

 This work is licensed under a
This work is licensed under a Abstract
46,XX disorders of sexual development (DSDs) occur rarely and result from disruptions of the genetic pathways underlying gonadal development and differentiation. We present a case of a young phenotypic male with 46,XX SRY-negative ovotesticular DSD resulting from a duplication upstream of SOX9 presenting with a painful testicular mass resulting from ovulation into an ovotestis. We present a literature review of ovulation in phenotypic men and discuss the role of SRY and SOX9 in testicular development, including the role of SOX9 upstream enhancer region duplication in female-to-male sex reversal.
Learning points:
In mammals, the early gonad is bipotent and can differentiate into either a testis or an ovary. SRY is the master switch in testis determination, responsible for differentiation of the bipotent gonad into testis.
SRY activates SOX9 gene, SOX9 as a transcription factor is the second major gene involved in male sex determination. SOX9 drives the proliferation of Sertoli cells and activates AMH/MIS repressing the ovary. SOX9 is sufficient to induce testis formation and can substitute for SRY function.
Assessing karyotype and then determination of the presence or absence of Mullerian structures are necessary serial investigations in any case of DSD, except for mixed gonadal dysgenesis identified by karyotype alone.
Treatment is ideal in a multidisciplinary setting with considerations to genetic (implications to family and reproductive recurrence risk), psychological aspects (sensitive individualized counseling including patient gender identity and preference), endocrinological (hormone replacement), surgical (cosmetic, prophylactic gonadectomy) fertility preservation and reproductive opportunities and metabolic health (cardiovascular and bones).
Background
DSDs result from disruptions to the delicate balance of the molecular pathways in the male and female sex-determining pathways. They can present at any age ranging from prenatal state, at birth (e.g., hypospadias, ambiguous genitalia, etc.) to early adulthood (delayed puberty, infertility). They can be extremely challenging owing to the associated diagnostic and ethical dilemmas. DSDs are rare and need a systematic approach to establish diagnosis through a multidisciplinary approach.
The case
A 17-year-old Caucasian male presented with a two-day history of a painful mass in left testis. There was no history of trauma, fever or weight loss. His pubertal development was normal. His medical history included right cryptorchidism with spontaneous descent at one year of age, Henoch-Schonlein purpura at four years, excision of a benign left extra adrenal ganglioneuroma at five years without recurrence, pyelonephritis at fourteen years and an upper lip capillary haemangioma.
He was the only child to a non-consanguineous couple who had three previous miscarriages. His father died aged 63 years from chronic lung disease secondary to asbestos exposure (Pedigree Chart, Fig. 1). Three first cousins had precocious puberty.
Figure 1.

Pedigree chart.
His height was 1.77 m (mother 1.75 m, father 1.68 m), weight 68.8 kg, with Tanner stage 4 pubertal development and small bilateral gynaecomastia, but no acne. By orchidometry, the left testis was 8 mL, containing a tender solid 2 cm mass and a large left hydrocele and the right testis was atrophic and 1 mL.
Investigations
Scrotal ultrasound demonstrated an atrophic right (3 mL) and a larger left testis containing a solid lesion 2 cm in diameter, suggestive of a neoplasm and a 22 mL left hydrocele. CT scan of the chest and abdomen did not show evidence of metastatic disease or abnormal lymph nodes; the seminal vesicles and prostate were normal.
He had elevated serum FSH (17.2 IU/L) and LH (11.7 IU/L), low serum testosterone (5.1 nmol/L) and normal serum SHBG (24 nmol/L). Serum αFP and HCG were negative. Semen analysis showed azoospermia. The working diagnosis was a testicular tumor on a background of possible Klinefelter syndrome.
Treatment and follow-up
Left partial orchidectomy was performed. Macroscopically the mass was a hematoma. Histopathology showed a hemorrhagic corpus luteal cyst within an ovotestis (Fig. 2A, B, C and D). The gonad, including the ovarian tissue, was contained within a tunica albuginea. The testicular tissue contained Sertoli-cell-only seminiferous tubules, interstitial Leydig cells and some Sertoli cell nodules. The ovarian tissue comprised ovarian stroma, a few primordial follicles (the only germ cells in the ovotestis), a hemorrhagic corpus luteum and several corpora albicans.
Figure 2.
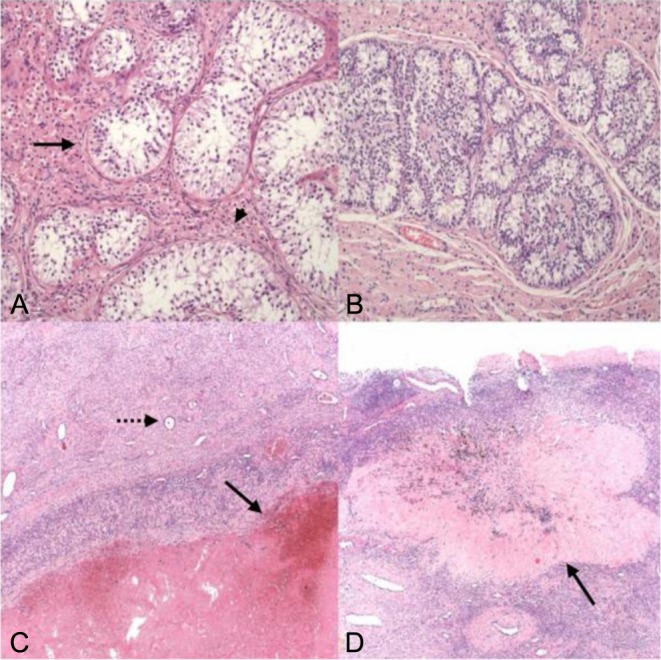
Ovo-testis histology. (A) Seminiferous tubules (arrow), devoid of germ cells or spermatogenesis, with interstitial Leydig cells (arrow head). (B) Sertoli cell nodule in testicular component (Leydig-rich background). (C) Ovarian tissue with an involuting haemorrhagic corpus luteum (arrow) and a primordial follicle (dotted arrow). (D) Ovarian tissue with an old corpus albicans (arrow), the adjacent ovarian stroma with a further primordial follicle. Image dimensions, A and B: 0.9 × 0.7 mm; C and D: 2.2 × 1.7 mm.
Karyotype was 46,XX with a notably absent SRY signal on FISH analysis. The ovarian and testicular tissue both had confirmed 46,XX chromosomes without Y signal. Radiological bone age (wrist) was consistent with chronological age. Post-operatively, both serum FSH (38.2 IU/L) and LH (22.7 IU/L) rose further with a serum total testosterone of 8.7 nmol/L.
Repeat genetic testing on DNA extracted from blood, confirmed the absence of SRY and a 46,XX karyotype (1). This classified the diagnosis as 46,XX SRY negative (−ve) ovotesticular DSD. Further analysis identified a duplication upstream of the SOX9 gene, which was confirmed by a second independent method (2).
The proband’s mother did not carry the duplication, however there was no paternal family history of DSD or infertility.
Within a multidisciplinary team, as the patient’s gender identity was unequivocally male, he was managed as a young man with hypogonadism requiring testosterone replacement and counseling regarding irreversible infertility associated with a missing Y chromosome. He was treated with injectable testosterone undecanoate (1000 mg, per 12 weeks), which achieved Tanner stage 5 virilization with gain of muscle bulk and acne. His most recent trough serum total testosterone concentration was 9.6 nmol/L with suppressed serum LH (<0.1 IU/L) and FSH (0.8 IU/L), indicating adequate replacement therapy.
Discussion
In mammals, the early gonad is bipotential and can differentiate into either a testis or an ovary. SRY is the master switch in testis determination (3) having evolved from the ancestral SOX3 (SRY type high-mobility group box gene) gene. SRY (sex-determining region on the Y chromosome) is specific to mammals (4). SRY upregulation of SOX9 induces differentiation of the bipotential gonad into a testis and, in its absence, the genital ridge develops by default into ovaries.
The only known function of SRY is to upregulate the evolutionarily more conserved SOX9 (5), an autosomal (chromosome 17) gene. SRY-induced SOX9 activation drives Sertoli cell differentiation and proliferation, a decisive step in testis development (4, 5). SRY is sufficient but not necessary for testis development, as increased SOX9 expression, is sufficient to induce testis formation, thus substituting for SRY function (6). Ectopic expression of SOX9 in an XX gonad induces testis development in transgenic XX mice (6) while SOX9 duplication can cause XX sex reversal in humans (7). Furthermore, experimentally replacing SOX9 for Sry, and Eif2s3x for Eif2s3y (spermatogonial proliferation factor) in mice produced phenotypic males with testes despite the absence of the Y chromosome. These two genes alone were sufficient to produce fertile haploid male gametes that could fertilize oocytes in vitro and produce offspring (8).
SOX9 is a pleiotropic gene with an array of gene dosage effects. Inactivation of both SOX9 alleles leads to the formation of ovaries in XY mice (9). In humans, SOX9 gene haploinsufficiency causes campomelic dysplasia (CD), an autosomal dominant skeletal dysplasia, as well as male-to-female sex reversal in about 75% of the 46,XY patients (10). SOX9 is also implicated in pancreatic development with pancreatic hypoplasia reported in patients with CD and in mouse models (11). SOX9 regulates neural crest development and ectopic expression of SOX9 in the neural tube progenitors results in neural crest-like properties (12). These links to neural crest development could explain the benign extra adrenal ganglioneuroma in this patient.
Our 46,XX SRY −ve patient had a novel duplication in the regulatory region upstream of SOX9, leading to a male phenotype (2). He had at least one ovotestis that produced sufficient testosterone to almost complete a phenotypically normal male puberty but not sufficient to suppress ovulation on more than one occasion, as shown by the apparent recent ovulation as indicated by a hemorrhagic corpus luteum as well as a corpus albicans, indicating prior ovulation. Given his male phenotype, no serum estradiol or progesterone concentrations were obtained pre-operatively nor did the hormonal levels (Table 1) confirm ovulation. It cannot be excluded that rather than true ovulation, the histological appearances of a corpus luteum and corpus albicans may have resulted from a collapsed, incompletely mature antral follicle without true LH surge-triggered ovulation. Apparent ovulation in phenotypic men has been reported previously in 46,XX, 46,XX/46,XY, 46,XX/47,XXY DSDs (Table 2, summary of case reports); however, this is the only case of a 46,XX SRY −ve ovotesticular DSD from SOX9 duplication presented in Table 2.
Table 1.
Hormonal profile pre-operatively, post-operatively and on testosterone replacement therapy.
| Results | Reference range | ||||||
|---|---|---|---|---|---|---|---|
| Hormonal panel | 15 days pre-op | 13 days pre-op | 2 months post-op | 6 months post T Rx | 14 months post T Rx | Male | Female |
| FSH (IU/L) | 13 | 17 | 38 | 3.3 | 0.8 | 1.0–12 | Follicular 3.5–12.5 |
| Mid-cycle 4.7–21.5 | |||||||
| Luteal 1.7–7.7 | |||||||
| LH (IU/L) | 8 | 12 | 23 | 16 | <0.1 | 0.6–12 | Follicular 2.4–12.6 |
| Mid-cycle 14–95 | |||||||
| Luteal 1–11.4 | |||||||
| Testosterone (nmol/L) | 4.5 | 5.1 | 8.7 | 9.6 | 9.7 | 11.5–32 | <2.8 |
| SHBG (nmol/L) | 24 | 15–80 | |||||
Table 2.
Clinical characteristics, histopathology of the gonad removed and hormonal profile, semen analysis and karyotype of phenotypic men with evidence of ovulation.
| Gallegos et al. (5) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Paper | Parvin (1) | Perez-Palacios et al. (2) | Ceci et al. (3) | Kanaka-Gantenbein et al. (4) | Case 1 | Case 2 | Case 3 | Our case |
| Age at presentation (year) | 32 | 16 | 22 | 13 | 15 | 13 | 11 | 18 |
| Presenting symptom | Pain in R testis | Bl gynaecomastia | Pain in R testis | L scrotal pain following injury | Familial, siblings | Pain and mass in L testis | ||
| Duration | 1 week | NR | 2 days | NR | NR | 2 days | ||
| Phenotype | Male | Male | Male | Male | Male | Male | Male | Male |
| H/o cryptorchidism | + | + | – | – | – | – | – | + |
| Fertility | + | NA | NR | NR | NA | NA | NA | Azoospermic |
| Relevant past history | Testicular pain age 24 years | Thelarche at 14 year | L hypospadias, b/l mammoplasty | L inguinal hernia at 6 months of age | Hypospadias, Bl gynaecomastia | Hypospadias, Bl gynaecomastia | Hypospadias | Benign extra adrenal ganglioneuroma, lip capillary haemangioma, Henoch-Schonlein purpura |
| Examination | Small, hard, tender R testis over pubic tubercle, normal Lf testis | Bl gynaecomastia, 5.5 cm penis, 2.5 mL R testis and absent Lf testis | Atrophic R testis and rubbery hard large left testis | Acne, gynaecomastia, pubic hair stage 5, normal penis, tender Lf scrotum, R testis 5–16 mL | Scrotal gonads | Scrotal gonads | Scrotal gonads | Tanner stage 4 puberty, small Bl gynaecomastia, L testis 8 mL with a tender solid 2 cm mass, large Lf hydrocele, R testis atrophic 1 mL |
| Primordial follicles | + | + | + | + | + | + | + | + |
| Corpus luteum | – | Hemorrhagic | + | Hemorrhagic | NR | NR | NR | + |
| Corpus albicans | + | – | + | NR | NR | NR | NR | + |
| Fallopian tube | + | + | + | + | – | – | – | – |
| uterus | – | + endometrial bleed | + | + | – | – | – | – |
| Ovotestis | + | + | + | – | + | + | + | + |
| Testicular tissue | – | + | Rudimentary epididymis | + in contralateral gonad | + | + | + | + |
| Leydig cells | – | + | – | – | + | + | ||
| Sertoli cells | – | + | – | + | + | + | + | |
| Seminiferous tubules | – | + | – | + | + | + | + | + |
| Spermatogenesis | – | Spermatogia | – | Spermatogia | – | – | – | – |
| FSH (IU/L) | 7.6 | 5.5 | 26.3 | 20* | 13 | |||
| LH (IU/L) | 7.4 | 11 | 9.8 | 8.2* | 8 | |||
| T (nmol/L) | 24.5 | 2.4 | 15.5 | 9.1* | 4.5 | |||
| E2 (pmol/L) | NR | 180 | 135 | 36.7* | – | |||
| P4 (nmol/L) | NR | 1.2 | NR | NR | – | |||
| Semen analysis | 15 × 106 sperm/mL 60% motile | NA | NR | NR | NA | NA | NA | Azoospermia |
| Karyotype peripheral blood | 46,XX/46,XY Chimera (81%, 19%) | 46,XX/47XXY (72%, 28%) | 46,XX/46,XY Chimera (11%, 89%) | 46,XX/47,XXY (70%, 30%) | 46,XX, no Y detected | 46,XX, no Y detected | 46,XX, no Y detected | 46, XX, SRY −ve |
| Karyotype gonadal tissue | _ | 46,XX/47XXY | _ | 46,XX/47XXY | 46,XX/47XXY | 46,XX/47XXY | 46,XX/47XXY | 46,XX, SRY −ve |
2 weeks post gonadectomy.
Bl, bilateral; E2, estradiol; L, left; NA, not applicable; NR, not reported; P4, progesterone; R, right; T, testosterone.
The presence of the Y chromosome is associated with risk of malignancies in a dysgenic gonad. The patient’s 46,XX karyotype on all cell lines tested, suggests his risk of gonadoblastoma is low. However, based on a single case report of a Sertoli cell tumor in phenotypic male with 46 XX ovotesticular DSD (13), his follow-up will include regular scrotal ultrasound surveillance.
His genetic diagnosis may have implications for the extended paternal family if the SOX9 duplication was inherited. The lack of paternal DNA precluded distinguishing between a de novo germline duplication in the patient or paternal transmission of the SOX9 duplication. The latter has been reported in a fertile 46,XY father carrying a SOX9 or upstream enhancer duplication with transmission to a child with 46,XX karyotype resulting in female-to-male sex reversal (14, 15, 16). As the paternal uncle and all his sons were fertile, there is no evidence to suggest this was paternally inherited. Previously reported SOX9 duplications are summarized in Table 3, our case is unique in his presentation with a testicular mass from ovulation and with evidence of probable multiple ovulations.
Table 3.
Summary of case reports with SOX9/upstream duplication.
| No | Paper | N | Age at presentation | Presenting complaint | Histology | Genetics | Comments |
|---|---|---|---|---|---|---|---|
| 1 | Huang et al. (6) | 1 | Infant | Ambiguous genitalia | Not performed | SOX9 duplication | – |
| 2 | Kojima et al. (7) | 5 | Infant | Ambiguous genitalia | Bl Wolffian structures + Germinal aplasia, Sertoli and Leydig cells + No ovarian tissue/uterus/tubes | SOX9 upregulation and DAX1 duplication | Sporadic cases |
| 3 | Cox et al. (8) | 3 | Adult | Infertile | Leydig, Sertoli cells + atrophies seminiferous tubulesNo spermatogenesis | ~178 kb duplication 600 kb upstream of SOX9 | 3 members (2 brothers and paternal uncle) from a family including a 46,XY father who carried the duplication |
| 4 | Vetro et al. (9) | 2 | Adult | Infertile | Germinal cell aplasia | 96 kb triplication 500 kb upstream of SOX9 | Brothers |
| 5 | Benko et al. (10) | 4 | Birth | Ambiguous genitalia | Case1: ovotestis and fallopian tubes | Case 1: ~605–695 kb duplication 353 kb upstream of SOX9 | All 3 were sporadic cases |
| Case 2: L ovary, fallopian tubes, rudimentary vagina and uterus | Case 2: ~148 kb duplication −595 to −447 kb upstream of SOX9 | Case 2: duplication paternally inherited. Duplication shared by two 46,XY brothers and not the sister | |||||
| Case 3: ovotestis, primordial follicles, seminiferous tubules, Sertoli cells, fallopian tube + | Case 3 and 4: ~762–780 kb duplication ~508 kb upstream of SOX9 | Case 3 and 4: Brothers inherited duplication from 46,XY father and 46,XY grandfather | |||||
| 6 | Xiao et al. (11) | 1 | Adult | Infertile | Not reported | ~74 kb duplication ~510–584 kb upstream of SOX9 | – |
| 7 | Lee et al. (12) | 1 | 4 years | Small testis | duplication upstream of SOX9 | SOX9 duplication | – |
| 8 | Kim et al. (13) | 3 | Case1: 25 years | Azoospermia | Not reported | 143 kb duplication ~516–659 kb upstream of SOX9 | – |
| Case 2: At birth | Ambiguous genitalia | L ovary, fallopian tubes+, R ovotestis primitive seminiferous tubules | Case 2: ~444 kb duplication ~259–703 kb upstream of SOX9 | Paternally inherited | |||
| Case 3: At birth | Ambiguous genitalia | R ovary, L dysgenic testis/ovotestis; vagina, rudimentary uterus + | Case 3: 480 kb duplication 264–744 kb upstream of SOX9 | – | |||
| 9 | Hyon et al. (14) | 3 | Adult | Infertile | Case 1 and 2: atrophic seminiferous tubules containing only Sertoli cells suggestive of testicular dysgenesis, Leydig cell hyperplasia; No spermatogenesis | Case 1: 83 kb duplication ~694 kb upstream of SOX9Case 2: 83 kb duplication ~694 kb upstream of SOX9Case 3: 140 kb duplication ~694 kb upstream of SOX9 | Case 1 and 2 were brothers |
| 10 | Vetro et al. (15) | 2 | Case 1: Adult | Infertile | N/A | Duplication upstream of SOX9 | – |
| Case 2: Infant | Ambiguous genitalia | Ovotestis, numerous oocytes, pre-pubertal seminiferous tubules | Duplication upstream of SOX9 | ||||
| 11 | Our Case | 1 | Adult | Mass in L testis | Ovotestis, hemorrhagic corpus luteum and corpus albicans, primordial follicles, Sertoli cell nodules, seminiferous tubules with no spermatogenesis | Duplication upstream of SOX9 |
Consensus statement on management of intersex disorders of the Paediatric Endocrine Society and European Society for Paediatric Endocrinology at Chicago International Consensus Conference on Intersex (Chicago consensus) of 2006 (17) defined DSDs as congenital conditions in which development of chromosomal, gonadal or anatomic sex is atypical. The proposed nomenclature emphasizes on the knowledge of karyotype, which is key in categorizing the DSDs as (1) sex chromosome DSDs (variation in the number of sex chromosomes e.g. Turner’s syndrome 45,X; Klinefelter’s syndrome 47,XXY; mixed gonadal dysgenesis, etc.), (2) 46,XY DSDs and (3) 46,XX DSDs as described in Table 4 (18). Table 4 also demonstrates categories of 46,XX DSDs that can cause virilization or male phenotype in 46,XX individuals as presented in our present case.
Table 4.
Classification of DSDs, in particular 46,XX DSDs that can result in virilization or male phenotype (18).
| DSD category | Examples | Karyotype or genes involved |
|---|---|---|
| Sex chromosome DSD | Klinefelter’s syndrome | 47,XXY and variants/mosaics |
| Turner’s syndrome | 45,XO and variants/mosaics | |
| Ovotesticular DSD | 46,XX/46,XY chimerism | |
| 46,XY DSD | Disorders of androgen action (androgen insensitivity syndrome) | AR |
| Disorders of androgen biosynthesis | CYP11A1, HSD3B2, CYP17A1, StAR, HSD17B3, SRD5A2, POR | |
| Disorders of testicular development (ovotesticular DSD, testis regression) | DHH, SRY, SF1, WT1, SOX9 | |
| Other syndromic associations of male genital development, isolated hypospadias, cryptorchidism | CXorf6, INSL3, GREAT | |
| 46,XX DSD | Disorders of ovarian development (ovotesticular DSD, testicular DSD) | SRY, RSPO1, dup SOX3, dup SOX9* |
| Disorders of androgen excess – fetal (congenital adrenal hyperplasia), feto placental (aromatase deficiency) and maternal (virilizing tumors e.g. luteomas) | HSD3B2, CYP21A2, CYP11B1, POR, CYP19 | |
| Other syndromic associations (e.g. cloacal anomalies), Mullerian agenesis/hypoplasia, uterine abnormalities, vaginal atresia (e.g. McKusick-Kaufman), labial adhesions | MKKS |
Gene duplication detected in the current case of 46,XX karyotype in a young phenotypic male.
Management of DSDs should integrate the medical aspects of care with the psychosocial needs of the patient. Currently, there are no recommendations for sex assignment in neonates who have DSD. Any surgical procedure in children that leads to irreversible change must be considered with utmost caution (18). Ideally, the surgical procedure should also aim to preserve fertility in the most optimal way. In general, individuals with 46,XY DSD have an increased risk of malignancy may need prophylactic gonadectomy if indicated. Treatment is ideal in a multidisciplinary setting with considerations to genetic (implications to family and including reproductive recurrence risks), psychological aspects (sensitive individualized counseling including patient gender identity and preference), endocrinological (hormone replacement), surgical (cosmetic, prophylactic gonadectomy) fertility preservation, reproductive opportunities and metabolic health (cardiovascular and bones).
There are still uncharacterized genes causing DSD. Such DSDs are very rare and require a careful systematic, sensitive approach to diagnosis and management of the diagnostic and ethical challenges. Clinical assessment with karyotype (including SRY expression) and subsequent determination of the presence or absence of Müllerian structures are essential early investigations in any case of DSD. The sophisticated genetic diagnoses now feasible should be undertaken as the basis for genetic counseling of the extended family in any more complex cases.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
A written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
The article was conceived and written by N Shankara Narayana under the supervision of D J Handelsman and S M Twigg. A M Kean, A Vasilaras, L Ewans and G Watson were involved in the clinical care and organized the relevant investigations and revision of the manuscript. T Ohnesorg, K L Ayers and A Sinclair performed the genetics investigations of the diagnosis and contributed to the manuscript.
References
- 1.Ohnesorg T, Turbitt E, White SJ. 2011. The many faces of MLPA. Methods in Molecular Biology 687 193–205. ( 10.1007/978-1-60761-944-4_13) [DOI] [PubMed] [Google Scholar]
- 2.Ohnesorg T, van den Bergen JA, Belluoccio D, Shankara-Narayana N, Kean A-M, Vasilaras A, Ewans L, Ayers KL, Sinclair AH. 2017. A duplication in a patient with 46,XX ovo-testicular disorder of sex development refines the SOX9 testis-specific regulatory region to 24 kb. Clinical Genetics. Epub ahead of print. ( 10.1111/cge.12976) [DOI] [PubMed] [Google Scholar]
- 3.Kashimada K, Koopman P. 2010. Sry: the master switch in mammalian sex determination. Development 137 3921–3930. ( 10.1242/dev.048983) [DOI] [PubMed] [Google Scholar]
- 4.Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. 1990. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346 240–244. ( 10.1038/346240a0) [DOI] [PubMed] [Google Scholar]
- 5.Jiang T, Hou C-C, She Z-Y, Yang W-X. 2013. The SOX gene family: function and regulation in testis determination and male fertility maintenance. Molecular Biology Reports 40 2187–2194. ( 10.1007/s11033-012-2279-3) [DOI] [PubMed] [Google Scholar]
- 6.Vidal VPI, Chaboissier M-C, de Rooij DG, Schedl A. 2001. Sox9 induces testis development in XX transgenic mice. Nature Genetics 28 216–217. ( 10.1038/90046) [DOI] [PubMed] [Google Scholar]
- 7.Huang B, Wang S, Ning Y, Lamb AN, Bartley J. 1999. Autosomal XX sex reversal caused by duplication of SOX9. American Journal of Medical Genetics 87 349–353. () [DOI] [PubMed] [Google Scholar]
- 8.Yamauchi Y, Riel JM, Ruthig VA, Ortega EA, Mitchell MJ, Ward MA. 2016. Two genes substitute for the mouse Y chromosome for spermatogenesis and reproduction. Science 351 514–516. ( 10.1126/science.aad1795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrionuevo F, Bagheri-Fam S, Klattig J, Kist R, Taketo MM, Englert C, Scherer G. 2006. Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biology of Reproduction 74 195–201. ( 10.1095/biolreprod.105.045930) [DOI] [PubMed] [Google Scholar]
- 10.Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovic M, Weissenbach J, Mansour S, Young ID, Goodfellow PN, et al. 1994. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372 525–530. ( 10.1038/372525a0) [DOI] [PubMed] [Google Scholar]
- 11.Seymour PA. 2014. Sox9: a master regulator of the pancreatic program. Review of Diabetic Studies 11 51–83. ( 10.1900/RDS.2014.11.51) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung M, Briscoe J. 2003. Neural crest development is regulated by the transcription factor Sox9. Development 130 5681–5693. ( 10.1242/dev.00808) [DOI] [PubMed] [Google Scholar]
- 13.Gunasegaram R, Mathew T, Ratnam S. 1981. Sertoli cell tumour in a true hermaphrodite: suggestive evidence for ectopic gonadotrophin production by the tumour. Case report. British Journal of Obstetrics and Gynaecology 88 1252–1256. ( 10.1111/j.1471-0528.1981.tb01207.x) [DOI] [PubMed] [Google Scholar]
- 14.Kim G-J, Sock E, Buchberger A, Just W, Denzer F, Hoepffner W, German J, Cole T, Mann J, Seguin JH, et al. 2015. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. Journal of Medical Genetics 52 240–247. ( 10.1136/jmedgenet-2014-102864) [DOI] [PubMed] [Google Scholar]
- 15.Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, Thomas S, Bruland O, David M, Nicolino M, et al. 2011. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. Journal of Medical Genetics 48 825–830. ( 10.1136/jmedgenet-2011-100255) [DOI] [PubMed] [Google Scholar]
- 16.Cox JJ, Willatt L, Homfray Tessa MB, Woods CG. 2011. A SOX9 duplication and familial 46,XX developmental testicular disorder. New England Journal of Medicine 364 91–93. ( 10.1056/NEJMc1010311) [DOI] [PubMed] [Google Scholar]
- 17.Lee PA, Houk CP, Ahmed SF, Hughes IA. 2006. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex Pediatrics 118 e488–e500. ( 10.1542/peds.2006-0738) [DOI] [PubMed] [Google Scholar]
- 18.Hughes IA, Houk C, Ahmed SF, Lee PA. 2006. Consensus statement on management of intersex disorders. Journal of Pediatric Urology 2 148–162. [DOI] [PubMed] [Google Scholar]