

Abstract
Protein−ligand interactions are frequently screened using nuclear magnetic resonance (NMR) spectroscopy. The dissociation constant (KD) of a ligand of interest can be determined via a spin-spin relaxation measurement of a reporter ligand, in a single scan when using hyperpolarization by means of dissolution dynamic nuclear polarization (D-DNP). Despite nearly instantaneous signal acquisition, a limitation of D-DNP for the screening of protein−ligand interactions is the required polarization time on the order of tens of minutes. Here, we introduce a multiplexed NMR experiment, where a single hyperpolarized ligand sample is rapidly mixed with protein injected into two flow cells. NMR detection is achieved simultaneously on both channels, resulting in a chemical shift resolved spin relaxation measurement. Spectral resolution allows the use of reference compounds for accurate quantification of concentrations. Simultaneous use of two concentration ratios between protein and ligand broadens the range of KD that is accurately measurable in a single experiment to at least an order of magnitude. In a comparison of inhibitors for the protein trypsin, the average KD values of benzamidine and benzylamine were found to be 12.6±1.4 μM and 207±22.3 μM from three measurements, based on KD = 142 μM assumed known for the reporter ligand 4-(trifluoromethyl)benzene-1-carboximidamide. Typical confidence ranges at 95% evaluated for single experiments were (8.3 μM, 20 μM) and (151 μM, 328 μM). The multiplexed detection of two or more hyperpolarized samples increases throughput of D-DNP by the same factor, improving the applicability to most multi-point measurements that would traditionally be achieved using titrations.
Keywords: Dissolution dynamic nuclear polarization, nuclear magnetic resonance, protein−ligand interaction
Introduction
Primary challenges for the application of nuclear magnetic resonance (NMR) spectroscopy in the determination of protein−ligand interaction in drug discovery include the achievable sensitivity and throughput.1,2 These limitations can partially offset the benefits of high resolution and chemical selectivity of this technique. Throughput can be increased by reliance on automation, which routinely permits the unsupervised acquisition of spectra from a large number of samples, but remains limited by the signal averaging time. Increases in the NMR sensitivity to reduce the needed acquisition time can be implemented in the form of high-field magnets, cold probe technology or nuclear spin hyperpolarization.3,4
A hyperpolarization method amenable to the production of spin polarized ligand solutions is a technique called dissolution dynamic nuclear polarization (D-DNP).5 In D-DNP, the hyperpolarization is first achieved in the solid state at a temperature of 1–2 K, and samples are subsequently dissolved for NMR measurement at room temperature. Signal gains of up to four orders of magnitude achieved in a single scan meet or exceed the requirements for detection of difficult targets in the sub-micromolar concentration range.
Several methods for employing the signal gains from D-DNP for characterizing protein−ligand interactions have been proposed. Among these are the use of direct 13C or 19F NMR detection of the hyperpolarized ligands,6,7 as well as the combination of ligand signal based determination of protein−ligand interactions with long-lived spin states to extend the lifetime of hyperpolarization.8 Transfer of ligand hyperpolarization via nuclear Overhauser effect to the protein or to a competing ligand can further provide a means to prove binding and potentially characterize the binding site.9,10 While the latter may already extend beyond the most basic screening application, a parameter that is often of interest in drug discovery is the dissociation constant of the ligand for the protein. Typically, the determination of binding affinities using NMR spectroscopy is achieved by a titration of protein or ligand concentration. In a D-DNP experiment, however, the measurement of each data point requires a new hyperpolarization and sample dissolution step. While it is possible to carry out the titrations using D-DNP,6 this requirement imposes a new limitation on the achievable throughput. A method to circumvent the need for titrations, based on the relaxation measurement of displaced reporter ligands,11–14 has been recently developed for use with D-DNP in our group.15 Even in this case, the measurement of relaxation parameters at two or three different concentrations can be desirable. DNP instruments that are capable of polarizing multiple samples have been developed.16,17 However, even with polarization of a single sample, the amount of hyperpolarized substance generated is often much larger than what can be used in a single experiment. The use of large aliquots of hyperpolarized substance has been recently demonstrated for magnetic resonance imaging (MRI) experiments, where several metabolic experiments were carried out simultaneously.18
Here, we describe an implementation of an NMR screening experiment that allows for parallel measurements of competitive protein−ligand interactions using a single hyperpolarized aliquot injected into separate detection coils located in a high-field NMR spectrometer. We demonstrate the measurement of 19F spin-spin relaxation rate (R2) of a hyperpolarized (HP) reporter ligand in the presence of non-HP solutions of protein and inhibitor at different mixing ratios in two channels, permitting the calculation of the dissociation constant KD in a larger range, and discuss its applications in drug discovery.
Experimental Section
Two-channel NMR probe
An NMR probe was designed for accommodating two flow cells needed for simultaneous acquisition of relaxometry data (Figure S1). The flow cells were manufactured in U-shape by joining borosilicate glass tubing (4 mm outer diameter, 0.5 mm inner diameter) for inlet and outlet with tubing of the same material (5 mm outer diameter, 3.6 mm inner diameter) for the measurement chamber. Inlet and outlet PEEK tubing was inserted into the ends of the flow cells and glued in place using acrylic adhesive. The flow cells were arranged perpendicular to each other in the probe, to reduce cross-talk. Flow cells were held by pieces printed from acrylonitrile butadiene styrene (ABS) plastic, and assembled on a support frame made of brass. The flow cell supports were designed to allow for modular stacking. A grounded copper foil (0.762 mm thickness) was placed between the cells, in contact with the probe shield, for radio-frequency (RF) shielding. Solenoidal RF coils with 6 turns were directly wound on each cell using a copper wire of 0.812 mm diameter (20 AWG), comprising a detection volume of approximately 100 μL. Due to space constraints, each coil was pre-tuned close to the 19F NMR frequency (376.4 MHz) with non-magnetic fixed value capacitors of 1.1 – 1.3 pF (Johanson Technology, Inc., CA). A transmission line (UT-141C semi-rigid coaxial cable, Amawave, Oceanside, CA, inside of the probe, and double-shielded flexible coaxial cable outside of the probe) with a total length of 3λ/2 ~ 86.5 cm connected each coil to a remote tuning circuit outside of the probe.19 Remote tuning was achieved using tuning and matching capacitors of 0.45 – 4 pF (Voltronics, Salisbury, MD). NMR cross-talk between the two channels was measured by pulsing on, and detecting signal from one channel, which was empty, while the other channel was filled with 130 mM trifluoroethanol.20 With this method, less than 1% of cross-talk was observed, which was consistent with the value S21 = −34 dB measured between the coils using a vector impedance analyzer (Model VIA Echo 1000 SF, AEA Technology Inc., Carlsbad, CA). The NMR probe was located in a standard bore magnet (40 mm accessible diameter within the shim stack) with 9.4 T field strength (Bruker Biospin, Billerica, MA).
Sample preparation
The non-HP sample component was prepared as a solution of 2 or 4 μM trypsin (AMRESCO, Road Solon, OH) and 100 μM or 2 mM of ligand of interest in buffer (50 mM Tris(hydroxymethyl)aminomethane, 10 mM KCl, 15 mM trifluoroacetic acid (TFA), pH 8). Benzylamine (TCI, Portland, OR) and benzamidine (Sigma Aldrich, St. Louis, MO) were the two ligands of interest used in the screening experiments. A volume of 10 μL HP sample, prepared in a mixture of D2O/DMSO-d6 (v/v 1:1), was composed of 4 mM reporter ligand 4-(trifluoromethyl)benzene-1-carboximidamide hydrochloride hydrate (TFBC·HCl; Maybridge, UK), 0.88 M KF and 15 mM 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL; Sigma Aldrich, St. Louis, MO). The concentrations of stock solutions were determined by UV/Vis and NMR spectroscopy. TFA and KF were used as reference compounds for non-HP and HP samples, respectively, to evaluate final protein and ligand concentrations in the flow cells using NMR spectroscopy.
Hyperpolarization
Hyperpolarization was generated on 19F spins in a HyperSense DNP polarizer (Oxford Instruments, Abingdon, UK) by irradiating with microwaves of 94.055 GHz and 100 mW power, at a temperature of 1.4 K. After 20 min of the polarization, the samples were dissolved in a stream of 4 mL of buffer that had been pre-heated until a pressure of 5 bar was reached at an approximate temperature of 385 K. Dissolved samples were automatically loaded into the injection loop.
Sample mixing and injection
Figure 1 shows the flow paths and accessories used to achieve the sample injection. The non-HP solutions of protein or protein and ligand of interest were preloaded into the separate sections of tubing designed to hold the samples for each channel (t4 and t4′, V = 70 μL), before commencing the experiments. Shut-off valves and plugs were used to block the inlets and outlets of these sections. High-pressure syringe pumps (pump A and B) containing water as a driving fluid started prior to dissolution of the HP sample in preparation for injection, with the fluid directed to a waste line by the injection valves.21 The samples were injected after the HP sample entered the injection loop (L1, V = 1 mL), which was indicated by an optical detector. At this time, the first injection valve (V1) was switched, and then the second injection valve (V2) after 370 ms to have the HP and non-HP samples push together into the flow cells. Before having the samples enter the probe, the HP sample was split into two streams by a splitter (IDEX Health & Science, Oak Harbor, WA). Each stream passed through a Y-mixer (IDEX Health & Science, Oak Harbor, WA) to which the tubing filled with the non-HP solution was connected, resulting in mixing the HP and non-HP solutions. To prevent back- or cross-flow due to pressure differentials, check valves (IDEX Health & Science, Oak Harbor, WA) were installed on the mixers. The streams leaving the flow cells merged again before connecting to the return port of the V1. This connection held the flow stopped in both cells at the same time after switching of the injection valves. In all the DNP experiments, the pumps A and B delivered the fluid at the flow rates of 160 mL/min and 120 mL/min, which resulted in peak pressures of 6.8 MPa and 3.0 MPa, respectively. A total delay time of 770 ms allowed for the samples to arrive in the flow cells. The experimental verification and optimization of the injection characteristics were performed by recording images of the flow cells using a dye and measuring NMR signals of the reference compounds after the injection.
Figure 1.
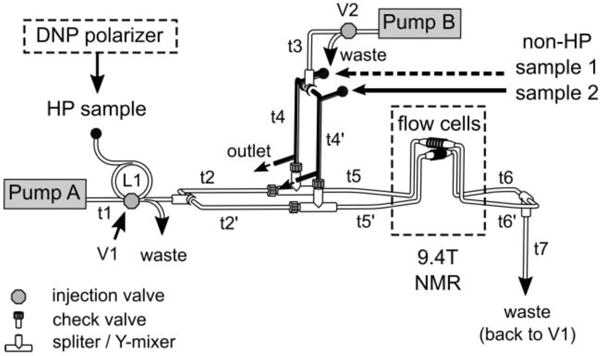
Flow path utilized for parallelized measurement of competitive protein-ligand interaction using a single hyperpolarized aliquot of ligand. The HP sample is injected through the sample loop (L1) into the flow path. The tubing (t4 and t4′, bold lines) is used to preload two non-HP solutions containing protein and a ligand of interest. The flow path connected to the bottom flow cell is distinguished from the top flow cell by primed notations. The tubing connections to the two flow cells are symmetric. Tubing length: t1: 152.4 cm; t2: 20 cm; t3: 23 cm; t4: 15 cm; t5 and t6: 60 cm; t7: 170 cm. L1: 50 cm. Tubing inner diameter: t1, t2, t3 and t5: 0.02”; t4, t6 and t7: 0.03”.
NMR experiments
For the NMR experiments, a home-built multiplexing NMR spectrometer for 19F detection was utilized. An NMR probe accommodating two solenoidal coils wound around the flow cells was installed in a 9.4 T NMR magnet (Bruker Biospin, Billerica, MA), with both coils tuned to 376.4 MHz. The spectrometer itself consisted of the RF generator and processing boards (SpinCore Technologies, Gainesville, FL). The RF signal was amplified by a power amplifier (model BT00250 Gamma, Tomco Technologies, Stepney, South Australia) before it was split and fed into both coils. The NMR signal from each channel was received separately. The NMR experiments were started 400 ms after having the flow of fluids stopped by switching the injection valves off. The experiments employed a Carr Purcell Meiboom Gill (CPMG) pulse sequence,22 where a train of spin-echoes was acquired using p1 − τ − [p2 − 2τ]n. Here, p1 is an excitation pulse with flip angle π/2 and phase x, p2 is a π pulse of phase y, τ = 4 ms and n = 860. A total of 280 complex data points was taken for 7 ms in the 2τ period indicated in the pulse sequence, resulting in a 40 kHz spectral width. Pulse strengths were γB1 = 45.5 kHz using 51.2 W forward power split equally on Channel 1 and 2 (top and bottom coils, respectively, shown in Figure 1). In this experiment, the transmitter offset frequency was set to the middle of the TFBC frequencies in Channel 1 and 2 due to a slight difference in magnetic fields at the two coils. The receiver frequency was set to the center of the TFBC and KF frequencies. After the decay of the hyperpolarization, a one-pulse NMR experiment was performed using 36 scans to determine the concentrations of the reference compounds.
Data analysis
For the data processing, each complex valued spin echo was multiplied by a sine-squared window function with the center of the echo set to t = 0, and Fourier transformed into a frequency-domain spectrum. A pure absorption spectrum was obtained for both the real and imaginary parts due to the time symmetric nature of the signal. The phase shift was corrected to maximize the real component while minimizing the imaginary one (Figure S2). Signal intensities were measured from the spectrum of each echo, and subsequently used for R2 determination by exponential fitting. Only the spectra after 160 ms (840 spectra) were used to avoid fluctuations arising during the sample settling immediately following the injection.
Under fast exchange, R2 of free and bound ligands (R2,f and R2,b, respectively) contribute to the overall R2 depending on their relative populations. An additional increase in the overall R2 can occur due to chemical exchange unless the chemical shift difference between the free and bound states (Δω) is negligibly small when compared to the exchange rate.23 If the fraction of bound ligands (pb) is small i.e., , its effect on the overall R2 is proportional to pb. The equation for the observed R2 (R2,obs) can then be approximated as24
| (1) |
where koff is the off-rate and . By using a reversible inhibitor of trypsin, TFBC (KD = 142 μM25) as a reporter ligand and keeping pb < 0.005 in all the experiments, the error in the exchange contribution due to the approximation was less than 1%. In the presence of protein and reporter ligand under binding without competition, pb in Equation (1) is given by26
| (2) |
where [P]tot and [R]tot are respectively the total protein and reporter ligand concentrations, and KD,r is the dissociation constant of the reporter ligand. When the reporter ligand is competing with the ligand of interest, pb is given in ref.27, additionally depending on the total concentration of the ligand of interest [C]tot and its dissociation constant KD,c (see Eq. S2 in the Supporting Information).
To determine KD,c from the competitive binding experiments, the reference parameter R2,f was measured from the CPMG experiments of free hyperpolarized TFBC, and R2,b* was obtained by fitting R2,obs of hyperpolarized TFBC in the presence of trypsin at different concentrations to Equation (1). The total protein and ligand concentrations were determined based on the signal intensities of the reference compounds in the flow cells measured after the hyperpolarized experiment (Figure S3).
The KD,c value was calculated first by obtaining the value of pb using the fitted R2,obs from the CPMG experiments, and then by solving the equations for the binding equilibrium for each channel individually (Eqs. S2−S6).15 Alternatively, the KD,c value was determined by fitting to the CPMG data sets from both channels at the same time. For this purpose, the decay rate of R2,obs was expressed as a function of pb given by Eq. S2, and a single value of optimized KD,c was obtained by minimizing the sum of squared residuals of the predicted values and the measured data from Channel 1 and 2.
The KD,c value is subject to the errors associated with the determinations of the values for the relaxation rates and the concentrations of proteins and ligands. This error was estimated using a Monte Carlo method. For each CPMG data set, 105 simulated groups of data were created, comprising normally distributed, randomized parameter values for R2,f, R2,b*, R2,obs, [P]tot, [R]tot, and [C]tot. For R2,f and R2,b*, averages and standard deviations were obtained from the separate CPMG experiments. The distribution of R2,obs was based on the fitted value and fitting error . The concentration values were used as determined from the NMR measurements and associated with an estimated 5% error. KD,r was a constant. Since at the competitive equilibrium, the measured decay rate should be between the rate of free reporter ligand and that under binding without competition, combinations with R2,obs values that are closer than to those limits were discarded. From the remaining groups of parameters, the resulting distribution of KD,c was calculated. All data analysis was conducted using the Matlab program (MathWorks, Natick, MA).
Results and Discussion
Two simultaneously acquired single-scan CPMG data sets using the hyperpolarized TFBC as a reporter ligand are shown in Figure 2. Here, the ligand of interest, benzylamine, was added to the protein solution in two different proportions. The ratios were chosen to enable the detection of competitive binding over a range of KD,c of up to two orders of magnitude. The hyperpolarized reporter was admixed during the sample injection at nominally equal ratio with respect to the protein. Each spectrum shown in the figure was obtained from an average of 20 successive spin-echoes acquired from the CPMG pulse train, thus indicating the time dependence of echo intensity due to R2 relaxation. With the inclusion of a single fluorinated site in the sample mixture, the echo signals in the absence of spectral resolution would in principle suffice to determine the relaxation rate. However, the spectra shown in the figure permit distinguishing the signals of the reporter TFBC at δ = −62.4 ppm from those of reference compounds, F− and TFA. TFA was not hyperpolarized and yielded signals close to the noise level, and the NMR cross-talk signal was not observed despite the signal enhancement over thousand-fold. The signal decay of the reporter is dependent on the fraction of protein-bound reporters. A slower relaxation in Channel 2, due to increased displacement of bound reporters in the presence of 20 times more of benzylamine, is clearly visible.
Figure 2.
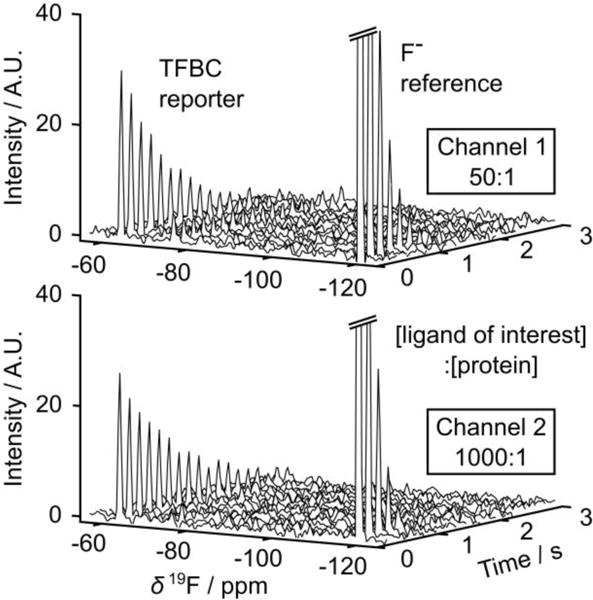
Single-scan CPMG spectra of hyperpolarized TFBC (δ = −62.4 ppm) and reference compounds, KF (δ = −118.7 ppm) and TFA (δ = −75.0 ppm) in the presence of 23.1 μM TFBC, 0.31 μM trypsin and 15.6 μM benzylamine (top, Channel 1), and 18.3 μM TFBC, 0.26 μM trypsin and 258 μM benzylamine (bottom, Channel 2). For presentation only, each spectrum shown represents the average of 20 acquired spectra.
The time dependence of the signals of TFBC competing with benzylamine, as well as in separate data sets competing with benzamidine, is shown in Figure 3. Additional repetitions are included in the supporting information (Figures S4–S5). The noise appears larger than in the spectra in Figure 2, because the peak intensity from each echo was plotted without averaging in Figure 3. In order to assess the binding affinity of the competing ligand, a measurable change in the population of bound reporters is required. This change can be quantified by comparing the difference between R2,obs and R2,f to that of the limiting values of R2,f and R2,obs in the absence of competition. It was previously shown that the ratio of these differences, here designated as α, is equal to the ratio of pb under competitive binding to that under binding without competition.15 In Figure 3, it is apparent that the R2,obs values from Channel 1 with benzamidine (Figure 3a) and Channel 2 with benzylamine (Figure 3d) appear mid-way between the two limits, which are reflected by the values of α close to 0.5. This indicates a partial displacement of the bound reporters due to binding competition. The other two data sets either show almost complete (Figure 3b) or almost negligible displacement (Figure 3c), indicated by the value of α close to zero or unity, respectively. The most accurate KD,c can be obtained when 50% displacement occurs, i.e., α ≈ 0.5.15,26 Hence, the multiplexed detection in both cases includes one set of concentrations expected to allow for reliable determination of KD,c.
Figure 3.
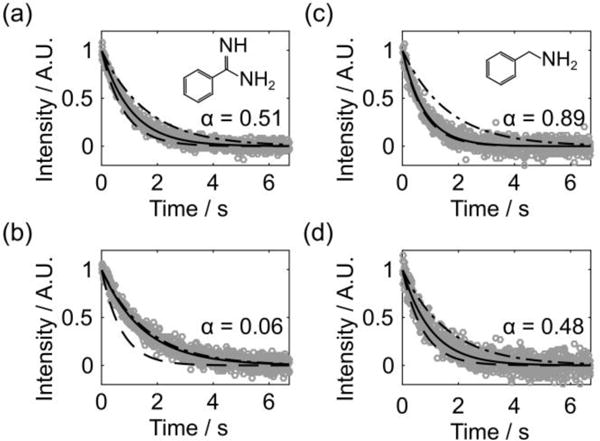
R2 relaxation decays of hyperpolarized TFBC competing with benzamidine (a and b) and benzylamine (c and d) obtained from the CPMG experiments. The first row (second row) displays the data acquired from Channel 1 (Channel 2). The maximum intensity of TFBC signals from 840 successive echoes are denoted by gray circles. The fitted R2,obs curves are indicated by solid lines. Signal decays of R2 limiting curves corresponding to free TFBC (R2,f = 0.62 s−1) (− – −) and maximally bound TFBC, calculated from R2,b*, (− −) are indicated. The α value shown in each graph reflects the ratio of pb with respect to pb in the absence of competition. The values of sample concentrations and fitted R2,obs are summarized in Tables S1−S2.
The limiting value of R2,obs for each data set was calculated using the values of R2,f = 0.62 ± 0.01 s−1 (Figure S6), and R2,b* = 388 ± 33 s−1 and 464 ± 34 s−1 for Channel 1 and 2, respectively (Figure S7 and Table S3). The difference in R2,b* of the two channels may be attributed to different RF-pulse (B1) field distributions of two coils and main field (B0) inhomogeneity. For KD,c determination, here, an individual R2,b* value was therefore applied to the data from each channel, but this difference could possibly be alleviated by using the compensated spin-echo pulse sequences.28–30
The measurement of data sets at two concentrations for each inhibitor allows for the possibility to fit a single KD,c parameter that minimizes residuals to both data sets, or alternatively to analyze each data set separately. Values for KD,c obtained by the two methods are shown on a logarithmic scale in Figure 4. Those by the group fitting were plotted using open symbols and those by the individual fitting using closed symbols. The horizontal and vertical coordinates of the closed symbols represent the KD,c values determined by the individual fitting for Channel 1 and 2. These values deviate from the diagonal, which indicates that the KD,c values from the two channels are different. Presumably due to the non-optimal value of α, the resulting KD,c for benzylamine from Channel 1 is underestimated.
Figure 4.
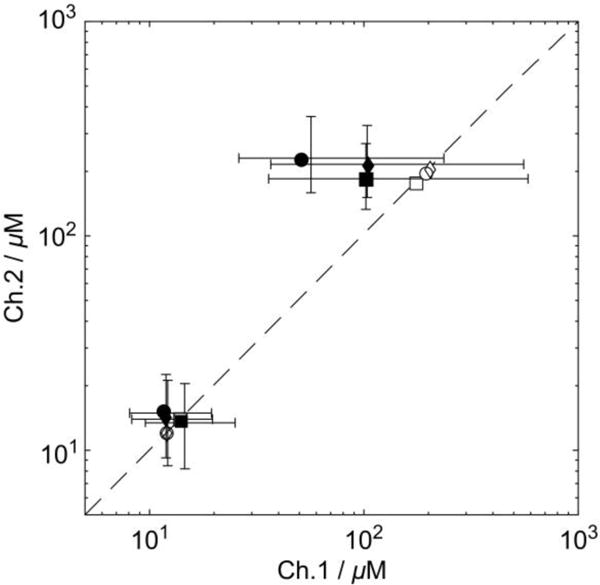
Numerical values of KD,c of benzamidine (lower left cluster) and benzylamine (upper right cluster) determined from three trials (‘◊’,‘□’,‘○’). Those obtained by the individual fitting are indicated using closed symbols. The error bars indicate the intervals containing 95% of the KD,c values simulated using Monte Carlo method. The empty symbols on diagonal represent the KD,c values obtained by the group fitting. All the values are summarized in Tables S4−S5.
To further assess the source of errors in KD,c, propagation due to uncertainty of the measured parameters were analyzed using Monte Carlo simulations. A source of error would be due to concentration variations between the different injections, which were found to be within approximately 10%. The accuracy of concentration values was, however, increased to 5% by determining the final concentrations using a one-pulse reference experiment after the decay of the hyperpolarization. Other sources of errors were determined as described in the Experimental Section. The result from the Monte Carlo simulations is indicated with horizontal and vertical bars at a confidence level of 95% for the resulting KD,c values from Channel 1 and 2. As expected, the errors evaluated for the KD,c values of benzylamine from Channel 1 were greater than those from Channel 2 where α was close to the optimal value. When a data set with near optimal α is available, generally, the individual fitting results in a more accurate KD,c, as shown by the error ranges. For these more optimal cases, the confidence intervals of benzamidine (Channel 1) and benzylamine (Channel 2) determined from a single data set were typically (8.3 μM, 20 μM) and (151 μM, 328 μM), respectively. The average KD,c values over the three separate experiments were 12.6 ± 1.4 μM and 207 ± 22.3 μM, respectively, where the ranges indicate standard deviation. The spread of values reflected in these standard deviations is consistent with the confidence intervals for the single measurement calculated above. The average KD,c values are also in good agreement with previously reported values (benzamidine, 16 ± 2 μM; benzylamine, 218 ± 43 μM at 50 mM Tris, pH 8.0, 304 K).15 Figure 4 further indicates that for the data sets obtained here, the group fitting method (open symbols) provided values of KD,c located within the confidence intervals of the individual fits, close to the individual values of the more accurate channel. If the data sets comprise no optimal sets of concentrations, the group fitting may be applied effectively to obtain a more reliable KD,c value.
Using the described multiplexing DNP-NMR system, two ligands of interest that have the binding affinities differing by an order of magnitude were successfully screened without changing the experimental conditions. The use of two channels broadens the target range of KD,c, reducing the possibility of missing the detection of binding due to non-optimal concentrations. One experiment taken to obtain a KD,c value required less than 25 min. Most of the time was used for achieving the hyperpolarization, while only a few minutes were required to prepare the dissolution by using the flow system, which obviates the steps of cleaning and loading NMR tubes between the sequential dissolutions. Considering that multiple experiments are often desired to obtain a reliable KD value, the overall experimental time can be reduced by two-fold owing to the capability of the parallelization. Besides simplifying the procedures, the flow system allows to achieve a wide range of volume ratios of non-HP to HP samples by adjusting the injection parameters. This ratio can be 1:1 or higher, which is not attainable with the gas-driven injection, where the volume of the secondary sample preloaded into the NMR tube is limited to about 10% of the final injection volume.31 Therefore, when injecting into the flow cell, the protein samples can be prepared less concentrated, avoiding protein aggregation or solubility issues. One drawback of this flow system is, on the other hand, a relatively low sample recovery fraction compared to the gas-driven injection method, likely caused by distribution in the tubing due to laminar flow. Here, the mass fraction of non-HP sample recovered was 20%, and the amount of HP sample was 5% in each flow cell. Sample recovery may be increased in the future by using a driving fluid that is immiscible with the samples.32 The latter number also indicates that the DNP polarizer has a capacity of polarizing a larger amount of sample than what is needed for the typical binding experiment. Adding several additional coils to the NMR probe could provide further time savings. Despite the use of multiple coils, the delivery of the sample required no more than two high-pressure pumps, for the HP and non-HP components, respectively. Using additional flow splitters, a larger number of cells could be filled without requiring additional pumps that would result in further expense and complexity.
Apart from the determination of KD,c as shown here, the ability to parallelize experiments would also be useful for other applications requiring titrations. For example, this design could be used to investigate inhibitors with allosteric properties, the importance of which have been highlighted in drug design among others targeting kinases.33,34
Conclusions
In conclusion, we have demonstrated a parallelized screening method using a multiplexing 19F NMR probe and spectrometer in combination with D-DNP. This method to acquire R2 relaxation rates from a single hyperpolarized aliquot of reporter ligands mixed with two different ratios of ligands of interest to target proteins can reduce the total experimental time and extend the detectable range of KD,c in a single scan. Since the affinity range of putative ligands is often unknown, the applicability of D-DNP NMR in drug discovery can be improved by the multiplexed screening experiments shown here.
Supplementary Material
Acknowledgments
The authors thank Hsueh-Ying Chen for useful discussions. Financial support from the National Institutes of Health (Grant R21-GM107927) and the Welch Foundation (Grant A-1658) is gratefully acknowledged.
References
- 1.Dalvit C. Drug Discov Today. 2009;14(21–22):1051–1057. doi: 10.1016/j.drudis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 2.Lepre CA, Moore JM, Peng JW. Chem Rev. 2004;104(8):3641–3676. doi: 10.1021/cr030409h. [DOI] [PubMed] [Google Scholar]
- 3.Kovacs H, Moskau D, Spraul M. Prog Nucl Magn Reson Spectrosc. 2005;46(2–3):131–155. [Google Scholar]
- 4.Lee JH, Okuno Y, Cavagnero S. J Magn Reson. 2014;241:18–31. doi: 10.1016/j.jmr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ardenkjær-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, Golman K. Proc Natl Acad Sci USA. 2003;100(18):10158–10163. doi: 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Zeng H, Ruedisser S, Gossert AD, Hilty C. J Am Chem Soc. 2012;134(42):17448–17451. doi: 10.1021/ja308437h. [DOI] [PubMed] [Google Scholar]
- 7.Lerche MH, Meier S, Jensen PR, Baumann H, Petersen BO, Karlsson M, Duus JØ, Ardenkjaer-Larsen JH. J Magn Reson. 2010;203(1):52–56. doi: 10.1016/j.jmr.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Buratto R, Bornet A, Milani J, Mammoli D, Vuichoud B, Salvi N, Singh M, Laguerre A, Passemard S, Gerber-Lemaire S, Jannin S, Bodenhausen G. ChemMedChem. 2014;9(11):2509–2515. doi: 10.1002/cmdc.201402214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Min H, Sekar G, Hilty C. ChemMedChem. 2015;10(9):1559–1563. doi: 10.1002/cmdc.201500241. [DOI] [PubMed] [Google Scholar]
- 10.Lee Y, Zeng H, Mazur A, Wegstroth M, Carlomagno T, Reese M, Lee D, Becker S, Griesinger C, Hilty C. Angew Chem Int Ed. 2012;51(21):5179–5182. doi: 10.1002/anie.201201003. [DOI] [PubMed] [Google Scholar]
- 11.Dalvit C, Flocco M, Knapp S, Mostardini M, Perego R, Stockman BJ, Veronesi M, Varasi M. J Am Chem Soc. 2002;124(26):7702–7709. doi: 10.1021/ja020174b. [DOI] [PubMed] [Google Scholar]
- 12.Dalvit C, Fagerness PE, Hadden DTA, Sarver RW, Stockman BJ. J Am Chem Soc. 2003;125(25):7696–7703. doi: 10.1021/ja034646d. [DOI] [PubMed] [Google Scholar]
- 13.Heath RE, Dykes GM, Fish H, Smith DK. Chem – Eur J. 2003;9(4):850–855. doi: 10.1002/chem.200390094. [DOI] [PubMed] [Google Scholar]
- 14.Jahnke W, Floersheim P, Ostermeier C, Zhang X, Hemmig R, Hurth K, Uzunov DP. Angew Chem Int Ed. 2002;41(18):3420–3423. doi: 10.1002/1521-3773(20020916)41:18<3420::AID-ANIE3420>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.Kim Y, Hilty C. Angew Chem Int Ed. 2015;54(16):4941–4944. doi: 10.1002/anie.201411424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu S, Larson PEZ, VanCriekinge M, Leach AM, Park I, Leon C, Zhou J, Shin PJ, Reed G, Keselman P, von Morze C, Yoshihara H, Bok RA, Nelson SJ, Kurhanewicz J, Vigneron DB. Magn Reson Imaging. 2013;31(4):490–496. doi: 10.1016/j.mri.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batel M, Krajewski M, Weiss K, With O, Däpp A, Hunkeler A, Gimersky M, Pruessmann KP, Boesiger P, Meier BH, Kozerke S, Ernst M. J Magn Reson. 2012;214(1):166–174. doi: 10.1016/j.jmr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Ramirez MS, Walker CM, Chen Y, Yi S, Sandulache VC, Lai SY, Bankson JA. J Magn Reson. 2015;260:20–27. doi: 10.1016/j.jmr.2015.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kodibagkar VD, Conradi MS. J Magn Reson. 2000;144(1):53–57. doi: 10.1006/jmre.2000.2052. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Wolters AM, Malawey PV, Sweedler JV, Webb AG. Anal Chem. 1999;71(21):4815–4820. doi: 10.1021/ac990855y. [DOI] [PubMed] [Google Scholar]
- 21.Chen H-Y, Hilty C. ChemPhysChem. 2015;16(12):2646–2652. doi: 10.1002/cphc.201500292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meiboom S, Gill D. Rev Sci Instrum. 1958;29(8):688–691. [Google Scholar]
- 23.Vulpetti A, Dalvit C. ChemMedChem. 2013;8(12):2057–2069. doi: 10.1002/cmdc.201300351. [DOI] [PubMed] [Google Scholar]
- 24.Vulpetti A, Hommel U, Landrum G, Lewis R, Dalvit C. J Am Chem Soc. 2009;131(36):12949–12959. doi: 10.1021/ja905207t. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y, Zeng H, Ruedisser S, Gossert AD, Hilty C. J Am Chem Soc. 2012;134(42):17448–17451. doi: 10.1021/ja308437h. [DOI] [PubMed] [Google Scholar]
- 26.Fielding L. Prog Nucl Magn Reson Spectrosc. 2007;51(4):219–242. [Google Scholar]
- 27.Wang Z-X. FEBS Lett. 1995;360(2):111–114. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 28.Gullion T, Baker DB, Conradi MS. J Magn Reson 1969. 1990;89(3):479–484. [Google Scholar]
- 29.Shaka AJ, Rucker SP, Pines A. J Magn Reson 1969. 1988;77(3):606–611. [Google Scholar]
- 30.Zweckstetter M, Holak TA. J Magn Reson. 1998;133(1):134–147. doi: 10.1006/jmre.1998.1437. [DOI] [PubMed] [Google Scholar]
- 31.Bowen S, Hilty C. Phys Chem Chem Phys. 2010;12(22):5766. doi: 10.1039/c002316g. [DOI] [PubMed] [Google Scholar]
- 32.Harris T, Bretschneider C, Frydman L. J Magn Reson. 2011;211(1):96–100. doi: 10.1016/j.jmr.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noble MEM. Science. 2004;303(5665):1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 34.Hantschel O, Grebien F, Superti-Furga G. Cancer Res. 2012;72(19):4890–4895. doi: 10.1158/0008-5472.CAN-12-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.