

Summary
Autoimmune hepatitis (AIH) is characterized by overwhelming effector immune responses associated with defective regulatory T cells (Tregs). Several lines of evidence indicate CD4 as the main effectors involved in autoimmune liver damage. Herein we investigate the in‐vitro effects of prednisolone, 6‐mercaptopurine, cyclosporin, tacrolimus, mycophenolic acid (MPA) and rapamycin, immunosuppressive drugs (ISDs) used in AIH treatment, on the expression of proinflammatory cytokines, co‐inhibitory molecules and ability to proliferate of CD4+CD25– cells, isolated from the peripheral blood of treatment‐naive patients with AIH. We note that in healthy subjects (HS) following polyclonal stimulation and in the absence of ISDs, the expression of interferon (IFN)‐γ, interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α by CD4 effectors peaks at 48 h and decreases at 96 h to reach baseline levels. In contrast, in AIH the expression of all these proinflammatory cytokines continue rising between 48 and 96 h. Levels of programmed cell death‐1 (PD‐1), T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3) and cytotoxic T lymphocyte antigen‐4 (CTLA‐4) increase over 96‐h culture both in HS and AIH, although with faster kinetics in the latter. Exposure to ISDs contains IFN‐γ and PD‐1 expression in AIH, where control over CD4+CD25– cell proliferation is also noted upon exposure to MPA. Treatment with tacrolimus and cyclosporin render CD4+CD25– cells more susceptible to Treg control. Collectively, our data indicate that in treatment‐naive patients with AIH, all ISDs restrain T helper type 1 (Th1) cells and modulate PD‐1 expression. Furthermore, they suggest that tacrolimus and cyclosporin may ameliorate effector cell responsiveness to Tregs.
Keywords: autoimmune hepatitis, co‐inhibitory molecules, effector T cells, immunosuppressive drugs, proinflammatory cytokines
Introduction
Autoimmune hepatitis (AIH) is a chronic liver disease caused by an aberrant immune response to hepatic autoantigens. Diagnostic features include hypergammaglobulinaemia, elevated serum aminotransferase levels, autoantibody positivity and histological evidence of interface hepatitis 1.
Mounting evidence heavily implicates CD4 T cells in both the initiation and perpetuation of the immune response in AIH. First, the majority of genes conferring susceptibility to AIH lie within the human leucocyte antigen (HLA) region, which is responsible for the presentation of antigenic peptides to CD4 T cells, eventually triggering an adaptive immune response 2. Secondly, several studies have demonstrated that the T helper type 1 (Th1)‐derived proinflammatory cytokine, interferon (IFN)‐γ, is essential for the development of AIH in murine models 3, 4, 5, 6. Moreover, we have shown previously that AIH patients display enhanced Th1 cell immunity 7 and have an increased frequency of circulating IFN‐γ+ CD4 T cells compared to healthy controls 8, 9.
In addition to the well‐established role of IFN‐γ and Th1 cell immunity in AIH liver damage, recent data have proposed that in experimental models of AIH, mice deficient in either interleukin (IL)‐17 10 or the IL‐17 receptor 11 are partially protected from hepatic injury. There is also evidence that Th17 cells are elevated in the circulation and the liver of patients with AIH 12.
Moreover, polymorphisms within the tumour necrosis factor (TNF)‐α gene confer predisposition to AIH 13, 14, 15, 16, indicating the participation of this proinflammatory cytokine in autoimmune liver damage. Further, recent data have demonstrated that cells of the Th17 lineage display heightened TNF‐α production in AIH compared to health 17.
In health, overzealous immune responses are kept in check by a number of protective mechanisms. For example, autoantigen‐specific effector T cells express high levels of co‐inhibitory receptors such as cytotoxic T lymphocyte‐associated protein‐4 (CTLA‐4) and programmed cell death‐1 (PD‐1) 18, the engagement of which modulates cell activation upon antigen exposure. Regulatory T cells provide another level of protective regulation. In AIH, regulatory T cells (Tregs) are present at lower frequencies compared to healthy controls 19, 20, 21, 22, 23, 24 and are less able to suppress CD4 T cell function 8, 21, 23, 24. Conversely, CD4 effector lymphocytes from AIH patients are also less susceptible to Treg suppression, due to reduced expression of the co‐inhibitory receptor T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3) 8.
The mainstay of treatment for AIH is immunosuppression, most commonly beginning with relatively high doses of prednisolone, which are gradually tapered as azathioprine is introduced. Prednisolone is a steroid which binds glucocorticoid receptors 25, while azathioprine, a pro‐drug of 6‐mercaptopurine (6‐MP), is a purine analogue which prevents the de‐novo synthesis of purine nucleosides 26. Additional drugs that have been used to treat AIH are: mycophenolate mofetil (MMF), a drug similar to azathioprine that inhibits the activity of inosine‐5'‐monophosphate dehydrogenase, an enzyme involved in de‐novo purine synthesis 27, 28, 29, 30, 31; cyclosporin 32, 33, 34 and tacrolimus 34, 35, that interfere with the T cell signalling molecule calcineurin, thereby inhibiting the nuclear factor of activated T cells (NFAT) and the transcription of IL‐2; and rapamycin, that inhibits IL‐2 transcription and cell‐cycle progression through the blockade of mammalian target of rapamycin (mTOR) activity 36, while enhancing the proliferation and suppressive capacity of Tregs 37.
In the present study, we examined in vitro the effects of these immunosuppressive drugs (ISDs) on the expression of the co‐inhibitory molecules CTLA‐4, TIM‐3 and PD‐1 and on the production of the proinflammatory cytokines IFN‐γ, IL‐17 and TNF‐α by CD4 effector cells in treatment‐naive patients with AIH.
Patients and methods
Patients and controls
Peripheral blood samples were obtained from six patients presenting with AIH before the initiation of immunosuppressive treatment (Table 1). All patients met the International Autoimmune Hepatitis Group (IAIHG) revised criteria for probable or definite AIH 1, with diagnosis confirmed by the presence of anti‐nuclear and/or anti‐smooth muscle antibody positivity and the histological presence of interface hepatitis. None of the six patients had bile duct changes characteristic of sclerosing cholangitis on retrograde or magnetic resonance cholangiography 38 nor had inflammatory bowel disease. Demographic and biochemical data from these patients at presentation are shown in Table 1. Eight healthy subjects (HS) served as normal controls [four females, median age 31 (range = 21–51) years]. The age difference between AIH patients and HS derived from ethical constraints in obtaining blood from healthy children. The study was approved by the ethical committee of King's College Hospital, London and written consent was obtained from each AIH patient or legal guardian and HS enrolled in the study.
Table 1.
Autoimmune hepatitis (AIH) patient demographic and biochemical data
| Patient ID | Age (years) | Sex | AST (nv ≤ 50 IU/l) | ALT (nv ≤ 50 IU/l) | GGT (nv ≤ 55 IU/l) | Total bilirubin (nv ≤20 μmol/l) | AP (nv ≤ 350 IU/l) | AP/AST ratio | IgG (nv: 6.5–17 g/l) | ANA titre | SMA titre |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 12 | F | 842 | 805 | 124 | 45 | 169 | 0·20 | 19·2 | 1/20 | n.a. |
| 2 | 15 | F | 378 | 446 | 250 | 21 | 128 | 0·34 | 26·4 | 1/80 | 1/160 |
| 3 | 51 | F | 1664 | na | 294 | 358 | 146 | 0·09 | 19·6 | 1/40 | n.a. |
| 4 | 35 | F | 955 | 1039 | 71 | 109 | 291 | 0·30 | 17·5 | n.a. | 1/1280 |
| 5 | 9 | F | 494 | 752 | 64 | 16 | 427 | 0·86 | 26·7 | 1/20 | 1/160 |
| 6 | 12 | F | 908 | 978 | 65 | 73 | 277 | 0·31 | 28·5 | 1/20 | n.a. |
| Median | 13.5 | 875 | 805 | 97.5 | 59 | 223 | 0.30 | 23 | 1/20 | 1/160 |
AST = aspartate aminotransferase; ALT = alanine aminotransferase; GGT = gamma glutamyl transferase; AP = alkaline phosphatase; IgG = mmunoglobulin G; ANA = anti‐nuclear antibody; SMA = smooth muscle antibody; nv = normal value; n.a. = not available.
Cell separation
Peripheral blood mononuclear cells (PBMCs) were isolated as described previously 19. Viability of mononuclear cells, determined by trypan blue exclusion, exceeded 98%.
CD4+CD25– effector T cells were isolated from the total PBMC population using immunomagnetic beads (Dynal Invitrogen, Oslo, Norway), as described previously 19, 20. CD4+CD25+ Tregs to be used in co‐culture experiments with CD4+CD25– effector cells were obtained by immunomagnetic bead separation, as reported previously 24. Purity of both cell populations consistently exceeded 95%.
Cell stimulation
CD4+CD25– effector T cells were seeded at 1 × 106 cells/ml in 96‐well round‐bottomed plates in RPMI‐1640 presupplemented with 2 mM L‐glutamine and 1% antibiotic–anti‐mycotic solution (both from Gibco, Invitrogen, Paisley, UK) and 10% fetal calf serum (FCS). Cells were exposed to a polyclonal stimulus consisting of anti‐CD3/anti‐CD28 T cell expander (ratio bead/cell: 1/2; Dynal Invitrogen) and recombinant human IL‐2 (30U/ml; EuroCetus; Amsterdam, the Netherlands), a protocol chosen on the basis of previous experiments 20.
To test and compare the effects of ISDs on the activation of effector T cells in vitro, the following ISDs were added alongside polyclonal stimulation at a final molarity of 10 nM; sirolimus (rapamycin, 9·1 μg/l), prednisolone (3·6 μg/l), tacrolimus (FK‐506; 8 μg/l), mycophenolic acid (MPA, the prodrug of which is MMF, 3·2 μg/l), 6‐MP (2·77 μg/l) and cyclosporin (12 μg/l) (all Sigma‐Aldrich, Dorset, UK). The same molarity was used for all drugs to allow interdrug comparisons. Corresponding concentrations and recommended therapeutic ranges for each drug are indicated in Table 2. With the exception of prednisolone and 6‐MP, for which monitoring is not performed routinely in our centre, the concentration of most other drugs was close to or fell within the therapeutic range recommended in our institution. The chosen molarity enabled the observation of inhibition of proliferation while avoiding significant cell death. Cells were cultured for 48 and 96 h before the assessment of phenotype and proliferation. Control wells, in which no ISDs were present, were also included.
Table 2.
Concentration and therapeutic range of immunosuppressive drugs (ISDs)
| Drug | Concentration* | Therapeutic range |
|---|---|---|
| Rapamycin | 9·1 μg/l | 4–12 μg/l |
| Prednisolone | 3·6 μg/l | n.a. |
| Cyclosporin | 12 μg/l | 20–100 μg/l |
| Tacrolimus | 8 μg/l | 1–12 μg/l |
| 6‐MP | 2·77 μg/l | n.a. |
| MPA | 3·2 μg/l | 1–3·5 mg/l |
*The indicated concentration is equivalent to 10 nM. MPA = mycophenolic acid; n.a. = not available.
Flow cytometry
At baseline, and after 48 and 96 h of culture, cells were washed with phosphate‐buffered saline (PBS) supplemented with 1% FCS, and stained with phycoerythrin (PE)‐conjugated anti‐TIM‐3 (BD Biosciences, Discovery Labware, Oxford, UK) and fluorescein isothiocyanate (FITC)‐conjugated anti‐PD‐1 (eBioscience, Hatfield, UK). The percentage of cells positive for intracellular CTLA‐4 was determined after fixation and permeabilization with Cytofix/Cytoperm™ (BD Biosciences) and the addition of allophycocyanin (APC)‐conjugated anti‐CTLA‐4 (BD Biosciences).
The percentage of IFN‐γ‐, IL‐17‐ and TNF‐α‐positive cells was determined after exposure to leucocyte activation cocktail with BD GolgiPlug for 4 h. Cells were then stained using PE‐conjugated anti‐IFN‐γ and anti‐TNF‐α (both BD Biosciences) and FITC‐conjugated anti‐IL‐17 (eBioscience). Flow cytometry was performed on a Becton Dickinson fluorescence activated cell sorter (FACS‐Canto™ II; Beckton Dickinson Immunocytochemistry Systems, San Jose, CA, USA). FlowJo software (Tree Star Inc., Ashland, OR, USA) was used for analysis.
Assessment of proliferation
Proliferation of CD4+CD25– cells, in the absence and presence of Tregs, was assessed using the CellTrace™ carboxy fluorescein succinimidyl ester (CFSE) cell proliferation kit (Molecular Probes, Paisley, UK). In co‐culture experiments, Tregs were added after CD4+CD25– responder cells had been already treated with ISDs for either 48 or 96 h. After exposure to ISDs, CD4+CD25– cells were washed prior to co‐culture with Tregs.
Statistical analysis
The normality of variable distribution was assessed by the Kolmogorov–Smirnov goodness‐of‐fit test; once the hypothesis of normality was accepted (P < 0·05), comparisons were performed using one‐ or two‐way analysis of variance (anova), followed by the Bonferroni post‐test. Data were analysed using GraphPad Prism® version 5 software (GraphPad, San Diego, CA, USA).
Results
Effect of ISDs on proinflammatory cytokines
Frequency of CD4+CD25– cells producing IFN‐γ, IL‐17 and TNF‐α was determined at baseline before polyclonal stimulation and at 48 and 96 h post‐stimulation in the absence or presence of ISDs. Analysis was made to compare the frequency of CD4+CD25– cells producing cytokines: (a) at different time‐points (i.e. baseline, 48 and 96 h); (b) at the same time‐point between cells untreated and treated with different ISDs; and (c) at the same time‐point in the presence of the same treatment between HS and AIH. Percentages of cytokine‐producing cells and P‐values are presented in Table 3.
Table 3.
Effect of immunosuppressive drugs (ISDs) on the proportion of interferon (IFN)‐γ+, interleukin (IL)‐17+ and tumour necrosis factor (TNF)‐α+ CD4+CD25– cells in healthy subjects (HS) and autoimmune hepatitis (AIH) patients.
| HS | AIH | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 48 h | 96 h | Pa | Pb | Pc | Baseline | 48 h | 96 h | Pd | Pe | Pf | |
| % IFN‐γ + cells | ||||||||||||
| No treatment | 5 ± 1·3 | 20·9 ± 1·8 § | 12·8 ± 2 | < 0·001 | 3·1 ± 0·7 | 12·6 ± 3·3h | 20·4 ± 6·3 | < 0·01 | < 0·05 | < 0·001 | ||
| Rapamycin | – | 15·1 ± 2·6 | 10·3 ± 2·4 | – | 10·4 ± 2·3 | 5·3 ± 2i | < 0·05 | |||||
| Prednisolone | – | 14·8 ± 1·4 | 09·2 ± 2·5 | – | 12·3 ± 1·3 | 04·9 ± 1·1i | < 0·001 | |||||
| Cyclosporin | – | 14·1 ± 3·1 | 10·6 ± 2·9 | – | 12·6 ± 1·2 | 02·9 ± 1·1ij | < 0·001 | < 0·01 | ||||
| Tacrolimus | – | 15·2 ± 3·1 | 12·8 ± 3·7 | < 0·05 | – | 08·8 ± 2·8 | 03·7 ± 1·3ij | |||||
| 6‐MP | – | 15·7 ± 3·0 | 08·9 ± 1·8 | < 0·05 | – | 09·2 ± 2·1 | 05·6 ± 0·8 | |||||
| MPA | – | 13·7 ± 1·7 | 13·4 ± 1·9 | – | 09·1 ± 2·3 | 03·6 ± 1·2ij | ||||||
| % IL‐17+ cells | ||||||||||||
| No treatment | 4·8 ± 1·6 | 13·6 ± 2 | 11·7 ± 1·8 | < 0·01 | < 0·05 | 6·8 ± 2·3 | 15·2 ± 5·5 | 19·1 ± 6·7 | ||||
| Rapamycin | – | 11·8 ± 2·5 | 10·9 ± 0·7 | < 0·05 | – | 12·3 ± 4·8 | 12·5 ± 7·3 | |||||
| Prednisolone | – | 12·5 ± 2·9 | 09·1 ± 1·7 | < 0·01 | – | 15 ± 4·8 | 12·7 ± 5·9 | |||||
| Cyclosporin | – | 12·5 ± 1·1 | 10 ± 1·4 | < 0·01 | – | 12·1 ± 3·2 | 6·4 ± 1·6 | |||||
| Tacrolimus | – | 13·5 ± 1·7 | 8·7 ± 2·2 | < 0·01 | – | 13·3 ± 3·3 | 6·1 ± 1·6 | |||||
| 6‐MP | – | 12·9 ± 1·6 | 9·5 ± 1·3 | < 0·01 | – | 12·7 ± 5·4 | 6·5 ± 0·6 | |||||
| MPA | – | 11·8 ± 2·9 | 11·4 ± 2·1 | < 0·05 | < 0·05 | – | 6·9 ± 1·9 | 8·3 ± 2·4 | ||||
| % TNF‐α+ cells | ||||||||||||
| No treatment | 4·7 ± 1·2 | 24·1 ± 2·6 ¶ | 12·6 ± 2·6 | < 0·001 | < 0·001 | 5·1 ± 0·2 | 13·3 ± 3·2 | 21·7 ± 6·5 | ||||
| Rapamycin | – | 18·2 ± 4·4 | 11·9 ± 3·7 | < 0·001 | – | 12·6 ± 3·6 | 17·1 ± 8·2 | |||||
| Prednisolone | – | 22·7 ± 4·9 | 11·6 ± 3·5 | < 0·001 | < 0·01 | – | 14·2 ± 2·0 | 15·5 ± 6·2 | ||||
| Cyclosporin | – | 8·3 ± 1·5g | 8·8 ± 1·8 | – | 9·9 ± 1·9 | 6·7 ± 1·9 | ||||||
| Tacrolimus | – | 8·8 ± 1·9g | 8·3 ± 0·9 | – | 11·1 ± 2·5 | 5·7 ± 1·9 | ||||||
| 6‐MP | – | 18·8 ± 3·4 | 13·2 ± 2·4 | < 0·001 | < 0·05 | – | 11·2 ± 2·9 | 16·7 ± 7·70 | ||||
| MPA | – | 19·1 ± 5·5 | 13·8 ± 2·9 | < 0·001 | < 0·05 | – | 12·8 ± 3·9 | 8·7 ± 3·3 | ||||
Analysis was performed using one‐ or two‐way analysis of variance (anova) test, followed by the Bonferroni post‐test. Only statistically significant differences are reported. §P‐value of anova test comparing the frequency of IFN‐γ+ cells under different treatments in HS at 48 h: < 0·05. ¶ P‐value of anova test comparing the frequency of TNF‐α+ cells under different treatments in HS at 48 h: 0·01. P‐values of post‐test comparing the frequency of cytokine‐positive cells between: Pa, baseline and 48 h in HS; Pb, 48 and 96 h in HS; Pc, baseline and 96 h in HS; Pd, baseline and 48 h in AIH; Pe, 48 and 96 h in AIH; Pf, baseline and 96 h in AIH; Pg, TNF‐α, ‘no treatment’ and treatment with cyclosporin (P < 0·05) and tacrolimus (P < 0·05) at 48 h in HS; Ph, IFN‐γ, HS and AIH patients in the absence of treatment at 48 h (P = 0·02); Pi = ‘no treatment’ and treatment with immunosuppressive drugs (ISDs) at 96 h in AIH (P < 0·001 in all cases); Pj = IFN‐γ+, HS and AIH patients in the presence of cyclosporin (P < 0·05), tacrolimus (P < 0·05) and mycophenolic acid (MPA) (P < 0·01) at 96 h.
Polyclonal stimulation increased the proportion of cells producing IFN‐γ at 48 h in both HS (P < 0·001) and AIH patients (P < 0.01; Fig. 1a and Supporting information, Fig. S1). While in HS, IFN‐γ production reached its peak level at 48 h, in AIH it continued rising between 48 and 96 h (P < 0·05; Fig. 1a and Supporting information, Fig. S1). In HS no change in the expression of IFN‐γ was observed in the presence of any of the ISDs tested at 48 and 96 h, but in AIH the increase in IFN‐γ observed between 48 and 96 h was reversed by each of the ISDs tested (P < 0·001 for all drugs), bringing IFN‐γ levels back to baseline values (Fig. 1a). The frequency of IFN‐γ‐ producing CD4+CD25– cells was lower in AIH than in HS at 48 h in the absence of drugs and at 96 h upon cell exposure to cyclosporin, tacrolimus and MPA.
Figure 1.
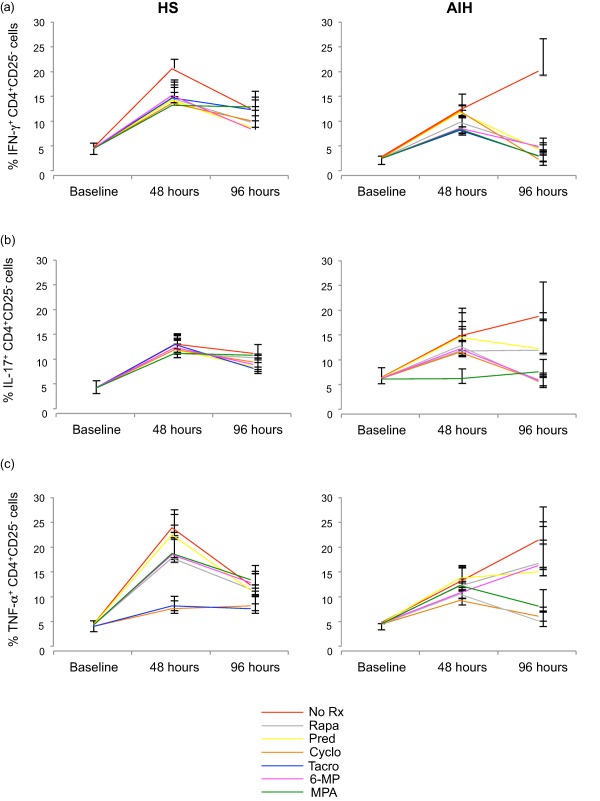
Effect of immunosuppressive drugs (ISDs) on the proportion of interferon (IFN)‐γ, interleukin (IL)‐17 and tumour necrosis factor (TNF)‐α‐producing CD4+CD25– cells. Frequency of IFN‐γ, IL‐17 and TNF‐α‐producing CD4+CD25– T cells was determined by flow cytometry at baseline, 48 and 96 h, in the absence and presence of ISDs. CD4+CD25– T cells were isolated from the peripheral blood of healthy subjects (HS) (n = 8) and autoimmune hepatitis (AIH) patients (n = 6). Mean (± standard error of the mean) frequency of (a) IFN‐γ, (b) IL‐17 and (c) TNF‐α‐producing CD4+CD25– cells over time in the presence of ‘no treatment’ (NT) or individual ISDs. [Colour figure can be viewed at wileyonlinelibrary.com].
In HS, polyclonal stimulation increased the proportion of IL‐17‐producing CD4+CD25– cells between baseline and 96 h (P < 0·05; Fig. 1b and Supporting information, Fig. S2), with the peak at 48 h (P < 0·001; Fig. 1b and Supporting information, Fig. S2). In AIH patients, IL‐17 production rose from baseline to 48 h, continuing to rise at 96 h, in a pattern similar to that observed for IFN‐γ. However, the change from baseline for IL‐17 at both 48 and 96 h was not significant, due presumably to the variability in the frequency of IL‐17‐producing cells. In AIH patients, the increase in IL‐17 production noted between 48 and 96 h in the absence of ISDs was contained, although not significantly, by prednisolone, rapamycin, tacrolimus and cyclosporin. No effect on IL‐17 production was observed at 96 h in the presence of 6‐MP and MPA. No differences in the frequency of IL‐17‐producing CD4+CD25– cells were noted between HS and AIH patients at 48 and 96 h in the absence or presence of ISDs.
In HS, polyclonal stimulation increased the frequency of TNF‐α‐producing CD4+CD25– cells between baseline and 96 h (P < 0·001; Fig. 1c and Supporting information, Fig. S3), with a peak at 48 h (P < 0·001; Fig. 1c and Supporting information, Fig. S3), mirroring the pattern already observed for IFN‐γ and IL‐17. In AIH patients, TNF‐α production rose from baseline at 48 h and continued to increase at 96 h (Fig. 1c and Supporting information, Fig. S3) without, however, reaching statistical significance. In HS the expression of TNF‐α was decreased in the presence of cyclosporin (P < 0·05) and tacrolimus (P < 0·05) at 48 h following polyclonal stimulation. No significant effect on TNF‐α production was noted in the presence of ISDs in HS at 96 h and in AIH patients at both 48 and 96 h. No difference in the proportion of TNF‐α‐producing CD4+CD25– cells was noted between HS and AIH patients at any of the time‐points tested.
Overall, these data show that, at variance with HS, CD4+CD25– cells from AIH patients undergo an increase in the production of IFN‐γ between 48 and 96 h of polyclonal stimulation, this increase being restrained by ISDs treatment.
Effect of ISDs on co‐inhibitory molecules
Frequency of CD4+CD25– cells positive for TIM‐3, PD‐1 and CTLA‐4 was also determined at baseline before polyclonal stimulation was started and then at 48 and 96 h, in the absence and presence of ISDs. Analysis was carried out as described above. Frequencies of cells and P‐values are indicated in Table 4.
Table 4.
Effect of immunosuppressive drugs (ISDs) on the proportion of T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3)+, programmed cell death‐1 (PD‐1)+ and cytotoxic T lymphocyte antigen‐4 (CTLA‐4)+ CD4+CD25‐ cells in healthy subjects (HS) and autoimmune hepatitis (AIH) patients
| HS | AIH | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 48 h | 96 h | Pa | Pb | Baseline | 48 h | 96 h | Pc | Pd | Pe | |
| % TIM‐3+ cells | |||||||||||
| No treatment | 5·4 ± 0·9 | 13·9 ± 3 § | 54·3 ± 8 ¶ | P < 0·001 | P < 0·001 | 3·7 ± 1 | 28·9 ± 8·9 | 49·8 ± 9·4 | P < 0·05 | P < 0·001 | |
| Rapamycin | – | 15·3 ± 3·5 | 52·2 ± 9·1 | P < 0·001 | P < 0·001 | – | 28·4 ± 8·1 | 51·6 ± 12·4 | P < 0·05 | P < 0·001 | |
| Prednisolone | – | 15·8 ± 4·6 | 49·3 ± 7·9 | P < 0·001 | P < 0·001 | – | 29·8 ± 9·6 | 54·5 ± 10·1 | P < 0·05 | P < 0·05 | P < 0·001 |
| Cyclosporin | – | 22·6 ± 6·1 | 40·3 ± 7·9 | P < 0·05 | P < 0·001 | – | 32·4 ± 10 | 44·6 ± 13·8 | P < 0·05 | P < 0·001 | |
| Tacrolimus | – | 20·9 ± 6·1 | 42·4 ± 8·5 | P < 0·01 | P < 0·001 | – | 32 ± 10·8 | 46·4 ± 13·3 | P < 0·05 | P < 0·001 | |
| 6‐MP | – | 14·5 ± 3·2 | 52·1 ± 7·5 | P < 0·001 | P < 0·001 | – | 25·2 ± 7·7 | 51·1 ± 11·1 | P < 0·05 | P < 0·001 | |
| MPA | – | 11·6 ± 3·3 | 34·6 ± 8·9 | P < 0·01 | P < 0·001 | – | 8·8 ± 4·6 | 35·9 ± 11·6 | P < 0·01 | ||
| % PD‐1 + cells | |||||||||||
| No treatment | 5·7 ± 1 | 15·3 ± 2 † | 42·8 ± 8 ∞ | P < 0·001 | P < 0·001 | 6·2 ± 1·6 | 35·5 ± 8 f | 56·9 ± 8·7 ∫ | P < 0·001 | P < 0·05 | P < 0·001 |
| Rapamycin | – | 11·7 ± 1·6 | 26·6 ± 7·1 | P < 0·01 | P < 0·001 | – | 42·3 ± 6·3 f | 45·8 ± 10·3 | P < 0·001 | P < 0·001 | |
| Prednisolone | – | 12·3 ± 1·8 | 32·9 ± 5·2 | P < 0·001 | P < 0·001 | – | 36·3 ± 7 f | 49·2 ± 7·8 | P < 0·001 | P < 0·001 | |
| Cyclosporin | – | 9·6 ± 2·1 | 21·9 ± 4·9 | P < 0·001 | – | 30·9 ± 5 f | 24·3 ± 3·5 | P < 0·01 | |||
| Tacrolimus | – | 8·9 ± 0·9 | 18·2 ± 3·7 | P < 0·001 | – | 25·9 ± 2·9 f | 27·8 ± 4·8 | P < 0·05 | |||
| 6‐MP | – | 11·7 ± 1·9 | 36·7 ± 6·2 | P < 0·001 | P < 0·001 | – | 34·3 ± 7·6 f | 42·8 ± 9·3 | P < 0·01 | P < 0·001 | |
| MPA | – | 9·4 ± 1·2 | 22·9 ± 4·3 | P < < 0·05 | P < 0·001 | – | 19·4 ± 2·4 f | 33·4 ± 7·2 | P < 0·01 | ||
| % CTLA‐4 + cells | |||||||||||
| No treatment | 6·9 ± 1·2 | 13·5 ± 3·6 ‡ | 29·4 ± 7 | P < 0·001 | P < 0·05 | 3·5 ± 0·9 | 15·1 ± 4 # | 26·8 ± 5·7 | P < 0·05 | ||
| Rapamycin | – | 16·6 ± 4 | 26·1 ± 5·4 | P < 0·01 | – | 14·3 ± 4·1 | 24 ± 4·1 | P < 0·05 | |||
| Prednisolone | – | 14 ± 3·5 | 32·5 ± 7·9 | P < 0·001 | P < 0·01 | – | 15·8 ± 4·5 | 36·1 ± 4·3 | P < 0·001 | ||
| Cyclosporin | – | 18 ± 4·3 | 25·2 ± 5·7 | P < 0·01 | – | 15·6 ± 4·7 | 18·6 ± 3·8 | P < 0·05 | |||
| Tacrolimus | – | 18·3 ± 5·9 | 28·2 ± 8·1 | P < 0·01 | – | 12·8 ± 3·8 | 21·5 ± 4·4 | P < 0·05 | |||
| 6‐MP | – | 14·7 ± 3·7 | 34 ± 7·1 | P < 0·001 | P < 0·01 | – | 13·8 ± 3·6 | 35·8 ± 7·7 | P < 0·001 | ||
| MPA | – | 14·3 ± 3·8 | 24·8 ± 7·2 | P < 0·01 | P < 0·05 | – | 14 ± 3·7 | 26·6 ± 6·5 | P < 0·01 | ||
Analysis was performed using one or two‐way analysis of variance (anova) test, followed by Bonferroni post‐test. Only statistically significant differences are reported.
P‐value of anova test comparing the frequency of: P§, TIM‐3+ cells under different treatments in HS at 48 h, < 0·05; P ¶, TIM‐3+ cells under different treatments in HS at 96 h, < 0·01; P †, PD‐1+ cells under different treatments in HS at 48 h, < 0·001; P ∞, PD‐1+ cells under different treatments in HS at 96 h, < 0·01; P ‡, CTLA‐4+ cells under different treatments in HS at 48 h, < 0·05; P ∫, PD‐1+ cells under different treatments in AIH at 96 h, < 0·05; P #, CTLA‐4+ cells under different treatments in AIH at 48 h, < 0·05.
P‐values of post‐test comparing the frequency of:
Pa, TIM‐3+, PD‐1+ and CTLA‐4+ cells between 48 and 96 h in HS; Pb, TIM‐3+, PD‐1+ and CTLA‐4+ cells between baseline and 96 h in HS; Pc, TIM‐3+, PD‐1+ and CTLA‐4+ cells between baseline and 48 h in AIH; Pd, TIM‐3+, PD‐1+ and CTLA‐4+ cells between 48 and 96 h in AIH; Pe, TIM‐3+, PD‐1+ and CTLA‐4+ cells between baseline and 96 h in AIH; Pf, PD‐1+ cells between HS and AIH patients at 48 h (‘no treatment’: P < 0·05; rapamycin, cyclosporin, tacrolimus: P < 0·001; prednisolone, 6‐MP, mycophenolic acid (MPA): P < 0·01).
In the absence of ISDs, TIM‐3 expression increased in both HS and AIH patients during the course of polyclonal stimulation (Fig. 2a and Supporting information, Fig. S4). However, while a significant increase in TIM‐3 expression compared to baseline was evident at 48 h in AIH patients (P < 0·05), this was not so for HS. TIM‐3 expression was elevated significantly compared to baseline by 96 h in both HS (P < 0·001) and AIH patients (P < 0·001). In both HS and AIH patients, rapamycin, prednisolone, cyclosporin and tacrolimus had little effect on the increase in TIM‐3 expression during the course of the experiment. This was also true of 6‐MP and MPA in HS. However, in AIH patients, the increase in TIM‐3 expression between baseline and 48 h was less marked in the presence of 6‐MP or MPA. No significant differences in the frequency of CD4+CD25– cells expressing TIM‐3 were noted between HS and AIH patients at baseline and, subsequently, at 48 and 96 h in the absence and presence of ISDs.
Figure 2.
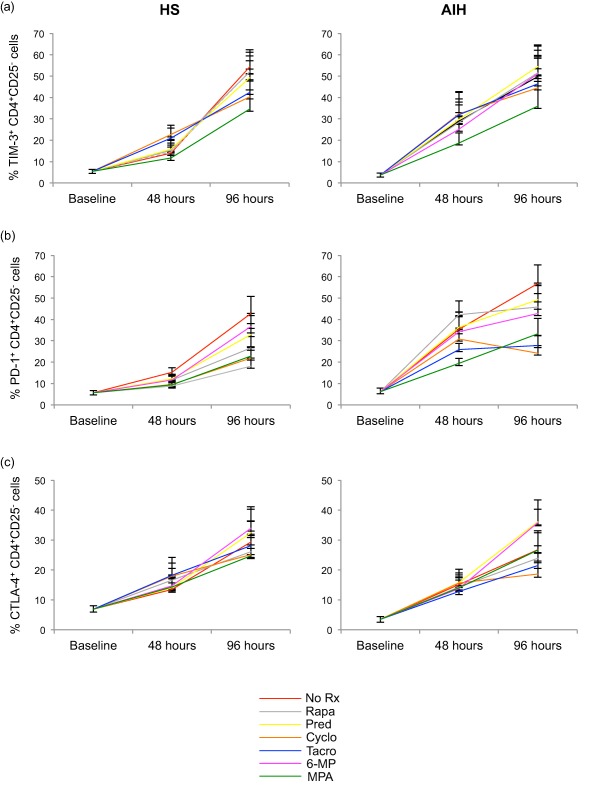
Effect of immunosuppressive drugs (ISDs) on the proportion of T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3), programmed cell death‐1 (PD‐1) and CTLA‐4 expressing CD4+CD25– cells. Frequency of CD4+CD25– T cells positive for TIM‐3, programmed cell death‐1 (PD‐1) and cytotoxic T lymphocyte antigen‐4 (CTLA‐4) was determined by flow cytometry at baseline, 48 and 96 h, in the absence and presence of ISDs. Data refer to eight healthy subjects (HS) and six autoimmune hepatitis (AIH) patients. Mean (± standard error of the mean) frequency of (a) TIM‐3+, (b) PD‐1+ and (c) CTLA‐4+ CD4+CD25– cells over time in the presence of ‘no treatment’ (NT) or individual ISDs. [Colour figure can be viewed at wileyonlinelibrary.com].
In the absence of ISDs, polyclonal stimulation increased the proportion of CD4+CD25– cells expressing PD‐1 from baseline to 96 h in both HS (P < 0·001) and AIH patients (P < 0·001; Fig. 2b and Supporting information, Fig. S5). PD‐1 expression increased more rapidly in AIH patients compared to HS, with PD‐1 expression greater at 48 h compared to baseline in AIH patients (P < 0·001) but not in HS [P = not significant (n.s.)]. Rapamycin, prednisolone and 6‐MP had little effect on PD‐1 expression throughout the course of the experiment. The rise in PD‐1 levels between baseline and 48 h seen in AIH patients was less evident in the presence of MPA and of tacrolimus. Cyclosporin had little effect at 48 h, but in AIH patients it dampened the rise in PD‐1 expression between baseline and 96 h. No differences were observed in the frequency of CD4+CD25– effector T cells expressing PD‐1 in HS and AIH patients at baseline. PD‐1 levels were higher in AIH patients than HS at 48 h in the absence (P < 0·05) and presence of all ISDs tested (P < 0·001 for rapamycin, cyclosporin and tacrolimus; P < 0·01 for prednisolone, 6‐MP and MPA), with this difference levelling off by 96 h.
When evaluating the frequency of CD4+CD25– cells expressing CTLA‐4, we observed that in the absence of ISDs, CTLA‐4 expression did not increase significantly from baseline to 48 h, but it increased between baseline and 96 h in both HS (P < 0·01) and AIH patients (P < 0·05; Fig. 2c and Supporting information, Fig. S6). ISDs did not affect significantly the increase in CTLA‐4 expression triggered by polyclonal stimulation both in HS and AIH patients at 48 and 96 h. Levels of CTLA‐4 expression were similar in HS and AIH patients at all time‐points tested.
Effect of ISDs on effector cell proliferation
We then evaluated the effects of ISDs on the proliferative capacity of CD4+CD25– cells in the absence and presence of Tregs (Fig. 3a,b). We found that in the absence of Tregs tacrolimus reduced significantly (P < 0·05) the percentage of CD4+CD25– proliferating cells in HS; treatment of CD4+CD25– cells in the presence of MPA resulted in a similar effect in AIH patients (P < 0·001). In the presence of Tregs, cyclosporin, tacrolimus and MPA markedly lowered the proportion of CD4+CD25– proliferating cells in AIH (P < 0·05 in all cases) (Fig. 3c); no effect was observed in HS in the presence of any of the ISDs tested (Fig. 3c).
Figure 3.
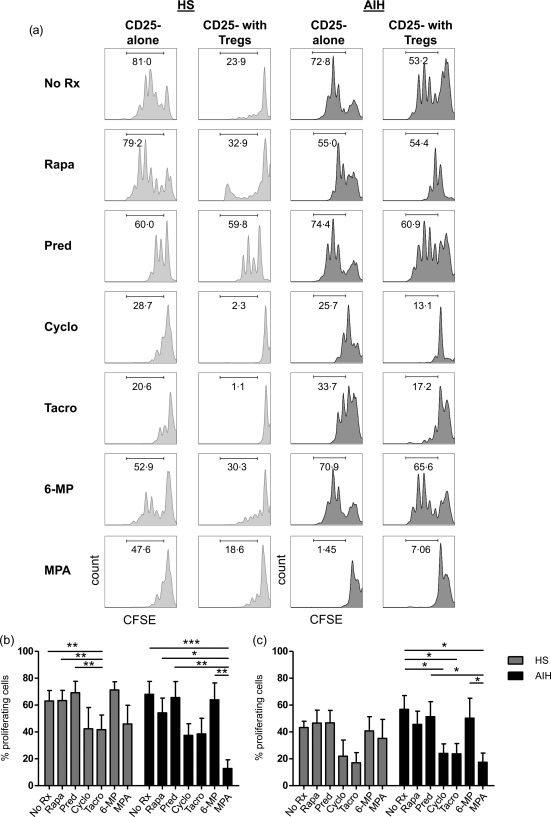
Effect of immunosuppressive drugs (ISDs) on CD4+CD25– cell ability to proliferate. CD4+CD25– cell proliferation in the absence and presence of ISDs and before and after regulatory T cell (Treg) addition was assessed by carboxyfluorescein succinimidyl ester (CFSE) staining and expressed as percentage of proliferating cells. (a) CFSE staining of CD4+CD25– cells alone or in the presence of Tregs in one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient. Mean (± standard error of the mean) % proliferating CD4+CD25– T cells in the presence of ‘no treatment’ (NT) or individual drugs in HS (n = 8) and AIH patients (n = 6) before (b) and after (c) Treg addition.
Discussion
By examining the effects of ISDs on proinflammatory cytokine production, co‐inhibitory molecule expression and on the proliferative capacity of CD4+CD25– effectors isolated from treatment‐naive AIH patients, we have made a number of interesting observations. We found that the expression kinetics of proinflammatory cytokines and co‐inhibitory molecules differs between HS and AIH. Further, in AIH ISDs affect predominantly IFN‐γ production and PD‐1 expression and some of these drugs, i.e. cyclosporin, tacrolimus and MPA, boost Treg function.
In health, the expression of proinflammatory cytokines, produced by CD4 effectors in the absence of ISDs, peaks at 48 h and falls at a later time‐point, possibly as result of the progressive increase in the expression of co‐inhibitory molecules. At variance with health, the levels of proinflammatory cytokines produced by effector cells in AIH continue rising after 48 h despite a parallel increase in the expression of co‐inhibitory molecules. This might be due to dysfunctional co‐inhibition that fails to keep effector cell activation under control. Altered PD‐1/PDL1 interactions were described previously by our group in patients with autoimmune liver disease and in their first‐degree relatives 39.
Of note, the increase in IFN‐γ and TNF‐α follows slower kinetics in AIH than in health, probably as result of in‐vivo cell exhaustion in the former. In contrast, the levels of PD‐1, TIM‐3 and CTLA‐4 undergo a much quicker rising in AIH, possibly in the attempt to restrain overwhelming inflammation in the disease setting.
Our data indicate IFN‐γ and PD‐1 as the main targets of ISDs in AIH. With regard to IFN‐γ, these findings may indicate that ISDs exert a preferential control over Th1 cell immunity. Of the three inhibitory molecules investigated, only PD‐1 had expression levels that were impacted upon exposure to ISDs. It remains unclear, however, whether this decrease in PD‐1 expression is a direct effect of the drugs on this molecule, or it is secondary to the decrease of the in‐vivo inflammatory milieu under laboratory conditions. The former hypothesis is supported by the finding of a direct association between increase in PD‐1 mRNA levels and intensity of liver inflammation in chronic hepatitis B 40.
When examining the production of TNF‐α by CD4 effectors, while no effect on TNF‐α‐producing cells was noted in AIH upon cell treatment with ISDs, in HS TNF‐α production was contained in the presence of cyclosporin and tacrolimus. This phenomenon may be linked to the presence of TNF‐α polymorphisms, found previously to be associated with predisposition to AIH and poor response to immunosuppressive treatment 14, 15, 16.
Analysis of cell proliferation showed that tacrolimus in HS and MPA in AIH had an inhibitory effect on the ability of CD4 effectors to proliferate. When we tested whether Treg addition could impact the effector proliferative response in the presence of ISDs, we found that in AIH addition of Tregs was accompanied by a marked inhibition over CD4 effector cell proliferation not only in the presence of MPA, but also in the presence of cyclosporin and tacrolimus. This control of cell proliferation could result subsequently in a better Treg suppressive function, as proposed in previously published work 41, in which it was suggested that Treg suppression at the peak of inflammation might be achieved upon appropriate control of the inflammatory environment.
The reason for a higher impact of ISDs on the proliferation, IFN‐γ production and PD‐1 expression of effector cells from AIH patients than from HS remains unclear, although it might derive from in‐vivo stimulation by proinflammatory mediators rendering effectors more susceptible to the action of ISDs in vitro.
Curiously, no effect over cell proliferation was noted following Treg addition in the presence of the mTOR inhibitor rapamycin, which was reported previously to enhance Treg properties 37, 42, 43. This may result from poor susceptibility of Tregs from AIH patients to rapamycin, as we reported previously 44.
In summary, we describe for the first time the influence of ISDs over CD4 T cell effectors in treatment‐naive patients with AIH. In this unique clinical setting, ISDs target predominantly IFN‐γ and PD‐1 expression, with MPA, cyclosporin and tacrolimus being particularly effective at restraining cell proliferation. Assessment of these immunological parameters during ISD treatment might represent an important complement to conventional biochemical testing by providing information on immune system activation. These data not only help in understanding the mechanisms through which ISDs restrain effector cells in this clinical context, but also provide valuable information that should be taken into consideration where modulation of the inflammatory environment is aimed to achieve clinical and immunological disease remission.
Disclosure
None.
Author contributions
R. G. performed and analysed the experiments and drafted the manuscript; B. S. H. and R. L. performed and analysed part of the experiments; M. A. H., Y. M., G. M. V. and D. V. reviewed the manuscript; M. S. L. designed the study and wrote the manuscript.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site
Fig. S1. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– interferon (IFN)‐γ expression. Histograms showing expression of IFN‐γ by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S2. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– interleukin (IL)‐17 expression. Histograms showing expression of IL‐17 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S3. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– tumour necrosis factor (TNF)‐α expression. Histograms showing expression of TNF‐α by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S4. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3) expression. Histograms showing expression of TIM‐3 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S5. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– programmed cell death‐1 (PD‐1) expression. Histograms showing expression of PD‐1 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S6. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– cytotoxic T lymphocyte antigen‐4 (CTLA‐4) expression. Histograms showing expression of CTLA‐4 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Acknowledgements
This work has been supported by an Alex P. Mowat Studentship, King's College Hospital Charity, UK to C. R. G.; a Doctoral Grant from the Science and Technology Foundation, Science of Higher Education Ministry, Portugal to R. L.; the Roger Dobson Fund, King's College Hospital Charity, UK to M. S. L., D. V. and G. M. V.; and a Clinician Scientist Fellowship from the Medical Research Council, UK to M. S. L.
References
- 1. Hennes EM, Zeniya M, Czaja AJ et al Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008; 48:169–76. [DOI] [PubMed] [Google Scholar]
- 2. Liberal R, Grant CR, Mieli‐Vergani G, Vergani D. Autoimmune hepatitis: a comprehensive review. J Autoimmun 2013; 41:126–39. [DOI] [PubMed] [Google Scholar]
- 3. Tagawa Y, Sekikawa K, Iwakura Y. Suppression of concanavalin A‐induced hepatitis in IFN‐gamma(–/–) mice, but not in TNF‐alpha(–/–) mice: role for IFN‐gamma in activating apoptosis of hepatocytes. J Immunol 1997; 159:1418–28. [PubMed] [Google Scholar]
- 4. Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell‐dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996; 111:462–71. [DOI] [PubMed] [Google Scholar]
- 5. Mizuhara H, Uno M, Seki N et al Critical involvement of interferon gamma in the pathogenesis of T‐cell activation‐associated hepatitis and regulatory mechanisms of interleukin‐6 for the manifestations of hepatitis. Hepatology 1996; 23:1608–15. [DOI] [PubMed] [Google Scholar]
- 6. Nicoletti F, Di Marco R, Zaccone P et al Murine concanavalin A‐induced hepatitis is prevented by interleukin 12 (IL‐12) antibody and exacerbated by exogenous IL‐12 through an interferon‐gamma‐dependent mechanism. Hepatology 2000; 32:728–33. [DOI] [PubMed] [Google Scholar]
- 7. Ma Y, Bogdanos DP, Hussain MJ et al Polyclonal T‐cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology 2006; 130:868–82. [DOI] [PubMed] [Google Scholar]
- 8. Liberal R, Grant CR, Holder BS et al The impaired immune regulation of autoimmune hepatitis is linked to a defective galectin‐9/tim‐3 pathway. Hepatology 2012; 56:677–86. [DOI] [PubMed] [Google Scholar]
- 9. Longhi MS, Mitry RR, Samyn M et al Vigorous activation of monocytes in juvenile autoimmune liver disease escapes the control of regulatory T‐cells. Hepatology 2009; 50:130–42. [DOI] [PubMed] [Google Scholar]
- 10. Lafdil F, Wang H, Park O et al Myeloid STAT3 inhibits T cell‐mediated hepatitis by regulating T helper 1 cytokine and interleukin‐17 production. Gastroenterology 2009; 137:2125–35 e1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagata T, McKinley L, Peschon JJ, Alcorn JF, Aujla SJ, Kolls JK. Requirement of IL‐17RA in Con A induced hepatitis and negative regulation of IL‐17 production in mouse T cells. J Immunol 2008; 181:7473–9. [DOI] [PubMed] [Google Scholar]
- 12. Zhao L, Tang Y, You Z et al Interleukin‐17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin‐6 expression. PLOS ONE 2011; 6:e18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qin B, Li J, Liang Y, Yang Z, Zhong R. The association between cytotoxic T lymphocyte associated antigen‐4, Fas, tumour necrosis factor‐alpha gene polymorphisms and autoimmune hepatitis: a meta‐analysis. Dig Liver Dis 2014; 46:541–8. [DOI] [PubMed] [Google Scholar]
- 14. Li S, Huang X, Zhong H et al Tumour necrosis factor alpha (TNF‐alpha) genetic polymorphisms and the risk of autoimmune liver disease: a meta‐analysis. J Genet 2013; 92:617–28. [PubMed] [Google Scholar]
- 15. Cookson S, Constantini PK, Clare M et al Frequency and nature of cytokine gene polymorphisms in type 1 autoimmune hepatitis. Hepatology 1999; 30:851–6. [DOI] [PubMed] [Google Scholar]
- 16. Czaja AJ, Cookson S, Constantini PK, Clare M, Underhill JA, Donaldson PT. Cytokine polymorphisms associated with clinical features and treatment outcome in type 1 autoimmune hepatitis. Gastroenterology 1999; 117:645–52. [DOI] [PubMed] [Google Scholar]
- 17. Liberal R, Grant CR, Ma Y et al CD39 mediated regulation of Th17‐cell effector function is impaired in juvenile autoimmune liver disease. J Autoimmun 2016; 72:102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mueller DL. Mechanisms maintaining peripheral tolerance. Nat Immunol 2010; 11:21–7. [DOI] [PubMed] [Google Scholar]
- 19. Longhi MS, Ma Y, Bogdanos DP, Cheeseman P, Mieli‐Vergani G, Vergani D. Impairment of CD4(+)CD25(+) regulatory T‐cells in autoimmune liver disease. J Hepatol 2004; 41:31–7. [DOI] [PubMed] [Google Scholar]
- 20. Longhi MS, Hussain MJ, Mitry RR et al Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol 2006; 176:4484–91. [DOI] [PubMed] [Google Scholar]
- 21. Ferri S, Longhi MS, De Molo C et al A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology 2010; 52:999–1007. [DOI] [PubMed] [Google Scholar]
- 22. Longhi MS, Ma Y, Mitry RR et al Effect of CD4+ CD25+ regulatory T‐cells on CD8 T‐cell function in patients with autoimmune hepatitis. J Autoimmun 2005; 25:63–71. [DOI] [PubMed] [Google Scholar]
- 23. Grant CR, Liberal R, Holder BS et al Dysfunctional CD39(POS) regulatory T cells and aberrant control of T‐helper type 17 cells in autoimmune hepatitis. Hepatology 2014; 59:1007–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liberal R, Grant CR, Holder BS et al In autoimmune hepatitis type 1 or the autoimmune hepatitis‐sclerosing cholangitis variant defective regulatory T‐cell responsiveness to IL‐2 results in low IL‐10 production and impaired suppression. Hepatology 2015; 62:863–75. [DOI] [PubMed] [Google Scholar]
- 25. Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax 2000; 55:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maltzman JS, Koretzky GA. Azathioprine: old drug, new actions. J Clin Invest 2003; 111:1122–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli‐Vergani G. Mycophenolate mofetil as rescue treatment for autoimmune liver disease in children: a 5‐year follow‐up. J Hepatol 2009; 51:156–60. [DOI] [PubMed] [Google Scholar]
- 28. Devlin SM, Swain MG, Urbanski SJ, Burak KW. Mycophenolate mofetil for the treatment of autoimmune hepatitis in patients refractory to standard therapy. Can J Gastroenterol 2004; 18:321–6. [DOI] [PubMed] [Google Scholar]
- 29. Hlivko JT, Shiffman ML, Stravitz RT et al A single center review of the use of mycophenolate mofetil in the treatment of autoimmune hepatitis. Clin Gastroenterol Hepatol 2008; 6:1036–40. [DOI] [PubMed] [Google Scholar]
- 30. Inductivo‐Yu I, Adams A, Gish RG et al Mycophenolate mofetil in autoimmune hepatitis patients not responsive or intolerant to standard immunosuppressive therapy. Clin Gastroenterol Hepatol 2007; 5:799–802. [DOI] [PubMed] [Google Scholar]
- 31. Richardson PD, James PD, Ryder SD. Mycophenolate mofetil for maintenance of remission in autoimmune hepatitis in patients resistant to or intolerant of azathioprine. J Hepatol 2000; 33:371–5. [DOI] [PubMed] [Google Scholar]
- 32. Alvarez F, Ciocca M, Canero‐Velasco C et al Short‐term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol 1999; 30:222–7. [DOI] [PubMed] [Google Scholar]
- 33. Cuarterolo M, Ciocca M, Velasco CC et al Follow‐up of children with autoimmune hepatitis treated with cyclosporine. J Pediatr Gastroenterol Nutr 2006; 43:635–9. [DOI] [PubMed] [Google Scholar]
- 34. Malekzadeh R, Nasseri‐Moghaddam S, Kaviani MJ, Taheri H, Kamalian N, Sotoudeh M. Cyclosporin A is a promising alternative to corticosteroids in autoimmune hepatitis. Dig Dis Sci 2001; 46:1321–7. [DOI] [PubMed] [Google Scholar]
- 35. Larsen FS, Vainer B, Eefsen M, Bjerring PN, Adel Hansen B. Low‐dose tacrolimus ameliorates liver inflammation and fibrosis in steroid refractory autoimmune hepatitis. World J Gastroenterol 2007; 13:3232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–40. [DOI] [PubMed] [Google Scholar]
- 37. Battaglia M, Stabilini A, Tresoldi E. Expanding human T regulatory cells with the mTOR‐inhibitor rapamycin. Methods Mol Biol 2012; 821:279–93. [DOI] [PubMed] [Google Scholar]
- 38. Gregorio GV, Portmann B, Karani J et al Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16‐year prospective study. Hepatology 2001; 33:544–53. [DOI] [PubMed] [Google Scholar]
- 39. Wang MS, Longhi G, Mieli‐Vergani D, Vergani YM. Genetically determined abnormality of the PD‐1/PD‐LS pathway may predispose to autoimmune liver disease. Hepatology 2007; 46:555A. [Google Scholar]
- 40. Germanidis G, Argentou N, Hytiroglou P et al Liver FOXP3 and PD1/PDL1 expression is down‐regulated in chronic HBV hepatitis on maintained remission related to the degree of inflammation. Front Immunol 2013; 4:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Korn T, Reddy J, Gao W et al Myelin‐specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med 2007; 13:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qu Y, Zhang B, Zhao L et al The effect of immunosuppressive drug rapamycin on regulatory CD4+CD25+Foxp3+T cells in mice. Transpl Immunol 2007; 17:153–61. [DOI] [PubMed] [Google Scholar]
- 43. Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol 2007; 178:320–9. [DOI] [PubMed] [Google Scholar]
- 44. Holder BS, Grant CR, Liberal R et al Retinoic acid stabilizes antigen‐specific regulatory T‐cell function in autoimmune hepatitis type 2. J Autoimmun 2014; 53:26–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site
Fig. S1. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– interferon (IFN)‐γ expression. Histograms showing expression of IFN‐γ by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S2. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– interleukin (IL)‐17 expression. Histograms showing expression of IL‐17 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S3. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– tumour necrosis factor (TNF)‐α expression. Histograms showing expression of TNF‐α by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S4. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– T cell immunoglobulin and mucin domain‐containing molecule‐3 (TIM‐3) expression. Histograms showing expression of TIM‐3 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S5. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– programmed cell death‐1 (PD‐1) expression. Histograms showing expression of PD‐1 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.
Fig. S6. Effect of immunosuppressive drugs (ISDs) on CD4+CD25– cytotoxic T lymphocyte antigen‐4 (CTLA‐4) expression. Histograms showing expression of CTLA‐4 by CD4+CD25– cells from one representative healthy subject (HS) and one autoimmune hepatitis (AIH) patient at baseline, 48 and 96 h in the absence and presence of ISDs.