
Fig. 7.
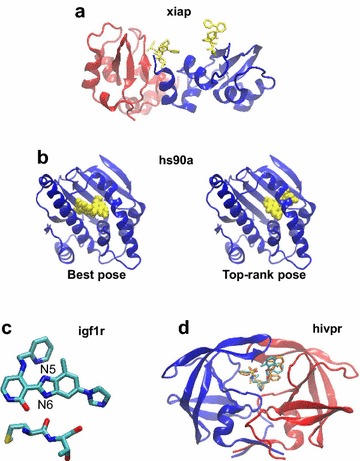
Representative structures to illustrate the top-ranking failure which is not due to scoring functions. a Structure of the crystallographic dimer of xiap, where chain A is in blue and chain B in red. The ligand is in yellow sticks. It is obvious that there are too few contacts between the protein and the ligand to allow the latter anchorage. b Structure of hs90a in blue with the ligand in yellow spheres. Left the ligand is in its best pose (RMSD = 0.32 Å), in the crystallographic binding pocket. Right the ligand is in the best off-center pose (RMSD = 9.54 Å), top-ranked by Glide, Gold and FlexX after the rescoring of the pool of poses. c Crystal structure of the ligand of igf1r with its closest protein environment. It shows the necessity of protonating the N6 atom to allow it to establish a hydrogen bond with a carbonyl from the protein backbone. The carbon atoms are in cyan, the nitrogen in blue, the oxygen in red and the sulfur in yellow. d hivpr, with chain A in blue and chain B in red. The crystal ligand is in the same color code as (c) and the top-rank pose is in orange. Their comparison shows that the discrepancies are located in the accessible to solvent part of the ligand, which has no consequence on the results. VMD software [45] was used for molecular visualization