

Abstract
Human inner ear tissue derived from pluripotent stem cells could provide a powerful platform for drug discovery or a source of sound- or motion-sensing cells for patients with hearing loss or balance dysfunction. Here we report a method for differentiating human pluripotent stem cells to inner ear organoids that harbor functional hair cells. Using a three-dimensional culture system, we modulate TGF, BMP, FGF, and Wnt signaling to generate multiple otic vesicle–like structures from a single stem-cell aggregate. Over two months, the vesicles develop into inner ear organoids with sensory epithelia that are innervated by sensory neurons. Additionally, using CRISPR/Cas9, we generate an ATOH1-2A-eGFP cell line to detect hair cell induction and demonstrate that derived hair cells exhibit electrophysiological properties similar to those of native sensory hair cells. Our culture system will be useful for elucidating mechanisms of human inner ear development and testing potential inner ear therapies.
The human inner ear contains ∼75,000 sensory hair cells that detect sound and movement via mechanosensitive stereocilia bundles1,2. Genetic mutations or environmental insults, such as loud noises and ototoxic drugs, can cause irreparable damage to these hair cells, leading to hearing loss or dizziness3,4. We previously demonstrated how to generate inner ear organoids from mouse pluripotent stem cells (PSCs) using timed manipulation of the TGFβ, BMP, FGF and Wnt signaling pathways in a 3D culture system5,6. We have shown that mouse inner ear organoids contain sensory hair cells that are structurally and functionally similar to native vestibular hair cells in the mouse inner ear7. Moreover, our past findings supported a working model of otic induction signaling cascades in which BMP signaling activation and TGFβ inhibition initially specify non-neural ectoderm, and subsequent BMP inhibition and FGF activation induce a pre-otic fate8,9. Despite several recent attempts, a developmentally faithful approach for deriving functional hair cells from human PSCs (hPSCs) has yet to be described10-15. Here, to generate human inner ear tissue from hPSCs, we first established a timeline of in vitro human inner ear organogenesis (Fig. 1a, b). The inner ear arises from the ectoderm layer and, in humans, produces the first terminally differentiated hair cells by ∼52 days post conception (dpc)16. Beginning with pluripotent cells in the epiblast, inner ear induction begins at ∼12 dpc with formation of the ectoderm epithelium. Then, the epithelium splits into the non-neural ectoderm (also known as surface ectoderm) and the neuroectoderm (Fig. 1a, b). The non-neural ectoderm ultimately produces the inner ear as well as the epidermis of the skin. Thus, in our initial experiments, we sought to establish a chemically defined 3D culture system for targeted derivation of non-neural ectoderm epithelia, from which we could derive inner ear organoids (Fig. 1a-c).
Figure 1.
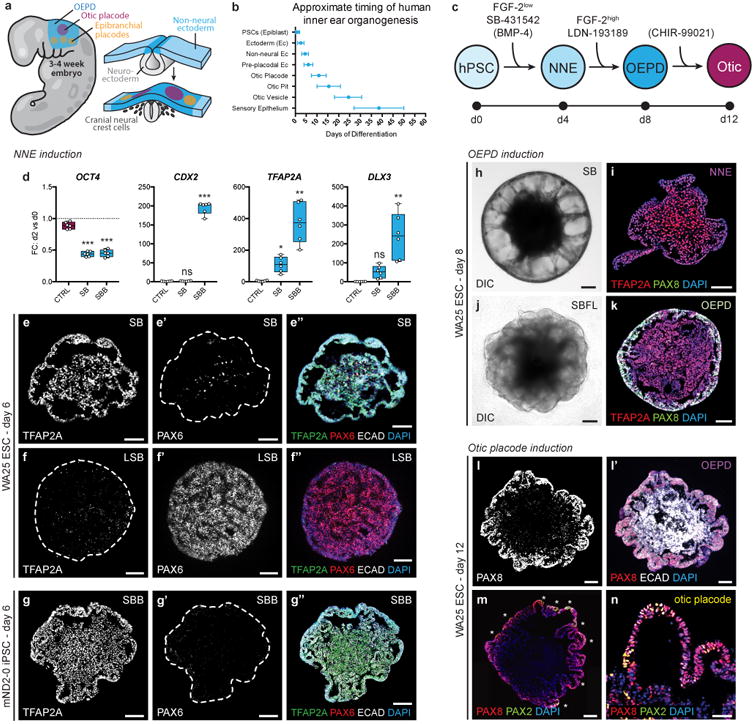
Step-wise induction of otic placode-like epithelia. a, Overview of mammalian ectoderm development in the otic placode cranial region. b, Timeline for key events of human otic induction. Day 0 on the timeline indicates the approximate stage of development represented by hPSC: ∼12 dpc. c, Differentiation strategy for non-neural ectoderm (NNE), otic-epibranchial progenitor domain (OEPD), and otic placode induction. Potentially optional or cell line-dependent treatments are denoted in parentheses. d, qPCR analysis on day 2 of differentiation of WA25 cell aggregates treated with DMSO (Control), 10 μM SB, or 10 μM SB + 10 ng/ml BMP4, denoted as SBB. Gene expression was normalized to undifferentiated hESCs; n = 3 biological samples, 2 technical repeats; *P<0.05, **P<0.01, ***P<0.001; error bars = max/min. e, f, Representative TFAP2A, ECAD, and PAX6 expression in WA25 aggregate treated with 10 μM SB or with 200 nM LDN + 10 μM SB for 6 days. g, TFAP2A, ECAD, and PAX6 expression in mND2-0 iPSCs treated with 10 μM SB + 2.5 ng/ml BMP4 (SBB) on day 6. h, i, Representative image of a SB-treated WA25 aggregate on day 8: live (h) and immunostained with PAX8 and TFAP2A antibodies (i). When comparing morphology in panels (h) and (i) note that the outer-epithelium crumples into the aggregate core during the cryosectioning process. j, k, Representative image of a SB-treated WA25 aggregate on day 8 after treatment with 50 ng/ml FGF-2 and 200 nM LDN (SBFL) on day 4: live (j) and immunostained with PAX8 and TFAP2A antibodies (k). l-n, WA25 SBFL-treated aggregates on day 12. The outer-epithelium contains PAX8+ ECAD+ cells (l) and occasional patches of PAX8+ PAX2+ otic placode-like cells (m, n). The specimens shown were treated with 25 μl of additional CDM on day 8. Scale bars, 100 μm (e-m), 50 μm (n).
We first confirmed that dissociated human embryonic stem cells (hESCs; WA25 cell line, WiCell) aggregate well in E8 Medium containing a ROCK inhibitor, Y-27632, and display superior uniformity and cell-survival compared to cells aggregated in a chemically defined differentiation medium (hereafter, CDM; Supplementary Fig. 1 and Table 1). Following a 2-day incubation in E8 Medium, we transferred aggregates to CDM containing a low concentration of Matrigel and FGF-2 to stimulate epithelization and ectoderm differentiation on the aggregate surface. We previously showed that a combination of BMP4 and the TGFβ inhibitor SB-431542 (hereafter, SB) can promote non-neural induction from mouse PSCs (mPSCs)6. We found that combining 10 ng/ml BMP4 and 10 μM SB (dual SB/BMP4 treatment referred to as SBB) induces not only non-neural marker genes, such as TFAP2A and DLX3, but also the extraembryonic marker CDX2 (Fig 1d; Supplementary Fig. 2)17. In contrast, SB treatment alone led to an increase in TFAP2A and DLX3 expression with no corresponding CDX2 expression (Fig. 1d). 100% of SB-treated aggregates generated TFAP2A+ E-cadherin (ECAD)+ epithelium with a surface ectoderm–like morphology by days 4-6 of differentiation—a time scale consistent with human embryogenesis (n = 15 aggregates, 3 experiments; Fig. 1b-e; Supplementary Fig. 2). Over a period of 20 days, the epithelium expanded into a cyst composed of TFAP2A+ Keratin-5 (KRT5)+ keratinocyte-like cells (Supplementary Fig. 3). From these findings, we concluded that treating WA25 cell aggregates with SB is sufficient to induce a non-neural epithelium.
To determine whether endogenous BMP activity is sufficient for non-neural specification, we performed a co-treatment with the BMP inhibitor LDN-193189 (hereafter, LDN; dual LDN/SB treatment referred to as LSB). As previously shown in hESC monolayer cultures18, LSB treatment of WA25 aggregates up-regulated neuroectoderm markers, such as PAX6 and N-cadherin (NCAD), and abolished TFAP2A and ECAD expression, suggesting that endogenous BMP signals drive non-neural conversion (Fig. 1f; Supplementary Fig. 4). To further validate our approach, we treated human iPSCs (mND2-0, WiCell) with SB and found, contrary to our results with WA25 hESCs, that SB-only conditions generated PAX6+ neuroectoderm and TFAP2A+ ECAD- neural crest-like cells (Supplementary Fig. 5). We reasoned that variation in endogenous BMP levels may underlie the different outcomes, and the BMP concentration may have to be fine-tuned for each cell line. Accordingly, a low concentration of BMP4 (2.5 ng/ml) in addition to SB (SBB) resulted in generation of TFAP2A+ ECAD+ non-neural epithelium from mND2-0 iPSCs (Fig. 1g; Supplementary Fig. 5). With either the SB or SBB approaches, the resulting epithelia closely resembled non-neural epithelia generated with mPSCs5,6. In contrast to our mouse culture, non-neural conversion occurs without off-target induction of Brachyury (BRA)+ mesendoderm cells (Supplementary Fig. 6). The following data were generated using primarily the SB approach on WA25 hESCs and were subsequently confirmed using the SBB approach on mND2-0 hiPSCs.
Next, we attempted to convert the non-neural epithelium into otic placode epithelium prior to keratinocyte commitment. Human cranial placodes arise at ∼18-24 dpc; thus, assuming hPSCs represent cells at ∼12 dpc, otic placodes would develop in our culture within the first 6-12 days of differentiation with proper signaling modulation (Fig. 1b). Drawing on our previous finding that FGF activation and BMP inhibition are essential for pre-placode and otic induction from mPSC cultures, we treated day 4 aggregates with a combination of FGF-2 and LDN (hereafter, SBFL). With SBFL treatment, the outer epithelium thickened relative to SB-treated samples and expressed a combination of posterior placode markers, such as PAX8, SOX2, TFAP2A, ECAD, and NCAD, indicating a phenotype similar to the otic-epibranchial progenitor domain (OEPD) from which the otic placode arises (Fig. 1h-k; Supplementary Fig. 7). When allowed to undergo self-guided differentiation in a minimal medium, we found that SBFL aggregates generated BRN3A+ TUJ1+ sensory-like neurons between days 10-30 (Supplementary Fig. 8). Since both the epibranchial placodes and the otic vesicles produce sensory neurons, we investigated which tissue type had developed. We did not detect expression of the otic marker PAX2 nor did we observe any vesicles in SBFL-treated aggregates, which would signify otic induction (data not shown). Thus, we concluded that SBFL treatment may be sufficient to induce epibranchial neurons, yet fails to initiate otic induction.
To promote PAX2 expression and vesicle formation, we tested various signaling modulators (Supplementary Fig. 9). Although none of the conditions we tested had a detectable effect on PAX2 gene expression using qPCR analysis, extensive immunostaining drew our attention to a small population of PAX2+ PAX8+ ECAD+ cells in the epithelia of aggregates of control samples on day 12, reminiscent of the otic placodes in vivo (Fig. 1l-n). Considering that extracellular matrix might provide structural support for vesicle formation, we transferred day 12 aggregates to Matrigel droplets in a minimal medium (Fig. 2a). In these cultures, we observed radial production of migratory cells but no vesicle-like structures or PAX2+ cells (Fig. 2b; Supplementary Fig. 10). Wnt activation seems to be essential for otic, but not epibranchial, development in vivo and can enhance the production of mouse inner ear organoids in vitro19-22. After Matrigel-embedded WA25 cell aggregates were treated with CHIR99021, a GSK3β inhibitor that activates the Wnt pathway23, 90.9 ± 5.2% (± S.D.) of the aggregates between days 12-16 (n = 84, 7 experiments) showed epithelial protrusions reminiscent of the otic pits that precede vesicle development in vivo (Fig 2c). These structures were PAX2+ PAX8+ SOX2+ SOX10+ JAG1+, confirming their otic identity (Fig. 2d-h; Supplementary Fig. 10). The pits were accompanied by migrating TFAP2A+ SLUG+ SOX10+ cranial neural crest–like cells that formed a mesenchyme around the otic pits, similar to the peri-otic mesenchyme in vivo (Fig 2c-f; Supplementary Fig. 10).
Figure 2.
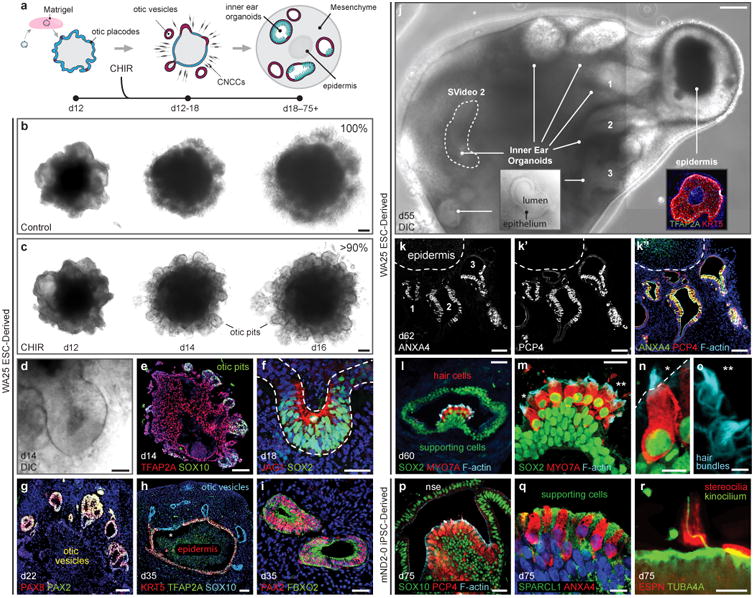
Wnt signaling activation initiates self-organization and maturation of inner ear organoids containing vestibular-like hair cells. a, Inner ear organoid induction strategy. Day 12 aggregates were embedded in Matrigel droplets to support vesicle formation. b-d, In CHIR-treated samples, but not DMSO (Control) samples, otic pit-like structures evaginate from the outer-epithelium (d). e-i, Between days 14-35, pits and vesicles expressed otic specific markers, such as SOX10, SOX2, JAG1, PAX8, PAX2, and FBXO2. The epithelium from which vesicles arise begins to express the epidermal keratinocyte marker KRT5 by day 35 (h). j, By day 40-60, the aggregates contain multiple organoids and, typically, a single epidermal unit visible under DIC imaging. Inner ear organoids are distinguishable by a defined epithelium with ∼25-40 μm apparent thickness and a lumen (j inset). k, Inner ear organoids are typically oriented around the epidermal unit and contain sensory epithelia with ANXA4+ PCP4+ hair cells. The luminal surface of organoids is actin-rich, as denoted by phalloidin staining (k″). l-o, Hair cells are MYO7A+ SOX2+, and supporting cells are SOX2+. F-actin-rich hair bundles protrude from the hair cells into the lumen (n, o; asterisks denote hair bundle location in m). p, q, mND2-0 iPSC-derived sensory epithelia have a similar morphology to WA25 hESC-derived sensory epithelia and contain PCP4+ ANXA4+ hair cells. SOX10 is expressed throughout the supporting and non-sensory epithelial cell populations, but not in hair cells (p). Supporting cells express the utricle supporting cell marker SPARCL1 (q). r, Hair cells in organoids have ESPN+ hair bundles with a single acetylated-tubulin (TUBA4A)+ kinocilium. Scale bars, 200 μm (j), 100 μm (b, c, e), 50 μm (d, g, h, k, l), 25 μm (f, i, m, p), 10 μm (n, q), 5 μm (r), 2.5 μm (o).
We cultured the aggregates in stationary droplets until day 18 and then transferred them to a 24-well plate on an orbital shaker or a spinner flask for further self-organized maturation. Both formats produced similar results. At 20-30 days in culture, vesicles remained visible through the surface of 71.7 ± 23.3% (± S.D.) WA25 cell aggregates examined (n = 37, 3 experiments; Supplementary Fig. 11). In each aggregate we immunostained, we found multiple otic vesicles surrounding a central core epithelium that expressed the basal keratinocyte markers TFAP2A and KRT5 (n = 15, 3 experiments; Fig. 2g-i; Supplementary Video 1). As late as day 35, we observed vesicles and otic placode–like epithelia that appeared to be partially attached or incorporated into the epidermal epithelium (Fig. 2h). In addition, older vesicles (>30 days) expressed the ubiquitin ligase FBXO2, which is highly specific to developing inner ear epithelia in mice (Fig. 2i)24,25. Each aggregate we analyzed between days 30-40 contained SLUG+ mesenchymal cells and cartilaginous masses with S100+ SOX9+ chondrocytes, which also stained positive for Alcian blue (n = 7, 3 experiments; Supplementary Fig. 10). These tissues likely arise from the cranial neural crest–like cells seen during vesicle formation on days 12-18. Thus, our culture system appears to derive multiple cranial tissues associated with the anatomic region surrounding the inner ear.
After 40-60 days of incubation, vesicles with complex multi-chambered morphologies were visible through the aggregate surface (Fig. 2j; Supplementary Video 2). Notably, a subset of vesicles in both WA25 and mND2-0 derived aggregates developed epithelia with cells expressing multiple hair cell markers, including MYO7A, PCP4, ANXA4, SOX2, and CALB2 (Fig. 2k-q; Supplementary Fig. 12). The sensory-like epithelia also contained SOX2+ SOX10+ SPARCL1+ cells, reminiscent of supporting cells in the mammalian utricle26. The luminal cells in these epithelia had elongated morphologies with F-actin-rich apical junctions characteristic of inner ear sensory epithelia (Fig. 2l-o). The cells expressing hair cell markers also had F-actin-rich and espin (ESPN)+ apical stereocilia bundles protruding into the vesicle lumen that were associated with an acetylated-alpha-Tubulin (TUBA4A)+ kinocilium (Fig. 2m-p, r). Together, these findings confirm that the hPSC-derived otic vesicles generate inner ear organoids with sensory epithelia containing hair cells and supporting cells. Moreover, the hair bundle morphology and the densely packed clusters of hair cells are reminiscent of vestibular end organs.
To facilitate live-cell imaging and electrophysiological experiments, we engineered an hESC reporter cell line to label nascent hair cells with enhanced green fluorescent protein (eGFP). We used the CRISPR/Cas9 system to insert a 2A-eGFP gene cassette at the stop codon of the ATOH1 gene, which is highly expressed during hair cell induction and early maturation (Fig. 3a; Supplementary Fig. 13)7. We verified inner ear organoid induction from two clones containing the bi-allelic insertion of the 2A-eGFP cassette using our established protocol (hereafter, ATOH1-2A-eGFP cells). As early as day 39, we observed eGFP+ hair cell-like cells emerging in inner ear organoids (Supplementary Fig. 13). The individual organoids often contained multiple discrete patches with hundreds of eGFP+ cells (Fig. 3b-d; Supplementary Video 3). Immunostaining with hair cell markers, such as BRN3C and ESPN (present on 99.4% of eGFP+ hair cell-like cells), confirmed the hair cell identity of eGFP+ cells (Fig. 3e, f). Occasionally, we observed eGFP expression in disorganized clusters of neuron-like cells, which were clearly distinct from hair cells (∼20% of aggregates, Supplementary Fig. 14). Between days 60-100, 19.7 ± 7.0% (± S.D.) of aggregates contained at least one organoid with eGFP+ hair cells (n = 167, 7 experiments). The seemingly low efficiency of hair cell induction may be due to our inability to detect organoids deep within the aggregates, or it could indicate that the endogenous signals required for sensory epithelia formation vary from aggregate to aggregate. Every WA25 and mND2-0 aggregate examined between days 60-100 contained organoids with PAX8+ PAX2+ SOX10+ non-sensory epithelia (n = 21, 5 experiments; Supplementary Fig. 12e), suggesting that although inner ear induction is highly reproducible, non-sensory or immature otic epithelia are preferentially induced. Using 3D reconstructions, we estimated that there were 68-779 hair cells per organoid (mean = 266, n = 12 organoids, 4 experiments). The stem cell–derived hair cells could be maintained for >150 days in floating culture and retained hair bundles even after dissection and sub-culturing (Fig. 3b-f; Supplementary Videos 3-4). In mice, Atoh1 is downregulated as hair cells mature; however, we did not detect any noticeable changes in eGFP expression in day 100-150 organoids, suggesting that maturation may be delayed or ATOH1 expression is more prolonged in human inner ear organogenesis.
Figure 3.
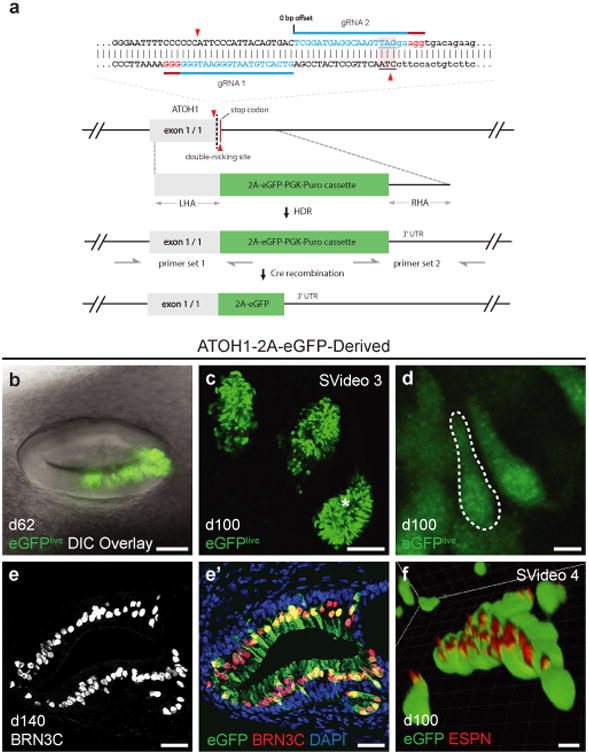
Development of an ATOH1 fluorescent reporter hESC line for tracking hair cell induction. a, ATOH1-2A-eGFP CRISPR design. The two guide RNAs (blue, with PAM sequence in red) direct Cas9n to make two nicks (red triangles) near the stop codon (underlined with pink background) of ATOH1. The resulting DNA double strand break is repaired by the donor vector, which has a 2A-eGFP-PGK-Puro cassette and 1kb left and right homology arms (LHA and RHA). The LoxP-flanked PGK-Puro sub-cassette is subsequently removed by Cre recombinase. In ATOH1 expressing hair cells, eGFP is transcribed along with ATOH1. b-d, Representative live cell images of eGFP+ hair cells in 62- and 100-day-old inner ear organoids. The asterisk in panel (c) denotes the approximate location of the hair cells in panel (d). e, Expression of BRN3C in 140-day-old eGFP+ hair cells. f, Expression of ESPN in the hair bundles of 100-day-old eGFP+ hair cells. Scale bars, 100 μm (c), 50 μm (b), 25 μm (e), 5 μm (d, f).
Next we examined whether hPSC-derived hair cells function similarly to native mammalian hair cells. Using aggregates generated from ATOH1-2A-eGFP cells, we dissected and flat-mounted inner ear organoids between differentiation days 63-70. The cells had large outwardly rectifying currents (Fig. 4a-c), typical of type II hair cells of human vestibular organs27, mouse vestibular organs28 and mouse organoid hair cells7. The amplitude of the currents ranged from several hundred picoAmps to several nanoAmps, presumably depending on the maturity of the cell. Small rapidly activating, rapidly inactivating inward currents were also present in 6 of 14 hPSC-derived hair cells (Fig. 4d), consistent with the small Na+ currents transiently expressed in developing mouse hair cells28 and mouse organoid hair cells7. Hyperpolarizing steps evoked slowly activating inward currents (Fig. 4e, f) that resembled Ih of human27 and mouse29 vestibular type II hair cells, but atypical of mammalian cochlear hair cells. Responses to step and sinusoidal current injection (Fig. 4g, h) resembled those of rodent vestibular and organoid hair cells7, with an initial peak followed by repolarization, and larger excursions for hyperpolarizing than depolarizing current injections. Resting potentials of the hPSC-derived organoid hair cells were slightly depolarized (−51 ± 2.4 mV, n = 6) relative to those of rodent vestibular cells. Negative resting potentials in mouse vestibular hair cells are maintained by prominent inward rectifier currents, which are carried by Kir2.1 channels30. Inward rectifier currents develop early in all rodent vestibular hair cells and mouse organoid hair cells but were absent in human organoid cells and native human vestibular cells27. The constricted lumen morphology seen in most >60-days-old organoids and in all of the organoids used for recording made hair bundle deflection and mechanotransduction analysis inaccessible (for example see Fig. 3b-e; Supplementary Video 3). Nonetheless, to our knowledge there have been no previous recordings of voltage-dependent currents and membrane response (Fig. 4a-h) from hPSC-derived human hair cells. Furthermore, the profile of current expression strongly suggests that the hPSC-derived organoid cells, like mouse inner ear organoid cells, have adopted a vestibular type II hair cell phenotype.
Figure 4.
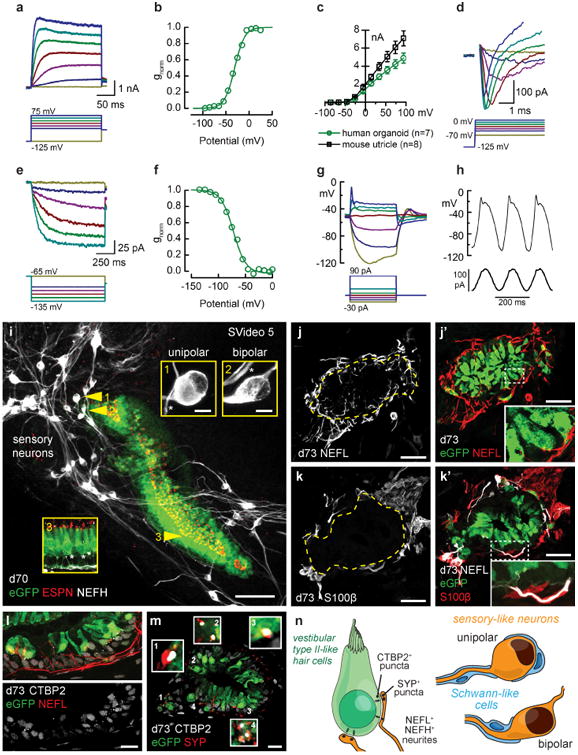
hESC-derived hair cells have similar electrophysiological properties as native hair cells and form synapse-like contacts with sensory neurons. a, Family of outward rectifier potassium currents recorded from a human organoid hair cell (d64), evoked by the series of voltage steps shown below. b, The outward currents had an activation range that was well-fitted by a Boltzmann equation (line) with voltage of half maximal activation of −31 mV. c, Mean (± S.E.) maximal current-voltage relationships for seven human organoid hair cells (d64-d67) and eight mouse utricle type II hair cells. For current-voltage relations, we averaged data from hair cells with large currents over 2 nA. d, Family of rapidly-activating, rapidly-inactivating, inward currents evoked by the depolarizing steps shown below. e, Family of slowly-activating, non-inactivating inward currents (d64) evoked by hyperpolarizing steps, shown below. f, Activation curve for the current family shown in panel e, fitted by a Boltzmann equation (line) with a voltage of half maximal activation of −71 mV. g, Family of membrane responses (d64) recorded in current-clamp mode, evoked by the current injection protocol shown below. h, Membrane response (d65) to three cycles of a 5-Hz sine wave stimulus (below). i, 3D projection of eGFP+ hair cells with ESPN+ hair bundles surrounded by clusters of sensory-like neurons. Insets 1 and 2 demonstrate the two neuron morphologies observed: unipolar and bipolar. Inset 3 demonstrates hair cell morphology and NEFH+ neurites in the sensory epithelium. j, Representative image of NEFL+ neurons innervating an organoid sensory epithelium. k, S100β+ Schwann-like cells associate with neuronal soma and appear to myelinate NEFL+ neuronal processes. l, NEFL+ neuronal processes infiltrate the epithelium and are closely associated with CTBP2+ puncta at the base of eGFP+ hair cells. m, CTBP2+ puncta are co-localized with SYP+ puncta. n, Summary of neurogenesis analysis. Scale bars, 100 μm (i), 25 μm (j, k), 10 μm (l, m), 5 μm (i insets).
Finally, we examined neurogenesis during inner ear organoid formation. We observed the first sign of TUJ1+ BRN3A+ neurons in aggregates between days 20-30 after the start of differentiation (Supplementary Fig. 15). Immunostaining for TUJ1 and neurofilaments (light chain, NEFL, and heavy chain, NEFH) between days 60-75 revealed a mix of unipolar and bipolar neurons surrounding inner ear organoids (Fig. 4i, j and Supplementary Video 5). Inner ear ganglia neurons are bipolar, whereas epibranchial and neural crest ganglia neurons typically mature from a bipolar to a unipolar morphology31,32; thus, the cells observed here may be derived from placode or neural crest. We also observed S100β+ cells, reminiscent of myelinating Schwann cells, associated with NEFL+ soma and processes (Fig. 4k and Supplementary Fig. 15). In each sensory epithelium we analysed, we found neurites infiltrating the epithelium and contacting hair cells (Fig 4j-l, n = 15 sensory epithelia, 6 separate experiments). A subset of hair cells displayed CTBP2+ puncta, indicating putative ribbon synapse–like structures. (Fig 4l, m and Supplementary Fig 15). Moreover, the CTBP2+ puncta were typically localized near synaptophysin (SYP)+ puncta, a post-synaptic marker of ribbon synapses (Fig. 4m and Supplementary Fig. 15)26. Together, these findings suggest that the human inner ear organoids assemble a sensorineural circuit between hair cells and sensory neurons, similar to in vivo organs and mouse inner ear organoids (Fig. 4n)6,7,31. However, it remains to be determined whether the neurons innervating hair cells are authentic vestibular afferent neurons and whether the putative synapses are functional.
In conclusion, we have established a robust differentiation system for guiding the development of human inner ear organoids in 3D culture (see Supplementary Fig. 16 and Supplementary Table 5 for a comparison to past human otic induction approaches). Our findings support our previous model of in vitro pre-otic induction and underscore the importance of carefully timed GSK3β inhibition for otic placode specification. Notably, the convoluted and multi-chambered morphologies of human inner ear organoids bear a striking resemblance to the inner ear's membranous labyrinth, which consists of a series of tubes and chambers containing sensory and non-sensory epithelia (Figures 2j-l, 3i and Supplementary Videos 2-5). In addition, much like mouse organoids, the hPSC-derived organoids appear to form vestibular sensory epithelia by default; thus, additional signaling manipulation will be needed to initiate cochlear organogenesis6,7. We expect that this culture system will provide a useful tool for uncovering mechanisms of human inner ear development, studying genetic disease of the inner ear, and testing potential therapies.
Online Methods
hPSC culture
Human PSCs (WA25 hESCs, passage 22-50; mND2-0 iPSCs, passage 28-46) were cultured in Essential 8 (E8) Medium (Invitrogen, cat. no. A1517001) or Essential 8 Flex Medium (E8f) (Invitrogen, cat. no. A2858501) supplemented with 100 μg/ml Normocin (Invivogen, cat. no. Ant-nr-1) on recombinant human Vitronectin-N (Invitrogen, cat. no. A14700)-coated 6-well plates according to an established protocol33. At 80% confluency or every 4-5 days, the cells were passaged at a split ratio of 1:10-1:20 using 0.5 mM EDTA in DPBS. Both cell lines were acquired from the WiCell Research Institute and arrived with a statement of verification and authenticity. For additional validation and testing information refer to the cell line webpages: https://www.wicell.org/home/stem-cell-lines/catalog-of-stem-cell-lines/wa25.cmsx, https://www.wicell.org/home/stem-cell-lines/catalog-of-stem-cell-lines/mirjt7i-mnd2-0.cmsx. Cell lines were determined to be mycoplasma contamination-free using the MycoAlert Mycoplasma Detection Kit (Lonza). Prior to differentiation, pluripotency was assessed using immunocytochemistry to detect stem-cell markers, SOX2, OCT4, and SSEA-1 (see Supplementary Figs. 1, 5, and 13). Routine karyotyping was not performed, however, the passage number was minimized to reduce the risk of spontaneous chromosomal duplications or genetic mutations.
hPSC differentiation
To start differentiation, hPSC cells were dissociated with StemPro Accutase (Invitrogen, cat. no. A1110501) and distributed, 5,000 cells per well, onto low-adhesion 96-well V-bottom plates in E8 medium containing 20 μM Y-27632 (Stemgent, cat. no. 04-0012-02) and Normocin. Following a 48 hour incubation, the aggregates were transferred to low-adhesion 96-well U-bottom plates in 100 μl of Chemically Defined Medium (CDM) containing 4 ng ml-1 FGF-2 (Peprotech, cat. no. 100-18B), 10 μM SB-431542 (Stemgent, cat. no. 04-0010-05), and, for some experiments, 2.5 ng ml-1 BMP4 (Stemgent, cat. no. 03-0007), and 2% Growth Factor Reduced (GFR) Matrigel (Corning, cat. no. 354230) to initiate non-neural induction—i.e. differentiation day 0. CDM contained a 50:50 mixture of F-12 Nutrient Mixture with GlutaMAX (Gibco) and Iscove's Modified Dulbecco's Medium with GlutaMAX (IMDM; Gibco) additionally supplemented with 0.5% Bovine Serum Albumin (BSA), 1X Chemically Defined Lipid Concentrate (Invitrogen), 7 μg ml-1 Insulin (Sigma), 15 μg ml-1 Transferrin (Sigma), 450 μM Mono-Thioglycerol, and Normocin (see Supplementary Table 1 for detailed formulation). After 4 days of incubation, 25 μl of CDM containing a 250 ng ml-1 FGF-2 (50 ng/ml final concentration) and 1 μM LDN-193189 (200 nM final concentration; Stemgent, cat. no. 04-0074-02) was added to the pre-existing 100 μl of media in each well. After an additional 4 days (8 days total), 25 μl of CDM was added to the media. For some experiments, CDM containing a 18 μM CHIR99021 (3 μM final concentration; Stemgent, 04-0004-02) was added to the pre-existing 125 μl of media in each well on day 8—we determined that this treatment is optional for inner ear organoid production, but may improve induction of otic placode-like cells. On differentiation day 12, the aggregates were pooled together and washed with freshly prepared Organoid Maturation Medium (OMM) containing a 50:50 mixture of Advanced DMEM:F12 (Gibco) and Neurobasal Medium (Gibco) supplemented with 0.5× N2 Supplement (Gibco), 0.5× B27 without Vitamin A (Gibco), 1× GlutaMAX (Gibco), 0.1 mM β-Mercaptoethanol (Gibco), and Normocin (see Supplementary Table 2 for detailed formulation). The aggregates were resuspended in ice cold undiluted GFR Matrigel and distributed in ∼25 μl droplets on the surface of a 100 mm culture plate. After at least 30 minutes of incubation at 37°C, the droplets were bathed in 10 ml of OMM containing 3 μM CHIR99021. For non-droplet otic induction, the aggregates were washed and plated individually into each well of a 24-well low cell adhesion plate in OMM containing 3 μM CHIR and 1% GFR Matrigel. For both culture formats, the medium was changed completely on day 15 with OMM containing fresh 3 μM CHIR. During days 12-18, aggregates were monitored using DIC imaging daily. Otic pit-like protrusions were identifiable as bright translucent bulges from the dark core of the aggregates. On day 15 or 16, a subset of at least three aggregates were fixed, cryosectioned, and immunostained using PAX2/PAX8 antibodies to positively identify otic pit-like structures. On day 18 of differentiation, the CHIR was removed from the medium by washing and the droplet aggregates were moved to a floating culture. Droplets were carefully dislodged using a wide-mouth P1000 tip and transferred to 75 ml of fresh OMM in a 125 ml disposable spinner flask (Corning). Spinner flasks were maintained on an in-incubator stir plate (Thermo Scientific) at 65 RPM for up to 180 days of differentiation. For some experiments, the aggregates were maintained in individual wells of 24-well low-cell adhesion plates in 1 ml of OMM on an in-incubator orbital shaker (Thermo Scientific) at 65 RPM for up to 140 days. Technical repeats entailed a different start date, different passage number, and typically a different lot of reagents (some experiments used the same reagent lots).
Choice of Media Components
We used two media components that may lead to variability in results due to lack of definition or poor compatibility with human cells: GFR Matrigel and BSA. GFR Matrigel contains, < 0.1 pg ml-1 FGF-2, < 0.5 ng ml-1 EGF, 5 ng ml-1 IGF-1, < 5 pg ml-1 PDGF, < 0.2 ng ml-1 NGF, and 1.7 ng ml-1 TGFβ. In particular, the TGFβ in GFR Matrigel may have impacted cell fate specification on day 12 or later because we did not include a TGFβ inhibitor in the media during that phase of culture. GFR Matrigel was chosen because it has been shown to be a reliable inducer of self-organizing epithelia from pluripotent stem cells in 3D culture34. A purified Laminin/Entactin complex (Corning) may be a suitable, fully defined, alternative35. In the CDM, BSA was chosen as a cost-effective and easy to dissolve alternative to Human Serum Albumin and Polyvinyl Alcohol (PVA), respectively. PVA has been shown to be a suitable chemically defined substitute for BSA in CDM36.
Signaling molecules and recombinant proteins
The following small molecules and recombinant proteins were used: recombinant human BMP4 (2.5-10 ng ml−1; Stemgent), human FGF-2 (25 ng ml−1; Peprotech), SB-431542 (10 μM; Stemgent), CHIR99021 (3 μM; Stemgent), and LDN-193189 (200 nM; Stemgent).
Quantitative PCR
Analysis was performed as previously described on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) or a Bio-Rad CFX96 quantitative PCR machine (Bio-Rad)6. Data were normalized to L27 expression (internal control) and the fold change was calculated relative to Ct values from d0 WA25 aggregates using the ΔΔCt method. Unless stated otherwise, data represent at least 3 separate biological samples from separate experiments. All indicators of statistical significance refer to comparisons between a given condition and the control group. Refer to Supplementary Table 3 for primer details.
Immunohistochemistry
Aggregates were fixed with 4% paraformaldehyde for 20 min at room temperature or at 4°C overnight. The fixed specimens were cryoprotected with a graded treatment of 15% and 30% sucrose and then embedded in tissue freezing medium. Frozen tissue blocks were sectioned into 12 μm cryosections on a Leica CM-1860 cryostat. For immunostaining, a 10% goat or horse serum in 0.1% Triton X-100 1X PBS solution was used for blocking, and a 3% goat or horse serum in 0.1% Triton X-100 1X PBS solution was used for primary/secondary antibody incubations. Alexa Fluor conjugated anti-mouse, rabbit, or goat IgG (Invitrogen) were used as secondary antibodies. ProLong Gold Antifade Reagent with DAPI (Thermo Scientific) was used to mount the samples and visualize cellular nuclei. For wholemount staining, a similar staining paradigm was used; however, the Triton X-100 concentration was increased to 0.5%, and the blocking and primary/secondary incubations were done at 37°C on a rotating shaker for 24 hours and 48 hours, respectively. Following each incubation, the samples were subjected to three 1-hour washes in 1X PBS containing 0.5% Triton X-100 at 37°C on a rotating shaker. Wholemount samples were mounted in ScaleS4 clearing solution for 1-2 days or ScaleSQ(5) clearing solution for 1-2 hours prior to imaging37. Microscopy was performed on a Leica DMi8 inverted microscope, a Nikon TE2000 inverted microscope, or an Olympus FV1000-MPE Confocal/Multiphoton Microscope. 3D reconstruction was performed using the Imaris 8 software package (Bitplane) housed at the Indiana Center for Biological Microscopy. For the segmentation analysis in Supplementary Video 3 and estimates of hair cell number per organoid, eGFP+ cells were processed using the ‘Spots’ module in Imaris. Classification was based on estimated size, quality and signal intensity. Objects touching the border of the image were excluded. The following build parameters were used to identify eGFP+ cell bodies: estimated XY diameter = 7.00 μm; estimated Z diameter = 10.00 μm; ‘Quality’ above 20.0; ‘distance to image border XYZ’ above 0.001 μm; ‘intensity center Ch = 1′ above 1,500. These parameters generated estimated, not absolute, counts and were adjusted to provide highly conservative values. For quality control, we performed slice-by-slice analysis of optical sections for three representative organoids. Movies were generated in Imaris from the raw image files and compiled in Adobe Premiere Pro to add titles and text. See Supplementary Table 4 for a list of antibodies.
Electrophysiological recordings
Human organoids were shipped at day 62 in cold Hibernate A medium supplemented with 1X GlutaMax, 1× B27 Supplement (without Vitamin A), 22 mM NaCl, and Normocin. They were replaced back into OMM on day 63 in an incubator at 5% CO2 and 37°C. On recording days, organoids were dissected out using sharp tungsten needles (Fine Science Tools) and pinned to glass coverslips. The eGFP+ signal was used to find areas with hair cells and to target hair cells for recording. Whole-cell patch clamp was performed on the semi-intact tissue with 4-5 MΩ glass electrodes. Data were acquired using an Axopatch 200B amplifier (Molecular Devices), filtered at 5000 Hz, then digitized at 20 kHz through a Digidata 1322A converter. The recording pipette solution contained (in mM): 135 KCl, 5 HEPES, 5 EGTA, 2.5 MgCl2, 2.5 K2-ATP, 0.1 CaCl2, adjusted with KOH to pH 7.4, ∼285 mmol kg-1. The external solution contained: 137 NaCl, 5.8 KCl, 0.7 NaH2PO4, 10 HEPES, 1.3 CaCl2, 0.9 MgCl2, 5.6 Glucose, and was supplemented with vitamins and essential amino acids (Invitrogen), adjusted to pH 7.4 with NaOH, ∼310 mmol kg-1. Recordings were compensated 40% and cells were held at −66 mV for voltage clamp. Averages are reported ± SEM.
Generation of ATOH1-2A-eGFP reporter cell line
gRNAs (5′-TCGGATGAGGCAAGTTAGGA-3′ and 5′-GTCACTGTAATGGGAATGGG-3′, offset = 0bp) targeting the stop codon region of ATOH1 were cloned into pSpCas9n(BB) vectors which express Cas9n under the control of CBh promoter (Addgene #48873)38. To construct the donor vector, a 2A-eGFP-PGK-Puro cassette (Addgene #31938)39 flanked by two 1kb homology arms PCR amplified from extracted WA25 hESC genomic DNA were cloned into a pUC19 backbone. The two gRNA vectors and the donor vector, as well as a vector expressing Cas9n under the control of CMV promoter (Addgene #41816)40 were transfected into WA25 hESCs with 4D Nucleofector (Lonza) using the P3 Primary Cell 4D-Nucleofector X kit and Program CB-150. After nucleofection, cells were plated in growth medium containing 1× RevitaCell (Thermo Fisher) for improved cell survival rate, and 1μM of Scr7 (Xcessbio) for higher HDR efficiency41. 0.5 μg ml-1 puromycin selection was performed for 10 days starting from 48 hours post-nucleofection. The PGK-Puro sub-cassette flanked by two LoxP sites was removed from the genome after puromycin selection by nucleofection of a Cre recombinase expressing vector (Addgene #13775). Clonal cell lines were established by low-density seeding (1 – 3 cells cm-2) of dissociated single hESCs followed by isolation of hESC colonies after 5-7 days of expansion. Genotypes of the clonal cell lines were analyzed by PCR amplification followed by gel electrophoresis, and by Sanger sequencing of total PCR amplicons or individual PCR amplicons cloned into TOPO vectors. Cell lines with bi-allelic eGFP integration were used for inner ear hair cell differentiation. Hair cell-bearing organoids were identified using an inverted epiflorescent microscope (Leica DMi8). We only counted eGFP+ cells observed in an organoid epithelium with defined apical and basal edges that were located toward the surface of the aggregate. Hair cell bearing organoids located in the interior of the aggregates were undetectable using this approach.
Statistical analysis
All statistics were performed using GraphPad Prism 7 software. A Shapiro-Wilk normality test was used prior to analysis to determine that the data had a normal distribution. Statistical significance was determined using a one-way analysis of variance (ANOVA) followed by a Dunnett's post-hoc test for multiple comparisons to a control group (e.g. vehicle treated). A Brown-Forsythe test was used to determine that the variation among sample groups was similar. No statistical test was used to predetermine sample size, the investigators were not blinded to the treatment groups, and the samples were not randomized.
Representative Data and Reproducibility
Unless stated otherwise, images are representative of specimens obtained from at least 3 separate experiments. For IHC analysis of aggregates between days 0-12, we typically sectioned 3-6 aggregates from each condition in each experiment. IHC analyses for later stages of the protocol were performed on at least 2 aggregates from each condition per experiment. The finalized culture method was successfully replicated 23 times by four independent investigators (K.R.K., J.N., E.L.M., and J.L.) using the WA25 (wild-type or ATOH1-2A-eGFP) cell line. The method, with noted modifications, was replicated 3 times using the mND2-0 iPSC line by K.R.K. A replication was deemed successful by confirming pit formation during days 12-18 and positively identifying hair cell-bearing inner ear organoids in at least one aggregate on days 50-100 of differentiation. If either of these criteria were not met, an experiment was deemed a failure and excluded from analysis. The investigators observed a failure rate of ∼21% (6/29 experiments), which was typically attributed to expiration/mishandling of a critical reagent or technical error.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health (NIH) grants R01 DC013294 (E.H. and J.R.H.) and R03 DC015624 (K.R.K.), Action on Hearing Loss International Project Grant (E.H.), and an Indiana Clinical and Translational Sciences Institute Core Grant (NIH UL1 TR001108; K.R.K). This work was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR020128-01 from the National Center for Research Resources, NIH. The authors would like to thank A. Mikosz, P-C. Tang, R. Nelson, S. Winfree, M. Kamocka for their comments and technical assistance and J. Bartles for the espin antibody.
Footnotes
Author Contributions: K.R.K. conceived, designed, and led the study, performed experiments, analyzed data, and drafted the manuscript with input from all authors. J.N. generated the ATOH1-2A-eGFP cell line, performed experiments, and wrote the manuscript. E.L.M. performed experiments, data analysis and wrote the manuscript. X-P.L. performed electrophysiology experiments and wrote the manuscript. J.L. performed experiments and data analysis. J.R.H designed and analyzed electrophysiology experiments and wrote the manuscript. E.H. designed and oversaw the study and wrote the manuscript.
Competing Financial Interests: K.R.K. and E.H., with the Indiana University Research and Technology Corporation, have applied for a patent on the cell culture method described in this manuscript.
References
- 1.Géléoc GSG, Holt JR. Sound strategies for hearing restoration. Science. 2014;344:1241062. doi: 10.1126/science.1241062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenhall U. Degenerative patterns in the aging human vestibular neuro-epithelia. Acta oto-laryngologica. 1973;76:208–220. doi: 10.3109/00016487309121501. [DOI] [PubMed] [Google Scholar]
- 3.Müller U, Barr-Gillespie PG. New treatment options for hearing loss. Nat Rev Drug Discov. 2015;14:346–365. doi: 10.1038/nrd4533. [DOI] [PubMed] [Google Scholar]
- 4.Sergeyenko Y, Lall K, Liberman MC, Kujawa SG. Age-Related Cochlear Synaptopathy: An Early-Onset Contributor to Auditory Functional Decline. 2013;33:13686–13694. doi: 10.1523/JNEUROSCI.1783-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koehler KR, Hashino E. 3D mouse embryonic stem cell culture for generating inner ear organoids. Nature Protocols. 2014;9:1229–1244. doi: 10.1038/nprot.2014.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature. 2013;500:217–221. doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu XP, Koehler KR, Mikosz AM, Hashino E, Holt JR. Functional development of mechanosensitive hair cells in stem cell-derived organoids parallels native vestibular hair cells. Nature Communications. 2016;7:11508. doi: 10.1038/ncomms11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung AW, Kent Morest D, Li JYH. Differential BMP signaling controls formation and differentiation of multipotent preplacodal ectoderm progenitors from human embryonic stem cells. Developmental biology. 2013;379:208–220. doi: 10.1016/j.ydbio.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon HJ, Bhat N, Sweet EM, Cornell RA, Riley BB. Identification of early requirements for preplacodal ectoderm and sensory organ development. PLoS genetics. 2010;6:e1001133. doi: 10.1371/journal.pgen.1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JR, et al. Effects of genetic correction on the differentiation of hair cell-like cells from iPSCs with MYO15A mutation. Cell Death & Differentiation. 2016 doi: 10.1038/cdd.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang ZH, et al. Genetic Correction of Induced Pluripotent Stem Cells From a Deaf Patient With MYO7A Mutation Results in Morphologic and Functional Recovery of the Derived Hair Cell-Like Cells. Stem Cells Transl Med. 2016;5:561–571. doi: 10.5966/sctm.2015-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohnishi H, et al. Limited hair cell induction from human induced pluripotent stem cells using a simple stepwise method. Neuroscience Letters. 2015;599:49–54. doi: 10.1016/j.neulet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 13.Ealy M, Ellwanger DC, Kosaric N, Stapper AP, Heller S. Single-cell analysis delineates a trajectory toward the human early otic lineage. PNAS. 2016;113:8508–8513. doi: 10.1073/pnas.1605537113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronaghi M, et al. Inner Ear Hair Cell-Like Cells from Human Embryonic Stem Cells. Stem Cells and Development. 2014;23:1275–1284. doi: 10.1089/scd.2014.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, et al. Restoration of auditory evoked responses by human ES-cell-derived otic progenitors. Nature. 2012;490:278–282. doi: 10.1038/nature11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim R, Brichta AM. Anatomical and physiological development of the human inner ear. Hearing research. 2016 doi: 10.1016/j.heares.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 17.Roberts RM, et al. Differentiation of trophoblast cells from human embryonic stem cells: to be or not to be? Reproduction. 2014;147:D1–12. doi: 10.1530/REP-14-0080. [DOI] [PubMed] [Google Scholar]
- 18.Chambers SM, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nature Biotechnology. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ladher RK, O'Neill P, Begbie J. From shared lineage to distinct functions: the development of the inner ear and epibranchial placodes. Development. 2010;137:1777–1785. doi: 10.1242/dev.040055. [DOI] [PubMed] [Google Scholar]
- 20.Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. 2012;139:245–257. doi: 10.1242/dev.067074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK. Wnt signals mediate a fate decision between otic placode and epidermis. Development. 2006;133:865–875. doi: 10.1242/dev.02271. [DOI] [PubMed] [Google Scholar]
- 22.DeJonge RE, et al. Modulation of Wnt Signaling Enhances Inner Ear Organoid Development in 3D Culture. PLoS ONE. 2016;11:e0162508. doi: 10.1371/journal.pone.0162508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kriks S, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman BH, Durruthy-Durruthy R, Laske RD, Losorelli S, Heller S. Identification and characterization of mouse otic sensory lineage genes. Frontiers in cellular neuroscience. 2015;9:79. doi: 10.3389/fncel.2015.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson RF, et al. Selective cochlear degeneration in mice lacking the F-box protein, Fbx2, a glycoprotein-specific ubiquitin ligase subunit. The Journal of Neuroscience. 2007;27:5163–5171. doi: 10.1523/JNEUROSCI.0206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns JC, Kelly MC, Hoa M, Morell RJ, Kelley MW. Single-cell RNA-Seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nature Communications. 2015;6:8557. doi: 10.1038/ncomms9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oghalai JS, et al. Ionic currents and electromotility in inner ear hair cells from humans. Journal of neurophysiology. 1998;79:2235–2239. doi: 10.1152/jn.1998.79.4.2235. [DOI] [PubMed] [Google Scholar]
- 28.Géléoc GSG, Risner JR, Holt JR. Developmental acquisition of voltage-dependent conductances and sensory signaling in hair cells of the embryonic mouse inner ear. The Journal of Neuroscience. 2004;24:11148–11159. doi: 10.1523/JNEUROSCI.2662-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horwitz GC, Risner-Janiczek JR, Jones SM, Holt JR. HCN channels expressed in the inner ear are necessary for normal balance function. The Journal of Neuroscience. 2011;31:16814–16825. doi: 10.1523/JNEUROSCI.3064-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levin ME, Holt JR. The function and molecular identity of inward rectifier channels in vestibular hair cells of the mouse inner ear. Journal of neurophysiology. 2012;108:175–186. doi: 10.1152/jn.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appler JM, Goodrich LV. Connecting the ear to the brain: Molecular mechanisms of auditory circuit assembly. Progress in neurobiology. 2011;93:488–508. doi: 10.1016/j.pneurobio.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grigaliunas A, Bradley RM, MacCallum DK, Mistretta CM. Distinctive neurophysiological properties of embryonic trigeminal and geniculate neurons in culture. Journal of neurophysiology. 2002;88:2058–2074. doi: 10.1152/jn.2002.88.4.2058. [DOI] [PubMed] [Google Scholar]
- 33.Beers J, et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nature Protocols. 2012;7:2029–2040. doi: 10.1038/nprot.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huch M, Koo BK. Modeling mouse and human development using organoid cultures. Development. 2015;142:3113–3125. doi: 10.1242/dev.118570. [DOI] [PubMed] [Google Scholar]
- 35.Nasu M, et al. Robust Formation and Maintenance of Continuous Stratified Cortical Neuroepithelium by Laminin-Containing Matrix in Mouse ES Cell Culture. PLoS ONE. 2012;7:e53024. doi: 10.1371/journal.pone.0053024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannan NRF, Segeritz CP, Touboul T, Vallier L. Production of hepatocyte-like cells from human pluripotent stem cells. Nature Protocols. 2013;8:430–437. doi: 10.1038/nprot.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hama H, et al. ScaleS: an optical clearing palette for biological imaging. Nature Neuroscience. 2015;18:1518–1529. doi: 10.1038/nn.4107. [DOI] [PubMed] [Google Scholar]
- 38.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hockemeyer D, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 2011 doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mali P, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maruyama T, et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature Biotechnology. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.