
Figure 1.
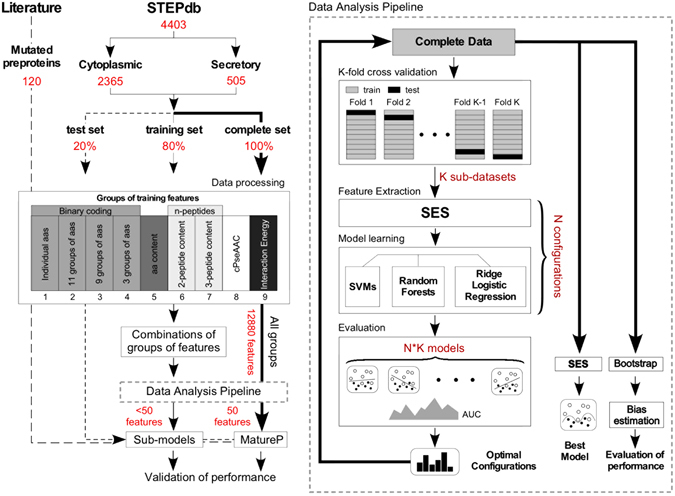
Bioinformatics pipeline of data analysis. Summary workflow of the machine learning process for the separation of secretory from cytoplasmic sequences. First secretory and cytoplasmic proteins from the E. coli K-12 proteome were collected based on the subcellular annotation in STEPdb1 (Table S1). In total, 2365 cytoplasmic and 505 secretory from eight sub-classes of the cell envelope were defined in STEPdb1 (Table S1a). 20% of the dataset (test set) along with 120 mutated preproteins that were collected from the literature, were left outside the training process and were used later on for validation. Raw data (sequences) where first processed and transformed into nine groups of training features (e.g. binary representation of amino acids, cPseAAC). The MatureP model was trained using all data and merging all training features. Data Processing pipeline: The sample set is partitioned to K folds. For each configuration (combination of algorithms and values of their hyper-parameters) and each excluded fold, a model is produced. The average performance of each configuration is then estimated and the optimal one is selected. Subsequently, the final model is trained on all the data using the best configuration. Next, using a boostrapped-based procedure the bias of the performance estimation of the final model is computed; the bias-corrected performance and the final models are returned by the pipeline.