophylactic repair of vascular aneurysms are less well established due to tendency to rupture without prerequisite aneurysm formation and high surgical mortality.3,39,43 Indeed, Pepin et al described a surgical mortality of just more than 40%, attributed to tissue fragility, poor wound healing, excess bleeding, fistulae formation, and adhesions seen in these patients.3,39 Surgery is therefore typically reserved for life-threatening vascular complications.13 In practice, a higher threshold for pursuing surgery is exercised along with the least traumatic approach when surgery is performed; this is in contrast to patients with LDS in whom the role of early prophylactic surgery is well established.13,43
There are no formal recommendations specific to the management of FTAAD due to significant clinical variability between and within its various genetic types and lack of sufficient data and experience regarding natural history to guide management. In practice, management involves baseline and surveillance imaging guided by the specific family history of vascular events.42
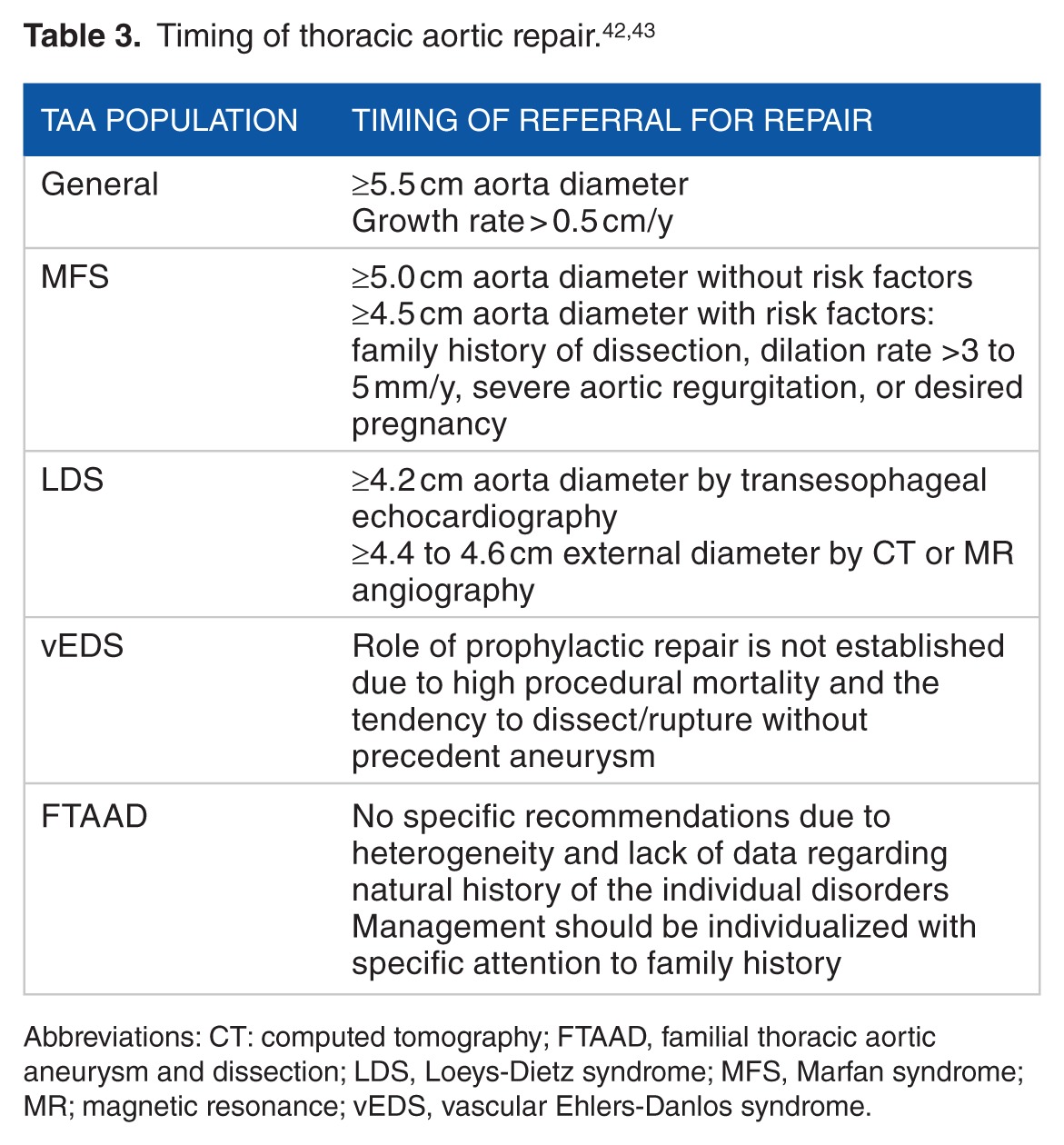
Table 3 summarizes the recommended timing of thoracic aortic aneurysm repair for the different populations discussed above.
Table 3.
Conclusions
The understanding of aortic aneurysmal disease has grown exponentially. Identification of specific genes associated with genetic aortopathies has delineated molecular mechanisms underlying aneurysm formation and allowed for investigations into rational targeted therapy. Clarification of the natural history of the various TAA entities has led to specific recommendations regarding surveillance and timing of prophylactic repair, significantly reducing mortality from catastrophic aortic complications. Despite the rapid growth of understanding of genetic aortopathies, there yet remain significant uncertainty and with it exciting avenues for future research.
The advent of next-generation sequencing and identification of new genes will undoubtedly enhance our understanding of pathogenetic mechanisms leading to aneurysm formation, thereby uncovering novel therapeutic targets.7 The conflicting data on currently available therapies will benefit from further study. Timing of prophylactic repair may soon be fine-tuned by exciting new approaches that go beyond size such as biomarkers of impending rupture or dissection, noninvasive assessments of aortic mechanical properties such as distensibility and wall tension, and positron emission tomography to assess the activity of an aneurysm.1 Management may become more personalized and specific to an individual’s predisposing mutation.
Clearly, there remain many unanswered questions in the management of genetic aortopathies. Sir William Osler’s observation that “There is no disease more conducive to clinical humility than aneurysm of the aorta” is as true today as it was more than 100 years ago.1,62
Footnotes
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 631 words, excluding any confidential comments to the academic editor.
FUNDING: The author(s) received no financial support for the research, authorship, and/or publication of this article.
DECLARATION OF CONFLICTING INTERESTS: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AG wrote the first draft of the manuscript. ARK contributed to the writing of the manuscript. MJC, JRR, and AM made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section.
REFERENCES
- 1.Elefteriades JA, Farkas EA. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J Am Coll Cardiol. 2010;55:841–857. doi: 10.1016/j.jacc.2009.08.084. [DOI] [PubMed] [Google Scholar]
- 2.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature. 2011;473:308–316. doi: 10.1038/nature10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2014;64:1725–1739. doi: 10.1016/j.jacc.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 4.Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation. 2006;114:2611–2618. doi: 10.1161/CIRCULATIONAHA.106.630400. [DOI] [PubMed] [Google Scholar]
- 5.Kuzmik GA, Sang AX, Elefteriades JA. Natural history of thoracic aortic aneurysms. J Vasc Surg. 2012;56:565–571. doi: 10.1016/j.jvs.2012.04.053. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz-Rixen T, Keese M, Hakimi M, et al. Ruptured abdominal aortic aneurysm-epidemiology, predisposing factors, and biology. Langenbecks Arch Surg. 2016;401:275–288. doi: 10.1007/s00423-016-1401-8. [DOI] [PubMed] [Google Scholar]
- 7.Rea G, Stewart FJ. Genetic biomarkers in aortopathy. Biomark Med. 2013;7:547–563. doi: 10.2217/bmm.13.74. [DOI] [PubMed] [Google Scholar]
- 8.Zarate YA, Sellars E, Lepard T, Tang X, Collins RT. Aortic dilation, genetic testing, and associated diagnoses. Genet Med. 2016;18:356–363. doi: 10.1038/gim.2015.88. [DOI] [PubMed] [Google Scholar]
- 9.Puranik R, Chow CK, Duflou JA, Kilborn MJ, McGuire MA. Sudden death in the young. Heart Rhythm. 2005;2:1277–1282. doi: 10.1016/j.hrthm.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Garg V, Ouzounian M, Peterson MD. Advances in aortic disease management: a year in review. Curr Opin Cardiol. 2016;31:127–131. doi: 10.1097/HCO.0000000000000267. [DOI] [PubMed] [Google Scholar]
- 11.Paterick TE, Humphries JA, Ammar KA, et al. Aortopathies: etiologies, genetics, differential diagnosis, prognosis and management. Am J Med. 2013;126:670–678. doi: 10.1016/j.amjmed.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 12.Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 13.Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005;42:98–106. doi: 10.1016/j.jvs.2005.03.053. [DOI] [PubMed] [Google Scholar]
- 14.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 15.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 17.Pannu H, Fadulu VT, Chang J, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Guo DC, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. doi: 10.1016/j.ajhg.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo DC, Regalado E, Casteel DE, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93:398–404. doi: 10.1016/j.ajhg.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 21.Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan syndrome and related disorders: 25 years of gene discovery. Hum Mutat. 2016;37:524–531. doi: 10.1002/humu.22977. [DOI] [PubMed] [Google Scholar]
- 22.Matt P, Schoenhoff F, Habashi J, et al. Circulating transforming growth factor-beta in Marfan syndrome. Circulation. 2009;120:526–532. doi: 10.1161/CIRCULATIONAHA.108.841981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki T, Bossone E, Sawaki D, et al. Biomarkers of aortic diseases. Am Heart J. 2013;165:15–25. doi: 10.1016/j.ahj.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki T, Trimarchi S, Sawaki D, et al. Circulating transforming growth factor-beta levels in acute aortic dissection. J Am Coll Cardiol. 2011;58:775. doi: 10.1016/j.jacc.2010.01.079. [DOI] [PubMed] [Google Scholar]
- 25.Franken R, den Hartog AW, de Waard V, et al. Circulating transforming growth factor-β as a prognostic biomarker in Marfan syndrome. Int J Cardiol. 2013;168:2441–2446. doi: 10.1016/j.ijcard.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 26.McKusick VA. The cardiovascular aspects of Marfan’s syndrome: a heritable disorder of connective tissue. Circulation. 1955;11:321–342. doi: 10.1161/01.cir.11.3.321. [DOI] [PubMed] [Google Scholar]
- 27.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elefteriades JA. Thoracic aortic aneurysm: reading the enemy’s playbook. World J Surg. 2008;32:366–374. doi: 10.1007/s00268-007-9398-3. [DOI] [PubMed] [Google Scholar]
- 29.Marfan AB. Un cas de deformation congenitale des quatre membres, plus prononcee aux extremites, caracterisee par l’allongement des os avec un certain degre d’amincissement. Bull Mem Soc Med Hop Paris. 1896:220–226. [Google Scholar]
- 30.Silverman DI, Burton KJ, Gray J, et al. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995;75:157–160. doi: 10.1016/s0002-9149(00)80066-1. [DOI] [PubMed] [Google Scholar]
- 31.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 32.Dietz HC, Pyeritz RE, Hall BD, et al. The Marfan syndrome locus: confirmation of assignment to chromosome 15 and identification of tightly linked markers at 15q15-q21.3. Genomics. 1991;9:355–361. doi: 10.1016/0888-7543(91)90264-f. [DOI] [PubMed] [Google Scholar]
- 33.McKusick VA. The defect in Marfan syndrome. Nature. 1991;352:279–281. doi: 10.1038/352279a0. [DOI] [PubMed] [Google Scholar]
- 34.Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 36.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 37.MacCarrick G, Black JH, Bowdin S, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16:576–587. doi: 10.1038/gim.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6:771–786. doi: 10.1038/nrcardio.2009.191. [DOI] [PubMed] [Google Scholar]
- 39.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi: 10.1056/NEJM200003093421001. [DOI] [PubMed] [Google Scholar]
- 40.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV) Genet Med. 2014;16:881–888. doi: 10.1038/gim.2014.72. [DOI] [PubMed] [Google Scholar]
- 41.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997 Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Erbel R, Aboyans V, Boileau C, et al. ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC) Eur Heart J. 2014;2014;35:2873–2926. doi: 10.1093/eurheartj/ehu281. [DOI] [PubMed] [Google Scholar]
- 43.Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. J Am Coll Cardiol. 2010;55:e27–e129. doi: 10.1016/j.jacc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Danyi P, Elefteriades JA, Jovin IS. Medical therapy of thoracic aortic aneurysms: are we there yet? Circulation. 2011;124:1469–1476. doi: 10.1161/CIRCULATIONAHA.110.006486. [DOI] [PubMed] [Google Scholar]
- 45.Boucek RJ, Gunja-Smith Z, Noble L, Simpson CF. Modulation by propranolol of the lysyl cross-links in aortic elastin and collagen of the aneurysm-prone turkey. Biochem Pharmacol. 1983;32:275–280. doi: 10.1016/0006-2952(83)90555-5. [DOI] [PubMed] [Google Scholar]
- 46.Halpern BL, Char F, Murdoch JL, Horton WB, McKusick VA. A prospectus on the prevention of aortic rupture in the Marfan syndrome with data on survivorship without treatment. Johns Hopkins Med J. 1971;129:123–129. [PubMed] [Google Scholar]
- 47.Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330:1335–1341. doi: 10.1056/NEJM199405123301902. [DOI] [PubMed] [Google Scholar]
- 48.Ladouceur M, Fermanian C, Lupoglazoff JM, et al. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome. Am J Cardiol. 2007;99:406–409. doi: 10.1016/j.amjcard.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 49.Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome: a meta-analysis. Int J Cardiol. 2007;114:303–308. doi: 10.1016/j.ijcard.2005.11.116. [DOI] [PubMed] [Google Scholar]
- 50.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34:3491–3500. doi: 10.1093/eurheartj/eht334. [DOI] [PubMed] [Google Scholar]
- 52.Lacro RV, Dietz HC, Sleeper LA, et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371:2061–2071. doi: 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milleron O, Arnoult F, Ropers J, et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2015;36:2160–2166. doi: 10.1093/eurheartj/ehv151. [DOI] [PubMed] [Google Scholar]
- 54.Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stein LH, Berger J, Tranquilli M, Elefteraides JA. Effect of statin drugs on thoracic aortic aneurysms. Am J Cardiol. 2013;112:1240–1245. doi: 10.1016/j.amjcard.2013.05.081. [DOI] [PubMed] [Google Scholar]
- 56.Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;47:166–172. doi: 10.1016/j.jvs.2007.09.016. discussion 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res. 2008;102:e73–e85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 58.Gott VL, Greene PS, Alejo DE, et al. Replacement of the aortic root in patients with Marfan’s syndrome. N Engl J Med. 1999;340:1307–1313. doi: 10.1056/NEJM199904293401702. [DOI] [PubMed] [Google Scholar]
- 59.Coady MA, Rizzo JA, Hammond GL, et al. What is the appropriate size criterion for resection of thoracic aortic aneurysms? J Thorac Cardiovasc Surg. 1997;113:476–491. doi: 10.1016/S0022-5223(97)70360-X. discussion 489–491. [DOI] [PubMed] [Google Scholar]
- 60.Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366:1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jondeau G, Detaint D, Tubach F, et al. Aortic event rate in the Marfan population: a cohort study. Circulation. 2012;125:226–232. doi: 10.1161/CIRCULATIONAHA.111.054676. [DOI] [PubMed] [Google Scholar]
- 62.Osler W. Notes on aneurism. JAMA. 1902;38:1483–1486. [Google Scholar]