

Abstract
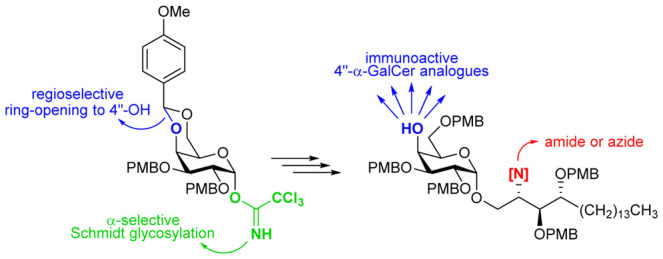
A synthesis strategy for the swift generation of 4″-modified α-galactosylceramide (α-GalCer) analogues is described, establishing a chemical platform to comprehensively investigate the structure–activity relationships (SAR) of this understudied glycolipid part. The strategy relies on a late-stage reductive ring-opening of a p-methoxybenzylidene (PMP) acetal to regioselectively liberate the 4″-OH position. The expediency of this methodology is demonstrated by the synthesis of a small yet diverse set of analogues, which were tested for their ability to stimulate invariant natural killer T-cells (iNKT) in vitro and in vivo. The introduction of a p-chlorobenzyl ether yielded an analogue with promising immunostimulating properties, paving the way for further SAR studies.
Keywords: α-Galactosylceramide, KRN7000, CD1d, Th1/Th2, cytokine secretion, immunomodulators
For over two decades, α-galactosylceramide (α-GalCer or KRN7000; 1) has been serving as a lead structure for the development of new glycosphingolipids targeting the immune system.1−4 α-GalCer is a synthetic glycolipid resulting from the structural optimization of agelasphins, a class of amphiphilic natural products isolated from the marine sponge Agelas mauritianus.5,6 It is composed of a polar d-galactose unit, α-anomerically linked to a lipophilic ceramide tail. This ceramide, in turn, is built from d-ribo-phytosphingosine, which is N-acylated at C2′ with cerotic acid (hexacosanoic acid).
α-GalCer binds to the major histocompatibility complex (MHC) class I-like glycoprotein CD1d, associated with the membrane of antigen-presenting cells (APCs).7 This binary CD1D-glycolipid complex is presented to the T-cell receptor (TCR) of invariant natural killer T-cells (iNKT cells), evoking simultaneous release of T-helper 1 (Th1) and T-helper 2 (Th2) cytokines.8,9 The pro-inflammatory Th1-cytokines, such as interferon-γ (IFN-γ), are involved in antitumor, antiviral, and antibacterial effects, whereas the anti-inflammatory Th2-cytokines, such as interleukin 4 (IL-4), counteract the development of autoimmune diseases. However, the antagonizing effect of both cytokine types severely hampers clinical potential of α-GalCer as an immunomodulator. This renders research toward new analogues with an improved immunological profile, slanting toward secretion of either Th1- or Th2-cytokines, highly relevant.
The list of new α-GalCer analogues continues to grow, with much attention being devoted to modifications of the hydroxyl groups of the galactose unit, thereby gradually revealing more of the SAR of this key glycolipid part. Modifications at the 2″-position lead to a complete disappearance of antigenicity, due to abrupt disturbance of a vital hydrogen bonding interaction with Gly96α of the TCR.10,11 A sulfate group can be substituted for the 3″-OH group.12 The SAR of the 6″-position has been extensively studied, mainly for two reasons. First, the 6″-OH is the only primary hydroxyl group of the sugar part, which from a chemoselective point of view implies that straightforward modification of this position is possible. Indeed, our laboratory has already explored the mild yet powerful regioselective benzylidene ring-opening route to access the 6″-position for the synthesis of fucosyl, galacturonic acid, triazole, carbamate, and urea analogues.13−15 Second, and more importantly, this hydroxyl group does allow for modifications due to the absence of any major interactions with either CD1d or the TCR,16 and this has given rise to an array of new immunoactive analogues.17−22
In contrast to this, only few 4″-analogues of α-GalCer are known (Figure 1), and as a consequence, the SAR of this position has been poorly investigated. Crystallographic studies have shown that the 4″-OH forms a hydrogen bond to the main chain carbonyl group of Phe29α of the TCR, indicating that it acts as a hydrogen bond donor.16 4″-Deoxygenation leads to a less active analogue (2) as compared to α-GalCer, yet recognition by the TCR is not severely disturbed.23 Derivatization of the 4″-OH as O-methyl ether (3) or O-ethanol ether (4) gives slightly less potent analogues, whereas the N-acetyl analogue (5) is a significantly weaker antigen.24 Additionally, a couple of active analogues bearing an aromatic ring on the 4″-position have been synthesized (6–8).25 Inversion of the 4″-OH gives α-glucosylceramide (α-GlcCer), which is slightly less active as compared to α-GalCer.7
Figure 1.

Structural formula of 1 and some reported 4″-analogues.
To explore the SAR more thoroughly, a reliable and scalable synthesis route is highly needed. Here, we present the synthesis of two powerful precursors (16 and 19) with a free 4″-OH, permitting the fast generation of new 4″-α-GalCer analogues in a late stage of the synthesis. This divergent strategy uses PMB ethers as hydroxyl protecting groups, in contrast to most other syntheses, which use benzyl groups. Although they are widely used because of their easy introduction and relative inertness toward a plethora of conditions, benzyl groups suffer from the major drawback that by deprotection via catalytic hydrogenolysis some medicinally interesting functionalities, such as alkenes, alkynes, thioethers, naphthalenes, (iso)quinolines, furans, thiophenes, cyclopropanes, and chloroarenes, might be (partly) reduced. This unavoidably restricts the structural diversity of the analogues. PMB ethers, however, can be cleaved under mild and widely tolerated conditions, therefore permitting a broader substrate scope.26
The previously reported trichloroacetimidate donor (13)27 was synthesized via an improved scalable route from cheap α-d-galactose pentaacetate (9) (Scheme 1). Introduction of the p-thiotolyl moiety as an anomeric protecting group and subsequent deacetylation under Zemplén conditions was followed by installation of the p-methoxybenzylidene acetal and protection of the remaining hydroxyl groups as PMB ethers to yield the fully protected intermediate 11. Attempts to remove the p-thiotolyl group with N-iodosuccinimide (NIS) in acetone/H2O 10:1 resulted in partial conversion of the p-methoxybenzylidene acetal to an isopropylidene acetal. This inconvenience was overcome by performing the reaction in acetonitrile/H2O 10:1, giving hemiacetal 12 as a mixture of anomers (α/β ≈ 6:4 at 25 °C in CDCl3) in a nearly quantitative yield. Finally, 12 was converted to the corresponding α-trichloroacetimidate under thermodynamic control, furnishing donor 13 in an overall yield of 75% over six steps. All intermediates, except for 13, can be purified by simple crystallization from a suitable solvent system.
Scheme 1. Synthesis of Trichloroacetimidate Donor 13.

Reagents and conditions: (a) p-thiocresol, BF3·Et2O, CH2Cl2, 0 °C (94%); (b) NaOMe, MeOH, rt (quant.); (c) p-anisaldehyde dimethylacetal, CSA, CH2Cl2, 4 Å MS, rt (87%); (d) NaH, PMBCl, TBAI, DMF, 0 °C to rt (98%); (e) NIS, MeCN, H2O, 0 °C (99%); (f) Cl3CCN, DBU, CH2Cl2, 0 °C (94%).
Efforts were undertaken to perform the glycosylation of the known acceptor 14(28) with the thioglycoside donor 11, as this would shorten the synthesis route. However, the application of neither benzenesulfinyl morpholine (BSM),29 nor NIS or copper(II) triflate30 as thiophilic promoters proved to be successful. These highly electrophilic conditions, typically used in thioglycosylations,31 were found to be incompatible with the p-methoxybenzylidene acetal. Indeed, p-methoxybenzylidene and p-thiotolyl cleavage were observed as the main side reactions.
Glycosylation was successful at the trichloroacetimidate stage through application of the “inverse protocol” (Scheme 2).32 By slow addition of donor 13 to a solution of acceptor 14 and BF3·OEt2 as the promoter at −20 °C, the desired glycoside 15 was obtained as α-anomer only in 85% yield. The use of trimethylsilyl trifluoromethanesulfonate (TMSOTf) as glycosylation promoter led to silylation of the acceptor, while the common protocol with BF3·OEt2, wherein the promoter is added to a solution of donor and acceptor, gave the glycoside in lower yields (30–50%) due to more extensive decomposition of the donor before glycosylation. The high stereoselectivity observed in this reaction is caused by a well-known conformational effect exhibited by the 4″,6″-O-acetal, forming a cis-decalin-like system with the galactose ring.33
Scheme 2. Glycosylation of 13 with 14 and Subsequent Regioselective Ring-Opening of Glycoside 15.

Reagents and conditions: (a) 14, BF3·Et2O, THF, Et2O, 4 Å MS, −20 °C (85%); (b) DIBALH, toluene, −80 °C (89%); (c) BaO, Ba(OH)2, MeI, DMF, rt (93%).
Next, attempts were made to regioselectively open the p-methoxybenzylidene acetal in order to liberate the 4″–OH function. Experiments involving BH3·THF/Cu(OTf)2,34 BH3·THF/nBu2BOTf35 or PhBCl2/TESH36 as reducing agents mainly gave rise to degradation of the p-methoxybenzylidene acetal and the glycosidic bond. A more successful approach was the use of diisobutylaluminum hydride (DIBALH) in toluene,37 while keeping the temperature at −80 °C to avoid azide reduction (Scheme 2). This afforded azido alcohol 16 as a single regioisomer in 89% yield. The structure of 16 was unambiguously proven by derivatization as the corresponding methyl ether (17) and analysis of the relevant cross peaks in the HSQC and HMBC spectra (see Supporting Information).
Azido alcohol 16 was subjected to azide reduction under the classical Staudinger conditions with PMe3, followed by EDC-mediated amide formation with hexacosanoic acid (cerotic acid). However, it was observed that the intermediate iminophosphorane was highly stable, and even with concentrated sodium hydroxide at elevated temperatures its hydrolysis proceeded sluggishly, providing amide 19 in a rather low yield (43% over 2 steps). To reduce the azide in a more efficient way, we turned our attention to hydrogen sulfide in a pyridine/H2O mixture. This readily furnished the amine, which was immediately acylated with N-succinimidyl hexacosanoate (NSHC, 18) under basic conditions to deliver amide 19 in 86% yield over 2 steps (Scheme 3). To drive the reaction to completion within a reasonable amount of time, the amide formation was carried out at 70 °C.
Scheme 3. Amide formation of 16.

Reagents and conditions: (a) (i) H2S, pyridine, H2O, rt; (ii) 18, Et3N, THF, 70 °C (86% over 2 steps).
Both 16 and 19 were now suited for late-stage diversification. Alkylation of the 4″-OH had to be performed at the azide stage since methylation of 19 with methyl iodide and BaO/Ba(OH)2 in DMF yielded an unseparable mixture of O- and N-alkylated products in a 4:1 ratio (as determined via 1H NMR spectroscopy). Thus, alkylation of 16 with p-chlorobenzyl bromide gave derivative 22 (Scheme 4). Both 17 and 22 were subjected to H2S-mediated azide reduction and subsequent amide formation. The final PMB ether cleavage was performed using HCl in 1,4-dioxane, swiftly delivering analogues 21 and 24. Anisole was added in large excess (10 equiv) as a scavenger for the highly reactive p-methoxybenzyl carbocation.
Scheme 4. Synthesis of O-Alkylated Analogues 21 and 24.

Reagents and conditions: (a) (i) H2S, pyridine, H2O, rt; (ii) 18, Et3N, THF, 70 °C (78% over 2 steps); (b) HCl, 1,4-dioxane, anisole, rt (32%); (c) p-chlorobenzyl bromide, BaO, Ba(OH)2, DMF, rt (92%); (d) (i) H2S, pyridine, H2O, rt; (ii) 18, Et3N, THF, 70 °C (66% over 2 steps); (e) HCl, 1,4-dioxane, anisole, rt (30%).
Besides alkylation, some other chemistries were explored to create a diverse set of analogues (Scheme 5). Carbamoylation of 19 with 1-naphthyl isocyanate delivered, after deprotection, naphthyl carbamate analogue 26. This analogue structurally resembles the 6″-naphthyl urea, which exerts a strong Th1-bias as a result of the naphthyl moiety occupying an additional binding pocket in CD1d.18 Oxidation of the alcohol in 16 using a Swern oxidation delivered the corresponding ketone, which, without intermediate purification, smoothly underwent a mild Julia–Kocienski olefination with 1-methyl-2-(methylsulfonyl)-1H-benzo[d]imidazole (MSBI, 27)38 to give the exocyclic methylene derivative. Following azide reduction, amide formation, and overall deprotection, analogue 29 was obtained. Finally, the OH group was converted into the corresponding azide 30 via a DPPA-mediated Mitsunobu reaction. The azide was reduced and derivatized as the corresponding naphthyl urea, eventually delivering analogue 32.
Scheme 5. Synthesis of Naphthyl Analogues 26 and 32, and Alkenyl Compound 29.

Reagents and conditions: (a) 1-naphthyl isocyanate, DMF, rt (89%); (b) HCl, 1,4-dioxane, anisole, rt (44%); (c) (i) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C; (ii) 27, KOtBu, DMF, rt; (iii) H2S, pyridine, H2O, rt; (iv) 18, Et3N, THF, 70 °C (61% over 4 steps); (d) HCl, 1,4-dioxane, anisole, rt (31%); (e) DPPA, DEAD, PPh3, THF, −20 °C to rt (74%); (f) (i) H2S, pyridine, H2O, rt; (ii) 1-naphthyl isocyanate, DMF, rt (22% over 2 steps); (g) HCl, 1,4-dioxane, anisole, rt (48%).
The set of new analogues (21, 24, 26, 29, and 32) was evaluated for its immunostimulating capacity, both in vitro and in vivo. C57BL/6 mice were injected intraperitoneally with 5 μg of the glycolipids (2.0 × 10–4 μg/kg), and the blood serum levels of the cytokines were measured by ELISA (Figure 2). IFN-γ was quantified 16 h after injection, whereas IL-4 was measured 4 h after injection. All of the analogues were able to stimulate the immune system, albeit at levels comparable to or lower than those elicited by α-GalCer (1). Although the 4″-OH acts as a hydrogen bond donor to interact with the TCR, derivatization as a simple ether is allowed. The antigenic effect is most dramatic when introducing a methyl group (21). The p-chlorobenzyl analogue 24 was found to polarize the cytokine response toward Th1, although its antigenicity was lower than that of α-GalCer. Yet, we believe that this analogue might be a good lead structure for future optimization toward highly potent Th1-polarizing α-GalCer analogues.
Figure 2.

IFN-γ and IL-4 secretion after intraperitoneal injection of 2.0 × 10–4 μg/kg of each analogues in C57BL/6 mice (5 μg/animal).
In the same way, introducing a gluco-naphthyl urea on the 4″-position (32) yields an analogue that is Th1-polarizing. Future structural studies might shed light on the mechanism underlying this Th1-polarization, but enhanced interaction with CD1d may account for this observation. The galacto-naphthyl carbamate (26), in which the naphthyl group is unlikely to interact with CD1d due to its axial configuration, is significantly less potent.
Remarkably, even when no heteroatom is present at C4″, as exemplified by the exocyclic alkene (29), immunostimulatory capacity is partly preserved. Apparently, the exocyclic alkene, which will partly flatten the galactose ring, induces a pyranose conformation that is still able to bridge between CD1d and the TCR. Further structural studies are required to gain more insight into this phenomenon.
To assess if these analogues also stimulate human iNKT cells, the latter were cocultured with HeLa CD1d cells with varying concentrations of the glycolipids. After 24 h of incubation the IFN-γ levels were determined by ELISA (Figure 3). These results reflect the observations in mice, namely, that all of the compounds are able to stimulate iNKT cells, with the methyl ether being most antigenic.
Figure 3.
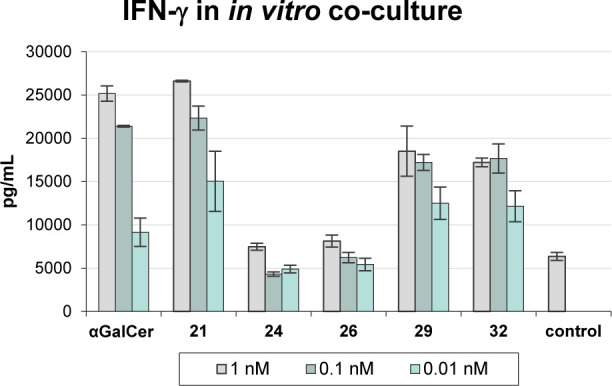
IFN-γ levels measured upon incubation of different concentrations of the analogues with human iNKT and HeLa CD1d cells.
In summary, we have developed a concise and scalable synthetic route toward two valuable precursors (16 and 19) to perform late-stage diversification of the 4″-OH position of α-GalCer. In this strategy, a triple role has been provided for the 4″,6″-O-p-methoxybenzylidene acetal: as a protecting group, as a stereocontrolling element during glycosylation, and as a structural element to enable a regioselective ring-opening. We have demonstrated the utility of this method by synthesizing a diverse set of analogues that was tested for the ability to stimulate iNKT cells both in vitro and in vivo. In both cases, the analogues were able to stimulate iNKT cells, some of them showing a clear pro-inflammatory activity due to Th1-polarization. Even though the 4″-OH is involved in binding to the TCR, we have shown that derivatization is allowed, which should inspire further biological and structural research to comprehensively explore the SAR of this glycolipid part.
Acknowledgments
Tim Courtin from the NMR & Structural Analysis Unit of Ghent University is thanked for recording the NMR spectra of the final compounds. Izet Karalic is gratefully acknowledged for purity analysis of the final compounds. Joren Guillaume and Dr. Martijn D. P. Risseeuw are acknowledged for valuable discussions.
Glossary
ABBREVIATIONS
- APC
antigen-presenting cell
- CSA
(1S)-(+)-10-camphorsulfonic acid
- DBU
1,8-diazabicyclo(5.4.0)undec-7-ene
- DEAD
diethyl azodicarboxylate
- DIBALH
diisobutylaluminum hydride
- DPPA
diphenylphosphoryl azide
- EDC
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide
- ELISA
enzyme-linked immunosorbent assay
- IFN-γ
interferon-γ
- IL
interleukin
- MHC
major histocompatibility complex
- MS
molecular sieves
- MSBI
1-methyl-2-(methylsulfonyl)-1H-benzo[d]imidazole
- NIS
N-iodosuccinimide
- NKT
natural killer T-cell
- NSHC
N-succinimidyl hexacosanoate
- PMB
p-methoxybenzyl
- PMP
p-methoxyphenyl
- SAR
structure–activity relationship
- TBAI
tetra-n-butylammonium iodide
- TCR
T-cell receptor
- TESH
triethylsilane
- Th
T-helper
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00107.
Experimental details and characterization data for the reported compounds, NMR spectra, and biological data (PDF)
Ghent University is gratefully acknowledged for financial support to this research, through a Special Research Fund grant to J.J. The work in the laboratory of M.T. is supported by the NIH (grant AI070258).
The authors declare no competing financial interest.
Supplementary Material
References
- Murphy N.; Zhu X.; Schmidt R. R. α-Galactosylceramide and analogues – important immunomodulators for use as vaccine adjuvants. Carbohydr. Chem. 2010, 36, 64–100. 10.1039/9781849730891-00064. [DOI] [Google Scholar]
- Banchet-Cadeddu A.; Hénon E.; Dauchez M.; Renault J.-H.; Monneaux F.; Haudrechy A. The stimulating adventure of KRN 7000. Org. Biomol. Chem. 2011, 9, 3080–3104. 10.1039/c0ob00975j. [DOI] [PubMed] [Google Scholar]
- Birkholz A. M.; Howell A. R.; Kronenberg M. The alpha and omega of galactosylceramides in T cell immune function. J. Biol. Chem. 2015, 290, 15365–15370. 10.1074/jbc.R115.647057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreño L. J.; Saavedra-Ávila N. A.; Porcelli S. A. Synthetic glycolipid activators of natural killer T cells as immunotherapeutic agents. Clin. Transl. Immunol. 2016, 5, e69. 10.1038/cti.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natori T.; Morita M.; Akimoto K.; Koezuka Y. Agelasphins, novel antitumor and immunostimulatory cerebrosides from the marine sponge Agelas mauritianus. Tetrahedron 1994, 50, 2771–2784. 10.1016/S0040-4020(01)86991-X. [DOI] [Google Scholar]
- Morita M.; Motoki K.; Akimoto K.; Natori T.; Sakai T.; Sawa E.; Yamaji K.; Koezuka Y.; Kobayashi E.; Fukushima H. Structure-activity relationship of α-galactosylceramides against B16-bearing mice. J. Med. Chem. 1995, 38, 2176–2187. 10.1021/jm00012a018. [DOI] [PubMed] [Google Scholar]
- Kawano T.; Cui J.; Koezuka Y.; Toura I.; Kaneko Y.; Motoki K.; Ueno H.; Nakagawa R.; Sato H.; Kondo E.; Koseki H.; Taniguchi M. CD1d-Restricted and TCR-mediated activation of Vα14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- Coquet J. M.; Chakravarti S.; Kyparissoudis K.; McNab F. W.; Pitt L. A.; McKenzie B. S.; Berzins S. P.; Smyth M. J.; Godfrey D. I. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4–NK1.1– NKT cell population. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11287–11292. 10.1073/pnas.0801631105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartabedian V. F.; Savage P. B.; Teyton L. The processing and presentation of lipids and glycolipids to the immune system. Immunol. Rev. 2016, 272, 109–119. 10.1111/imr.12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri L.; Costantino V.; Fattorusso E.; Mangoni A.; Aru E.; Parapini S.; Taramelli D. Immunostimulatory α-galactoglycosphingolipids: synthesis of a 2′-O-methyl-α-Gal-GSL and evaluation of its immunostimulating capacity. Eur. J. Org. Chem. 2004, 2004, 468–473. 10.1002/ejoc.200300512. [DOI] [Google Scholar]
- Barbieri L.; Costantino V.; Fattorusso E.; Mangoni A.; Basilico N.; Mondani M.; Taramelli D. Immunomodulatory α-galactoglycosphingolipids: synthesis of 2′-fluoro-2′-deoxy-α-galactosylceramide and an evaluation of its immunostimulating properties. Eur. J. Org. Chem. 2005, 2005, 3279–3285. 10.1002/ejoc.200500053. [DOI] [Google Scholar]
- Xing G.-W.; Wu D.; Poles M. A.; Horowitz A.; Tsuji M.; Ho D. D.; Wong C.-H. Synthesis and human NKT cell stimulating properties of 3-O-sulfo-α/β-galactosylceramides. Bioorg. Med. Chem. 2005, 13, 2907–2916. 10.1016/j.bmc.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Pauwels N.; Aspeslagh S.; Vanhoenacker G.; Sandra K.; Yu E. D.; Zajonc D. M.; Elewaut D.; Linclau B.; Van Calenbergh S. Divergent synthetic approach to 6″-modified α-GalCer analogues. Org. Biomol. Chem. 2011, 9, 8413–8421. 10.1039/c1ob06235b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels N.; Aspeslagh S.; Elewaut D.; Van Calenbergh S. Synthesis of 6″-triazole-substituted α-GalCer analogues as potent iNKT cell stimulating ligands. Bioorg. Med. Chem. 2012, 20, 7149–7154. 10.1016/j.bmc.2012.09.063. [DOI] [PubMed] [Google Scholar]
- Guillaume J.; Pauwels N.; Aspeslagh S.; Zajonc D. M.; Elewaut D.; Van Calenbergh S. Synthesis of C-5″ and C-6″-modified α-GalCer analogues as iNKT-cell agonists. Bioorg. Med. Chem. 2015, 23, 3175–3182. 10.1016/j.bmc.2015.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg N. A.; Wun K. S.; Kjer-Nielsen L.; Wilce M. C. J.; Pellicci D. G.; Koh R.; Besra G. S.; Bharadwaj M.; Godfrey D. I.; McCluskey J.; Rossjohn J. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 2007, 448, 44–49. 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- Zhou X.-T.; Forestier C.; Goff R. D.; Li C.; Teyton L.; Bendelac A.; Savage P. B. Synthesis and NKT cell stimulating properties of fluorophore- and biotin-appended 6″-amino-6″-deoxy-galactosylceramides. Org. Lett. 2002, 4, 1267–1270. 10.1021/ol025565+. [DOI] [PubMed] [Google Scholar]
- Trappeniers M.; Van Beneden K.; Decruy T.; Hillaert U.; Linclau B.; Elewaut D.; Van Calenbergh S. 6′-derivatised α-GalCer analogues capable of inducing strong CD1d-mediated Th1-biased NKT cell responses in mice. J. Am. Chem. Soc. 2008, 130, 16468–16469. 10.1021/ja8064182. [DOI] [PubMed] [Google Scholar]
- Hsieh M.-H.; Hung J.-T.; Liw Y.-W.; Lu Y.-J.; Wong C.-H.; Yu A. L.; Liang P.-H. Synthesis and evaluation of acyl-chain- and galactose-6″-modified analogues of α-GalCer for NKT cell activation. ChemBioChem 2012, 13, 1689–1697. 10.1002/cbic.201200004. [DOI] [PubMed] [Google Scholar]
- Jervis P. J.; Graham L. M.; Foster E. L.; Cox L. R.; Porcelli S. A.; Besra G. S. New CD1d agonists: synthesis and biological activity of 6″-triazole-substituted α-galactosyl ceramides. Bioorg. Med. Chem. Lett. 2012, 22, 4348–4352. 10.1016/j.bmcl.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung J.-T.; Sawant R. C.; Chen J.-C.; Yen Y.-F.; Chen W.-S.; Yu A. L.; Luo S.-Y. Design and synthesis of galactose-6-OH-modified α-galactosyl ceramide analogues with Th2-biased immune responses. RSC Adv. 2014, 4, 47341–47356. 10.1039/C4RA08602C. [DOI] [Google Scholar]
- Compton B. J.; Tang C.-w.; Johnston K. A.; Osmond T. L.; Hayman C. M.; Larsen D. S.; Hermans I. F.; Painter G. F. Synthesis and activity of 6″-deoxy-6″-thio-α-GalCer and peptide conjugates. Org. Lett. 2015, 17, 5954–5957. 10.1021/acs.orglett.5b02836. [DOI] [PubMed] [Google Scholar]
- Wun K. S.; Borg N. A.; Kjer-Nielsen L.; Beddoe T.; Koh R.; Richardson S. K.; Thakur M.; Howell A. R.; Scott-Browne J. P.; Gapin L.; Godfrey D. I.; McCluskey J.; Rossjohn J. A minimal binding footprint on CD1d-glycolipid is a basis for selection of the unique human NKT TCR. J. Exp. Med. 2008, 205, 939–949. 10.1084/jem.20072141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florence W. C.; Xia C.; Gordy L. E.; Chen W.; Zhang Y.; Scott-Browne J. P.; Kinjo Y.; Yu K. O. A.; Keshipeddy S.; Pellicci D. G.; Patel O.; Kjer-Nielsen L.; McCluskey J.; Godfrey D. I.; Rossjohn J.; Richardson S. K.; Porcelli S. A.; Howell A. R.; Hayakawa K.; Gapin L.; Zajonc D. M.; Wang P. G.; Joyce S. Adaptability of the semi-invariant natural killer T-cell receptor towards structurally diverse CD1d-restricted ligands. EMBO J. 2009, 28, 3579–3590. 10.1038/emboj.2009.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Xia C.; Nadas J.; Chen W.; Gu L.; Wang P. G. Introduction of aromatic group on 4′-OH of α-GalCer manipulated NKT cell cytokine production. Bioorg. Med. Chem. 2011, 19, 2767–2776. 10.1016/j.bmc.2010.11.061. [DOI] [PubMed] [Google Scholar]
- Greene T. W.; Wuts P. G. M.. Protective Groups in Organic Synthesis, 2nd ed.; John Wiley & Sons: New York, 1991. [Google Scholar]
- Yamamoto K.; Sato Y.; Ishimori A.; Miyairi K.; Okuno T.; Nemoto N.; Shimizu H.; Kidokoro S.; Hashimoto M. Synthesis of d-trigalacturonic acid methylglycoside and conformational comparison with its sulfur analogue. Biosci., Biotechnol., Biochem. 2008, 72, 2039–2048. 10.1271/bbb.80160. [DOI] [PubMed] [Google Scholar]
- Du W.; Gervay-Hague J. Efficient synthesis of α-galactosyl ceramide analogues using glycosyl iodide donors. Org. Lett. 2005, 7, 2063–2065. 10.1021/ol050659f. [DOI] [PubMed] [Google Scholar]
- Lu Y.-S.; Li Q.; Zhang L.-H.; Ye X.-S. Highly direct α-selective glycosylations of 3,4-O-carbonate-protected 2-deoxy- and 2,6-dideoxythioglycosides by preactivation protocol. Org. Lett. 2008, 10, 3445–3448. 10.1021/ol801190c. [DOI] [PubMed] [Google Scholar]
- Hou D.; Lowary T. L. 2,3-Anhydrosugars in glycoside bond synthesis. Application to 2,6-dideoxypyranosides. J. Org. Chem. 2009, 74, 2278–2289. 10.1021/jo900131a. [DOI] [PubMed] [Google Scholar]
- Lian G.; Zhang X.; Yu B. Thioglycosides in Carbohydrate Research. Carbohydr. Res. 2015, 403, 13–22. 10.1016/j.carres.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Schmidt R.; Toepfer A. Glycosylation with highly reactive glycosyl donors: efficiency of the inverse procedure. Tetrahedron Lett. 1991, 32, 3353–3356. 10.1016/S0040-4039(00)92704-7. [DOI] [Google Scholar]
- Yule J. E.; Wong T. C.; Gandhi S. S.; Qiu D.; Riopel M. A.; Koganty R. R. Steric control of N-acetylgalactosamine in glycosidic bond formation. Tetrahedron Lett. 1995, 36, 6839–6842. 10.1016/0040-4039(95)01442-K. [DOI] [Google Scholar]
- Shie C.-R.; Tzeng Z.-H.; Kulkarni S. S.; Uang B.-J.; Hsu C.-Y.; Hung S.-C. Cu(OTf)2 as an efficient and dual-purpose catalyst in the regioselective reductive ring opening of benzylidene acetals. Angew. Chem., Int. Ed. 2005, 44, 1665–1668. 10.1002/anie.200462172. [DOI] [PubMed] [Google Scholar]
- Hernández-Torres J. M.; Achkar J.; Wei A. Temperature-controlled regioselectivity in the reductive cleavage of p-methoxybenzylidene acetals. J. Org. Chem. 2004, 69, 7206–7211. 10.1021/jo048999m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilhas A.; Bonnaffé D. PhBCl2 promoted reductive opening of 2′,4′-O-p-methoxybenzylidene: new regioselective differentiation of position 2′ and 4′ of α-l-iduronyl moieties in disaccharide building blocks. Tetrahedron Lett. 2004, 45, 3643–3645. 10.1016/j.tetlet.2004.03.046. [DOI] [Google Scholar]
- Sarpe V. A.; Kulkarni S. S. Desymmetrization of trehalose via regioselective DIBAL reductive ring opening of benzylidene and substituted benzylidene acetals. Org. Biomol. Chem. 2013, 11, 6460–6465. 10.1039/c3ob41389f. [DOI] [PubMed] [Google Scholar]
- Ando K.; Kobayashi T.; Uchida N. Practical methylenation reaction for aldehydes and ketones using new Julia-type reagents. Org. Lett. 2015, 17, 2554–2557. 10.1021/acs.orglett.5b01049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.