

Muscles express giant polypeptides with kinase domains, but the in vivo significance of their catalytic activity has been unknown. Analysis of a mutant nematode that expresses the giant protein twitchin with a catalytically inactive kinase indicates that twitchin kinase inhibits muscle activity and is favored by selection.
Abstract
Muscle sarcomeres contain giant polypeptides composed of multiple immunoglobulin and fibronectin domains and one or two protein kinase domains. Although binding partners for a number of this family’s kinase domains have been identified, the catalytic necessity of these kinase domains remains unknown. In addition, various members of this kinase family are suspected pseudokinases with no or little activity. Here we address catalytic necessity for the first time, using the prototypic invertebrate representative twitchin (UNC-22) from Caenorhabditis elegans. In in vitro experiments, change of a conserved lysine (K) that is involved in ATP coordination to alanine (A) resulted in elimination of kinase activity without affecting the overall structure of the kinase domain. The same mutation, unc-22(sf21), was generated in the endogenous twitchin gene. The unc-22(sf21) worms have well-organized sarcomeres. However, unc-22(sf21) mutants move faster than wild-type worms and, by optogenetic experiments, contract more. Wild-type nematodes exhibited greater competitive fitness than unc-22(sf21) mutants. Thus the catalytic activity of twitchin kinase has a role in vivo, where it inhibits muscle activity and is likely maintained by selection.
INTRODUCTION
Sarcomeres, the fundamental units of muscle contraction, contain giant polypeptides (>700,000 Da) composed primarily of multiple copies of immunoglobulin (Ig) and fibronectin type 3 (Fn) domains, one or two protein kinase domains, and, in some proteins, highly elastic unique regions (Kontrogianni-Konstantopoulos et al., 2009). Members of this family include titin and obscurin in mammals; twitchin, UNC-89, and TTN-1 in nematodes; twitchin in molluscs; and projectin, Unc-89, sallimus, and stretchin in insects. Despite the different sources of these proteins and differences in their overall lengths, nearly all have a conserved kinase region with the same arrangement of domains surrounding the protein kinase domain, namely, Fn-NL-kinase-CRD-Ig (NL and CRD denote N- and C-terminal regulatory kinase tail domains, respectively; Mayans et al., 2013). The crystal structure of the largest segment containing a kinase domain from any of these giant polypeptides is from Caenorhabditis elegans twitchin kinase, namely, Fn-NL-kinase-CRD-Ig (von Castelmur et al., 2012). In this, both NL and CRD segments pack against the kinase domain, inhibiting its catalysis (von Castelmur et al., 2012). The 60-residue CRD tail is wedged between the two kinase subdomains, blocking sites for ATP binding, substrate recognition, and catalysis; the 45-residue NL forms a “crown” resting on the back of the hinge between the two kinase lobes.
Much is known about these proteins, especially for titin, with regard to their binding partners and roles in sarcomere assembly, muscle elasticity, and sensing and transduction of mechanical signals (Gieseler et al., 2016; Krüger and Kötter, 2016; Krüger and Linke, 2011). However, the in vivo substrates and physiological functions of the kinase domains in these proteins are largely unknown. In vitro, Aplysia twitchin kinase can phosphorylate regulatory myosin light chains (Heierhorst et al., 1996). In vitro, the C-terminal protein kinase of mouse obscurin can phosphorylate N-cadherin (Hu and Kontrogianni-Konstantopoulos, 2013). However, it is unclear whether these substrates are physiologically relevant. Moreover, it is even unknown whether some of these kinase domains might be catalytically active in vivo. In this respect, there is evidence that titin kinase (Bogomolovas et al., 2014) and kinase PK1 from Drosophila Unc-89 (Mayans et al., 2013) may be inactive pseudokinases. In addition, a number of scaffolding interactions have been reported for this family of giant kinases, which leads to the question of whether it is scaffolding and not catalysis that is of functional relevance for these kinase domains. For example, in cultured cardiomyocytes, partially open forms of titin kinase differentially bind to several proteins (autophagosome receptors p62 and Nbr1 and the E3 ubiquitin ligase MuRF2), and this leads to activity-dependent translocation of serum response factor, resulting in increased sarcomeric gene expression (Lange et al., 2005). Both protein kinase domains of nematode UNC-89 bind to SCPL-1, a CTD-type phosphatase (Qadota et al., 2008). Nematode twitchin kinase binds to and is phosphorylated by MAPKAP kinase 2 in vitro (Matsunaga et al., 2015). In Drosophila Unc-89, kinase domain 1 interacts with Ballchen (a protein kinase), and both kinase domains 1 and 2 interact with MASK (an ankyrin repeat protein; Katzemich et al., 2015).
To bring light to this debate, we explore for the first time the catalytic significance of a muscle giant kinase, twitchin kinase, in vivo. Using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene-editing technology, we created a C. elegans strain that expresses, as its sole source of twitchin, one that contains an intact but catalytically inactive protein kinase. Although nematodes expressing twitchin with a catalytically inactive kinase domain have normal sarcomere organization, remarkably, they move faster than wild-type nematodes. Our results demonstrate for the first time that twitchin kinase activity is required for normal muscle function and is involved in a pathway regulating muscle contraction.
RESULTS AND DISCUSSION
Conversion of catalytic lysine to alanine abolishes protein kinase activity and does not affect the structure of the protein kinase domain in vitro
The catalytic protein kinase domain of twitchin (TwcK) is active in in vitro phosphortransfer assays against a model peptide substrate (Lei et al., 1994; von Castelmur et al., 2012). A highly conserved lysine in the small lobe of protein kinases coordinates ATP and helps transfer the γ-phosphate. Mutation of this lysine into alanine or several other amino acids inactivates many known kinases (e.g., Iyer et al., 2005b). We purified bacterially expressed recombinant twitchin TwcK in wild-type form and as a mutant in which this lysine (residue 6290 in full-length twitchin, UNC-22B) was converted to an alanine (K6290A). As shown in Figure 1A, in an in vitro kinase assay, TwcK K6290A shows barely detectable phosphotransferase activity; it displays ∼0.1% of wild-type activity.
FIGURE 1:
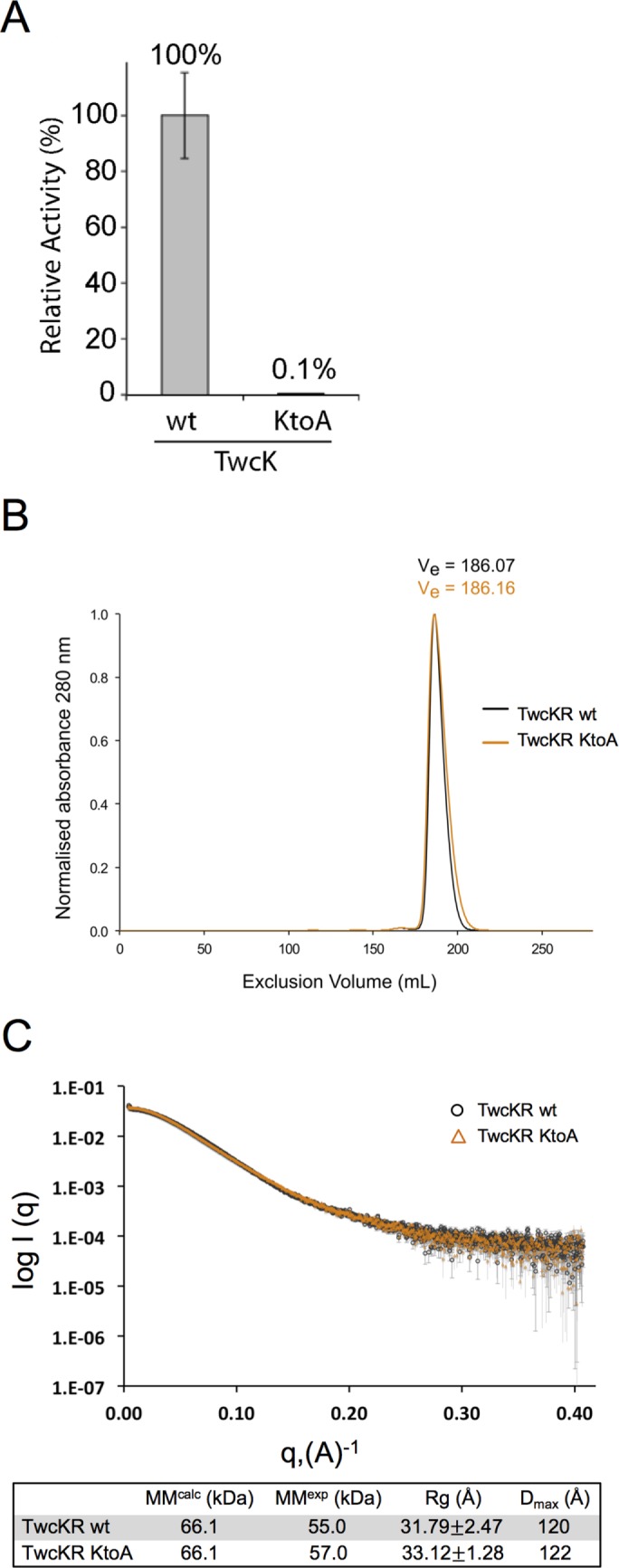
Mutation of a lysine involved in ATP coordination/phosphotransfer to alanine (K6290A) eliminates catalytic activity but has no effect on the overall structure of twitchin kinase in vitro. (A) The wild-type (wt) and the K6290A mutant forms of the catalytic domain of twitchin kinase (TwcK) were each expressed and purified from E. coli. The ability to phosphorylate a model peptide was measured with [γ-32P]ATP. As shown, the K6290A mutation almost entirely eliminates catalytic activity. (B) Size exclusion chromatograms of wt and mutant forms of a larger construct, TwcKR (Fn-NL-kin-CRD-Ig). TwcKR wt and TwcKR KtoA samples are coincident within experimental error, supporting the structural similarity of the samples. (C) SAXS analysis shows that the K6290A mutation does not affect the overall structure of the twitchin kinase region (TwcKR), so that the arrangement of assembled domains remains unaltered. Experimental scattering curves from TwcKR wt (black circle; capped error bars) and TwcKR K6290A (orange triangles; uncapped error bars) were scaled using DATADJUST (Petoukhov et al., 2012). The table below the graph lists molecular parameters calculated from the scattering data. MM, Rg, and Dmax denote molecular mass, radius of gyration, and maximal particle size, respectively. MMcalc is the theoretical MM of the constructs computed from primary sequence. Rg was calculated using AUTORG (Petoukhov et al., 2007).
Conversion of the catalytic lysine to alanine is not known to influence the overall fold of a kinase domain. Nevertheless, we investigated whether the K6290A mutation in the twitchin kinase domain would have an effect on its structure as well as on the overall assembled conformation of the kinase region, thereby validating that the NL and CRD tails remained bound to the kinase and that long-range alterations of the area had not taken place. For this purpose, we studied a larger segment of twitchin that included flanking domains, Fn-NL-kin-CRD-Ig (TwcKR). The expression, yield, and solubility of recombinant TwcKR K6290A were nearly identical to those of TwcKR wild type. In addition, the wild-type and K6290A mutant forms of recombinant TwcKR behaved indistinguishably on size exclusion chromatography, suggesting that the samples share an overall shape and surface chemistry (Figure 1B). The overall molecular features of twitchin kinase in its wild-type and KtoA mutant forms were further studied using small-angle x-ray scattering (SAXS). The experimental scattering curves from these samples (Figure 1C), as well as their corresponding radius of gyration (Rg) and maximum dimension (Dmax) values (Figure 1C), are in agreement within the error of this technique. This suggests that both TwcKR forms share a similar global conformation and that the introduction of the K6290A mutation does not detectably affect the conformation of twitchin.
Differential scanning fluorimetry (DSF) was used to assess the effect of the K6290A substitution on TwcKR thermal stability. Results revealed Tm values of 44 and 41°C for TwcKR wild-type and TwcKR K6290A, respectively (Supplemental Figure S1). The decrease in thermal stability (ΔTm) of −3°C observed for TwcKR K6290A is comparable to that recorded for the exchange KtoL in titin kinase (ΔTm = −2.2°C; Bogomolovas et al., 2014) and noticeably smaller than that of an equivalent KtoA mutation in titin kinase (ΔTm = –6.6°C; Bogomolovas et al., 2014) or a KtoH mutation in cAMP-dependent protein kinase (PKA; ΔTm = −10.7°C; Iyer et al., 2005a). None of these other exchanges was known to have functional consequences—even the most destabilizing KtoH substitution in PKA, although it abolished the catalytic activity of PKA, did not affect substrate or regulatory subunit binding (Iyer et al., 2005b). Furthermore, structural studies on mutated protein variants showing decreased thermal stabilities of up to ∼10°C found that fold alterations are rarely detectable in such cases. As an example, a nuclear magnetic resonance study of a variant of domain I10 from titin carrying the exchange T2850I (ΔTm ≈ −11°C) did not reveal any significant structural alteration—globally or locally (Bogomolovas et al., 2016). In the crystal structure of TwcKR (von Castelmur et al., 2012), the ε-amino group of the substituted K side chain interacts with the main-chain carboxyl group of residue Y6560 in the CRD. This interaction is abolished in TwcKR K6290A, possibly accounting for the slight decrease in thermal stability. However, SAXS experiments demonstrate that the K6290A substitution causes no detectable change to the conformation of TwcKR. In addition, the TwcKR K6290A Tm of 41°C remains significantly above the environmental temperature of C. elegans (15–25°C). Taken together, these data indicate that the K6290A substitution is unlikely to lead to artifactual structural effects in this in vivo study and that observations can be attributed to the lack of catalysis in this mutant.
Nematodes expressing twitchin with a catalytically inactive kinase domain (unc-22(sf21)) have normal sarcomere structure and normal response to nicotine and do not twitch
We next used CRISPR/Cas9 gene editing to create the same K6290A kinase domain mutation in the endogenous unc-22 (twitchin) gene (Supplemental Figure S2). We outcrossed this mutant, designated unc-22(sf21), three times to wild type to remove most of the possible off-target mutations generated by CRISPR. We also sequenced the unc-22 gene from this unc-22(sf21) strain and verified that no other mutations were present in the unc-22 gene. We characterized the unc-22(sf21) phenotype as follows. In contrast to all previously characterized unc-22 mutants (Moerman and Baillie, 1979; Moerman et al., 1988; Matsunaga et al., 2015), unc-22(sf21) animals do not twitch and show a wild-type response to nicotine (Supplemental Figure S3). Moreover, by Western blot, the unc-22(sf21) mutant expresses normal levels of various twitchin isoforms of the appropriate size (Supplemental Figure S4). The locomotion of nematodes arises from the alternating contraction and relaxation of body-wall striated muscles on its dorsal and ventral sides. By immunostaining with several antibodies to known sarcomeric proteins and visualization of thin filaments by phalloidin, we saw that the unc-22(sf21) mutant displays normal sarcomere structure in its body-wall muscle cells (Figure 2). This assessment includes the localization of twitchin, thick filament myosins (MYO-3, UNC-54), M-lines and dense bodies (UNC-95), and F-actin. This is in contrast to the loss of function unc-22(e66) and null unc-22(ct37) animals, which show disorganized sarcomeres (Supplemental Figure S5; unc-22(e66) is a 2–base pair deletion of coding sequence for Ig1, resulting in a frame shift and premature stop codon [unpublished data]; unc-22(ct37) is an intragenic deletion; and unc-22(ct37) animals show no detectable twitchin by Western blot [ Moerman et al., 1988]). The normal sarcomeric organization of the unc-22(sf21) mutant is a feature shared in common with the unusual allele unc-22(e105) (Matsunaga et al., 2015; Supplemental Figure S5), which has a missense mutation in Ig7, changing a highly conserved glycine to an arginine (Matsunaga et al., 2015). Nevertheless, unc-22(e105) is different from unc-22(sf21) in that, like all other known unc-22 mutants, unc-22(e105) twitches and is resistant to nicotine (Supplemental Figure S3). The sarcomeric organization of unc-22(sf21) nematodes was examined at higher resolution by transmission electron microscopy (EM). As shown in Figure 3, the myofilament lattice of unc-22(sf21) appears indistinguishable from wild type; there are regularly organized A- and I-bands, normal-appearing dense bodies and M-lines, and normal-appearing thick and thin filaments.
FIGURE 2:

Nematodes expressing twitchin with the K6290A mutation in its kinase domain show normal muscle sarcomere structure, including normal localization of twitchin, by immunofluorescence microscopy. Wild-type (WT) and unc-22(sf21) mutant nematodes were fixed and immunostained with antibodies to the indicated sarcomeric proteins: twitchin, myosin heavy chain A (MYO-3), and myosin heavy chain B (UNC-54) of the A-bands and UNC-95 of M-lines and dense bodies. Phalloidin staining of F-actin of I-bands is also shown. As indicated, unc-22(sf21) shows normal localization of each sarcomeric protein tested, including twitchin. Scale bars, 20 μm.
FIGURE 3:

The unc-22(sf21) mutant shows normal sarcomere structure by EM. Transmission EM of a cross-section of a body-wall muscle cell from a wild-type (WT) animal and from an unc-22(sf21) mutant animal. Top, across most of the width of a single cell near the vulva, there is uniform depth of the contractile apparatus along the body wall and regular spacing of the filament attachment structures, and the thick and thin filaments are all cut in cross-section, demonstrating correct orientation of the filaments parallel to the body axis. Bottom, higher-magnification views show proper morphology and organization of attachment structures and interdigitating thick and thin filaments. Arrows point to dense bodies, and arrowheads point to M-lines. The largest black dots are cross-sections of thick filaments in the A-bands; the smallest dots are cross-sections of thin filaments. Scale bars, 0.5 μm.
To summarize, the unc-22(sf21) mutant expresses full-length twitchin isoforms that are stable and normally incorporated into sarcomeres, displays normal organization of sarcomeres, and neither twitches nor is resistant to nicotine.
unc-22(sf21) worms move faster than wild type and display abnormal muscle kinetics
Despite having no effect on muscle structure, the unc-22(sf21) mutation results in a remarkable effect on muscle function. Nematodes have different locomotion patterns, depending on the medium: in liquid, they thrash back and forth in a C-shaped pattern; on a semisolid surface like agar, they crawl in a sinusoidal pattern. We measured the ability of adult nematodes to move back and forth in a droplet of aqueous buffer M9 (Epstein and Thomson, 1974). As shown in Figure 4A, when normalized to wild-type movement, worms with loss-of-function allele unc-22(e66) swam less well than wild type, and this is typical of nearly all unc-22 alleles. However, unc-22(sf21) mutant worms, unexpectedly, moved ∼30% faster than wild type; a similar increase in motility (up to 50% increase) was observed for unc-22(e105). We also measured the ability of adult worms to crawl on an agar surface. As indicated in Figure 4B, these results are similar to those obtained with the swimming assay, in that unc-22(e66) nematodes crawl more slowly than wild type, but unc-22(e105) and especially unc-22(sf21) animals crawl faster than wild type.
FIGURE 4:

Nematodes lacking twitchin kinase activity move faster and show a greater degree of muscle contraction. (A) Results of a swimming assay in which the number of times an animal moves back and forth in liquid are counted. The slower motility of animals with the loss-of-function allele unc-22(e66) is typical of unc-22 mutants. Both unc-22(sf21) and unc-22(e105) animals display, unexpectedly, faster motility. (B) Measurement of crawling velocity of nematodes on the surface of agar; results are normalized to the length of the animal. Results are similar to those obtained for swimming. (C) Typical results of an optogenetic experiment to measure contraction and relaxation of wild-type nematodes. Relative body area is a measure of the contraction state of body-wall muscle. (D) Optogenetic experiments on wild type and three unc-22 mutants. Compared with wild-type animals, unc-22(sf21) and unc-22(e105) worms display a greater degree of muscle contraction, whereas unc-22(e66) worms show less muscle contraction and an inability to maintain the contracted state. (E, F) Rate constants for contraction (“E” in C) and for relaxation (“F” in C) derived from fitting curves to data shown in D. (G) Relative body area at steady state, which is the average relative body area during 5 s before the relaxation (“G” in C). These numbers emphasize that unc-22(sf21) and unc-22(e105) nematodes contract more than wild type. For A, B, and E–G, n > 40; *p < 0.01, **p < 0.001, and ***p < 0.0001.
Recently we adapted the use of optogenetics to obtain certain measures of contraction/relaxation kinetics of body-wall muscle in C. elegans (Hwang et al., 2016). Channelrhodopsin-2 (ChR2) is expressed in motor neurons, and on exposure to blue light, contraction of all the body-wall muscles is induced at the same time. As a proxy of this contraction, we measure the relative body area of the worm, as shown in Figure 4C; by fitting curves, rate constants for contraction and relaxation can be derived. Figure 4D shows results of this assay on wild-type and three unc-22 mutant alleles. As reported previously (Hwang et al., 2016), unc-22(e66) nematodes (red line in Figure 4D) contract less well and cannot maintain a maximally contracted state. In contrast, both unc-22(e105) and especially unc-22(sf21) animals contract more deeply than wild type. Another measure from the optogenetic experiment in Figure 4D is the relative body area when muscles are maximally contracted (Figure 4G). In this measure, both unc-22(e105) and unc-22(sf21) nematodes contract more than wild type, whereas unc-22(e66) contracts less than wild type. Although the reduced contractility of unc-22(e66) nematodes may be explained by the disorganization of its myofilament lattice (Supplemental Figure S5), results for unc-22(e105) and unc-22(sf21) animals are puzzling. Either unc-22(e105) or unc-22(sf21) animals contract absolutely more, or they appear to contract more because they begin at a more relaxed state. Additional experiments will be required to distinguish these possibilities. In addition, all of the unc-22 mutant strains show faster rates of contraction and relaxation (Figure 4, E and F). Given that the unc-22(e66) strain has the fastest rates for contraction and relaxation, we might expect that it would also swim and crawl at the highest velocity. However, this is not the case: unc-22(e66) animals swim and crawl at decreased velocities compared with wild type. Most likely, the slower movement of the unc-22(e66) nematodes is explained by the fact that they have disorganized sarcomeres so that the output is less efficient or the coordination of the muscle cells is impaired.
There is selective pressure to maintain kinase activity of twitchin
The fact that loss of twitchin kinase activity results in increased velocity of swimming and crawling, more muscle contraction, and faster rates of contraction and relaxation suggests that the normal function of twitchin kinase activity is to inhibit the contraction/relaxation cycle. Among invertebrates (C. elegans, Drosophila, Aplysia), twitchin and the similar protein projectin have active protein kinase domains, as determined by in vitro kinase assays (Lei et al., 1994; Ayme-Southgate et al., 1995; Heierhorst et al., 1995, 1996; von Castelmur et al., 2012). Therefore it seems that among invertebrates, there has been selection to maintain twitchin kinase activity. To test this hypothesis, we set up a “competitive fitness assay.” We cultured 100 green fluorescent protein (GFP)–tagged test strain (JK2735) animals with 100 experimental animals (either unc-22(sf21) 3X oc or the N2 wild-type strain) and measured the frequency of the experimental strain after four generations of growth and reproduction in competition. If the strains exhibited equal levels of fitness, we would expect the 50% frequency to be maintained. Both the wild-type strain and unc-22(sf21) outcompeted the GFP-tagged tester strain (Figure 5A). However, the competitive fitness of unc-22(sf21) was significantly reduced relative to the wild-type strain (Figure 5A; χ21 = 9.14, p = 0.0025). Thus, at least in a laboratory setting, there is selective pressure to maintain the kinase activity of twitchin, despite its effect of reducing the overall locomotion of the animals. In support of our competition assay results, analysis of the sequences of twitchin orthologues from 47 nematode species, including C. elegans, showed that the six residues or segments crucial for catalysis, including the catalytic lysine (Endicott et al., 2012; Mayans et al., 2013), are conserved (Supplemental Figure S6).
FIGURE 5:
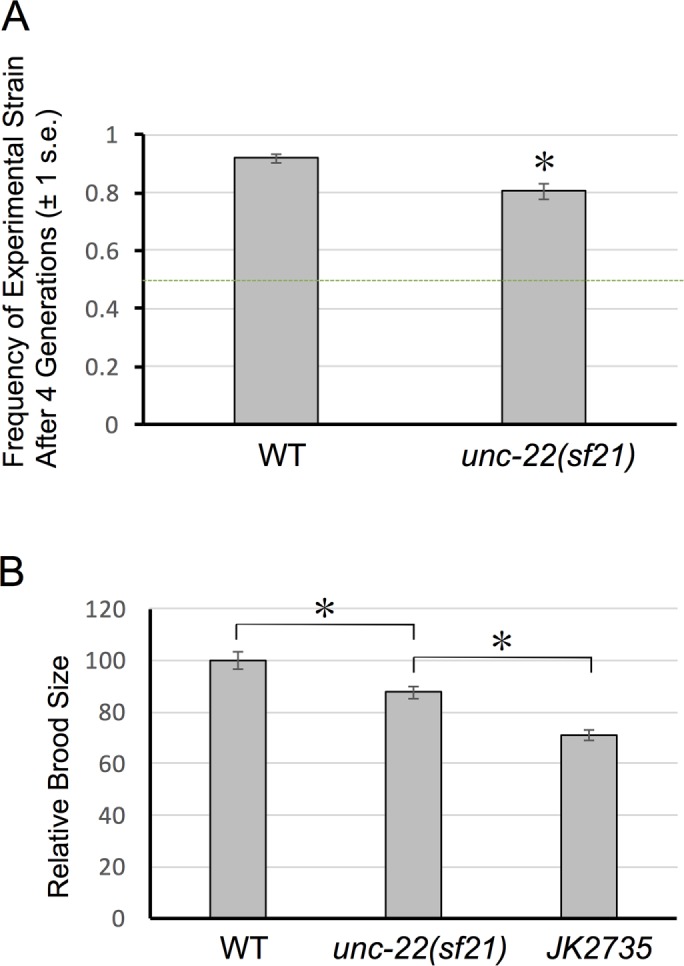
A competition assay demonstrates that the unc-22(sf21) mutant has a selective disadvantage relative to wild type. (A) Competition assay results. Equal numbers of GFP-tagged wild-type (“tester” strain, JK2735) and the indicated strains were cultured on plates with E. coli food, and after four generations, the frequency of each strain was determined. (B) Relative brood sizes of wild-type strain N2, unc-22(sf21), and GFP-tagged strain (JK2735). *Significance with p < 0.05.
We speculated that the poorer performance of unc-22(sf21) relative to wild-type N2 might be attributable to a reduction in brood size. As shown in Figure 5B, measurement of brood sizes indicate that, indeed, unc-22(sf21) has a smaller brood size than wild-type strain N2. One possibility for this reduced brood size is that twitchin is normally expressed in pharyngeal muscles (required for feeding and nutrition) and/or vulval or uterine muscles required for egg laying. Indeed, our promoter analysis (Supplemental Figure S7) indicates that several of the nine predicted twitchin mRNAs are expressed in pharyngeal and/or vulval muscles in addition to being expressed in body wall muscle. In addition, all of the isoforms are predicted and/or verified to include the kinase domain. It also must be noted that the GFP-tagged strain (JK2735) has a smaller brood size than either N2 or unc-22(sf21). The reduced brood size of the GFP strain explains why in the competition experiment, both N2 and unc-22(sf21) outcompeted the GFP strain. (The reduced brood size of JK2735 might be due to having GFP expressed in the pharynx, gut, and early embryo, thus interfering with feeding, nutrition, and/or development of this strain.)
Molecular dissection of twitchin functions
Until recently, all the unc-22 mutants were reported to display several phenotypic characteristics, which vary in severity with mutant allele: 1) “twitching,” which is abnormal constitutive fine movement along several portions of the body surface occurring approximately one to two times per second and originating from the underlying body-wall muscle; 2) resistance to the paralyzing effects of nicotine; 3) reduced locomotion; and 4) disorganization of sarcomeres (Moerman and Baillie, 1979; Moerman et al., 1988). Based on these phenotypes, twitchin is likely to have two functions: one in the assembly or maintenance of sarcomeres and the other in regulating muscle contraction. Whether these two functions are separate or related could not be discerned until now: all of the previously isolated alleles had some degree of each of the four phenotypes. Although the null allele, ct37, is an intragenic deletion (Moerman et al., 1988), our sequence analysis of 10 additional unc-22 mutants revealed that they all result in stop codons, either because they are simple nonsense mutations or because they are insertions/deletions that result in frameshifts and eventual stop codons (unpublished data). Until the study presented here, the one exception was unc-22(e105), which twitches mildly and is resistant to nicotine but has normal sarcomere structure and moves faster than wild type (Matsunaga et al., 2015). e105 is a missense mutation in a highly conserved glycine (to arginine) in Ig7 and results in normal expression and localization of twitchin. Thus, because we can assume that all the Ig and Fn domains are present in e105 mutant twitchin, we can conclude that the organization of sarcomeres and ability to locomote at normal (or better) velocity depends on the presence of these Ig and Fn domains. In the present study, we show that unc-22(sf21) animals, unlike e105, fail to twitch and have normal sensitivity to nicotine; however, like the e105 worm, unc-22(sf21) has normal muscle structure and moves faster than normal. Thus we can conclude that one normal function of twitchin is to inhibit the muscle contraction/relaxation cycle, and this depends on the catalytic activity of the kinase domain. We do not know how to account for the phenotype of e105; we can speculate that perhaps twitchin’s Ig7 inhibits the kinase domain in a twitchin dimer.
At this time, the mechanism by which twitchin kinase activity inhibits the contraction/relaxation cycle is unknown. A clue to this mechanism should arise from determining the substrate(s) for twitchin kinase, which we can now approach by using unc-22(sf21) mutant animals.
MATERIALS AND METHODS
Plasmids for expression of wild-type and K6290A mutant twitchin kinase
The plasmid for expression of histidine (His)-tagged wild-type twitchin kinase catalytic domain (TwcK) was described previously (von Castelmur et al., 2012). The same catalytic domain (TwcK) with the K6290A mutation was amplified by PCR using primers TwiK-3 and TwiK-4 from an Fn-Kin-Ig K6290A template described in Matsunaga et al. (2015); the DNA sequence of this fragment was confirmed and then cloned into the pETM11 vector for expression as a His-tagged protein. The wild-type cDNA encoding Fn-NL-kin-CRD-Ig (TwcKR) was cloned into pET28 as described previously (Greene et al., 2008) for expression as a His-tagged protein. The K6290A version of Fn-NL-kin-CRD-Ig (TwcKR K6290A) was created as described (Matsunaga et al., 2015) and cloned into pET28.
Recombinant protein production
All twitchin kinase constructs (TwcK, TwcKR, wild type, and mutant at 6290) were expressed in Escherichia coli Rosetta 2 (DE3) (Merck Millipore) and grown in Luria–Bertani medium containing 25 µg/ml kanamycin and 34 µg/ml chloramphenicol. Growth was at 37°C to OD600 = 0.6–0.8, followed by induction of protein expression with 0.5 mM isopropyl β-d-1-thiogalactopyranoside and further growth at 18°C for ∼18 h. Cell pellets were harvested by centrifugation and resuspended in 50 mM Tris-HCl, 500 mM NaCl, 1 mM dithiothreitol (DTT), pH 7.9 (lysis buffer), supplemented with 20 µg/ml DNase I (Sigma-Aldrich) and a complete EDTA-free protease inhibitor (Roche). Cell lysis was by sonication on ice, followed by clarification of the lysate by centrifugation.
In preparation for phosphotransfer assays, cell lysates were applied to Ni2+-nitriloacetic acid (NTA) resin (Qiagen) equilibrated in lysis buffer containing 20 mM imidazole. Elution was with 200 mM imidazole, and samples were buffer exchanged into lysis buffer using PD-10 desalting columns (GE Healthcare). The His6-tag removal was by TEV protease cleavage overnight at 4°C, followed by subtractive Ni2+-NTA purification. Size exclusion chromatography was carried out using a Superdex 200 16/60 column (GE Healthcare) in 50 mM Tris-HCl, 50 mM NaCl, and 1 mM DTT, pH 7.9.
In preparation for SAXS and DSF, cell lysates were applied to a 5-ml HisTrap HP column (GE Healthcare) equilibrated in lysis buffer containing 20 mM imidazole. Elution of His6-tagged protein was by continuous imidazole gradient, followed by buffer exchange into 50 mM Tris-HCl, 50 mM NaCl, and 1 mM DTT, pH 7.9 (anion exchange buffer), His6-tag cleavage by TEV protease, and subtractive Ni2+ purification. Anion exchange was performed using a 5-ml QFF column equilibrated in anion exchange buffer (GE Healthcare) with elution by continuous NaCl gradient. Size exclusion chromatography was carried out using a Superdex 200 26/60 column (GE Healthcare) in 50 mM Tris-HCl, 50 mM NaCl, and 0.5 mM tris (2-carboxyethyl)phosphine (TCEP), pH 7.9.
In vitro phosphotransfer assay
In vitro phosphotransfer assays were set up in 40-µl reactions containing 30 ng of TwcK and 20 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 0.2 mg/ml bovine serum albumin, and 0.2 mg/ml peptide substrate. Reactions were initiated by addition of 200 µM [γ32P]ATP (2 µCi per reaction). The peptide substrate has the sequence KKRARAATSNVFS and is a model peptide substrate derived from chicken myosin light chain (kMLC11-23) as described (Heierhorst et al., 1996; von Castelmur et al., 2012). Control reactions were included in all assays containing peptide substrate in the absence of TwcK and with TwcK in the absence of peptide substrate. Incubation of samples was at 25°C for 20 min before blotting on P81 phosphocellulose paper and submergence in 100 mM phosphoric acid to terminate reactions. Blotted samples were washed four times in 100 mM phosphoric acid for 5 min/wash before being transferred to acetone and air dried. Quantification was by Cherenkov counting using a Beckman Coulter scintillation counter.
Small-angle x-ray scattering
SAXS data were collected at the B21 beamline of the Diamond Light Source synchrotron (Didcot, United Kingdom) using the integrated SEC-SAXS setup, including the HPLC device Agilent 1260C. Approximately 1–1.2 mg of sample (TwcKR wild type, TwcKR KtoA) was loaded in 45 μl of buffer (50 mM Tris, pH 7.9, 50 mM NaCl, 0.5 mM TCEP) onto a preequilibrated Shodex KW403 column at a flow rate of 0.16 ml/min. Frames were collected for the entire eluate using a exposure time of 3 s/frame and a sample cell thermostated to 20°C. The x-ray scattering was recorded on a Pilatus 2 M detector (Dectris) at a sample-to-detector distance of 3.9 m and a wavelength of 1 Å. Data processing used ScÅtter (www.bioisis.net/tutorial/9; Diamond, unpublished). Scattering curves were analyzed with PRIMUS (Konarev et al., 2003) and the ATSAS software suite (Petoukhov et al., 2012) to determine Rg, Dmax, and the pair distribution function, P(r). The experimental molecular mass was calculated using ScÅtter (Rambo and Tainer, 2013).
Differential scanning fluorimetry
DSF assays were performed using a CFX connect Real-Time PCR machine (Bio-Rad) with a thermal ramping of 1°C/min in the temperature range 25–95°C. Proteins were tested at a concentration of 7.5 µM in 50 mM Tris-HCl, pH 7.9, 50 mM NaCl, and 0.5 mM TCEP. SYPRO-orange dye (Invitrogen) was diluted to a concentration of 1:1000 in the same buffer. Assays were carried out in a volume of 25 µl and in triplicate. Data were processed using CFX Manager software (Bio-Rad), and Tm values were calculated from the first derivative of the melting curves.
Nematode strains
N2 (Bristol) is the primary wild-type strain, and standard growth conditions were used (Brenner, 1974). The following alleles were used in this study: unc-22(e66)IV, unc-22(e105)IV, unc-22(ct37)IV, and zxIs6[unc-17p::ChR2(H134R):::YFP; lin15+]V (Liewald et al., 2008). unc-22(sf21); zxIs6 was generated by crossing. To check for mutations of mutant animals, we carried out single-worm PCR. After purification of PCR products, we examined a restriction enzyme digestion (PstI) and DNA sequence analysis to verify the mutation.
Creation of unc-22(sf21) animals
The K6290A mutation in the coding sequence of the endogenous unc-22 gene was generated using a CRISPR technique as described (Paix et al., 2015). For targeting to unc-22, a sequence, 5′-actccacatgaatctgaca-3′ was cloned into pDD162 (Addgene plasmid 47549) using a Q5 site-directed mutagenesis kit (New England BioLabs). To modify the genome using Cas9 triggered homologous recombination, the following DNA mixtures were injected: 50 ng/µl pDD162 with targeting sequences, 30 ng/µl single-strand oligo 5′-aactggaaacaactttgctgcagcattcgtaatgactccacatgaatccgacaaagaaactgtcagaaaagagattcaaaccatgtcagttttgagacac-3′ as repair template, and 30 ng/µl pTG96 (Yochem et al., 1998) as coinjection marker. For screening of the mutants, we picked single F1 animals to PCR tubes and performed single-worm PCR using the primers 5′-tgaaccaccaagattcccgattatc-3′ and 5′-ccaacgctccacatatcggtg-3′. After purification of PCR products, we examined a restriction enzyme digestion (PstI) and performed DNA sequence analysis to verify the mutation. The homozygous mutant was outcrossed three times to wild type before further analysis and designated unc-22(sf21).
Whole-genome sequencing
Genomic DNA from unc-22(sf21) was prepared and provided to the Emory Integrated Genomics Core (EIGC) for Illumina sequencing. The Genomic Services Laboratory at the HudsonAlpha Institute for Biotechnology performed quality control and constructed standard Illumina sequencing libraries following the manufacturer’s instructions. Sequencing was performed on an Illumina Hiseq version 3 with 100–base pair paired-end reads. Raw data were returned to the EIGC for bioinformatics analysis. Raw reads were mapped against the C. elegans reference genome (WS190/ce6) with the PEMapper software package. PEMapper maps short reads to a reference genome by first decomposing those reads in k-mers (16-mers in this case) and then performing a hash-based “rough mapping” of each read. After roughly mapping the read, the fine-scale position of the read is determined by a local Smith–Waterman alignment. Genotypes were determined using PECaller. Intuitively, we envisioned the six channels of data (number of A, C, G, T, deletion, and insertion reads) as being multinomially sampled, with some probability of drawing a read from each of the channels, but the probability varies from experiment to experiment and is itself drawn from a Dirichlet distribution. PECaller combines data across samples in order to identify the genotype with the highest likelihood at each sequenced base. Both PEMapper and PECaller are part of a custom software package developed at Emory University for mapping and identifying variant sites from Illumina raw sequencing data (D. J. Cutler and M. E. Zwick, personal communication). Analysis then focused on the ∼60-kb unc-22 gene. PECaller identified a single homozygous mutation specifying the desired KtoA change and no other mutations in the unc-22 gene.
Immunolocalization
Adult nematodes were fixed as described previously (Nonet et al., 1993; Wilson et al., 2012). Primary antibodies were used at the following dilutions: anti–ATN-1 (α-actinin; MH35; Francis and Waterston, 1991) at 1:200, anti–myosin heavy chain A (MHC A; mouse monoclonal 5-6; Miller et al., 1983) at 1:200, anti-paramyosin (UNC-15; monoclonal 5-23; Miller et al., 1983) at 1:200, anti–PAT-6 (Warner et al., 2013) at 1:200, anti-twitchin (I I II; Benian et al., 1996) at 1:200, and anti–UNC-89 (Benian et al., 1996; rabbit polyclonal EU30) at 1:200. MH35 antibodies were kindly provided by Pamela Hoppe (Western Michigan University). The 5-6 and 5-23 antibodies were obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA). For anti-twitchin and anti-UNC-89, the secondary antibody was anti-rabbit Alexa 488 (Invitrogen). For anti-ATN-1, anti-MHC A, and anti-paramyosin, the secondary antibody was anti-mouse Alexa 594 (Invitrogen). For anti-PAT-6, the secondary antibody was anti-rat Alexa 594 (Invitrogen). Each secondary antibody was used at 1:200 dilution. Images were captured at room temperature with a Zeiss confocal system (LSM510) equipped with an Axiovert 100 M microscope and an Apochromat 63×/1.4 numerical aperture oil objective in 2.5× zoom mode. The color balances of the images were adjusted by using Adobe Photoshop. We examined the staining of at least three worms from each sample.
Electron microscopy
Nematodes were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) for 3 h. Samples were then cut in half to allow additional fixation overnight. After washing with 0.1 M cacodylate buffer and postfixing with 1% osmium tetroxide in the same buffer for 1 h, samples were washed with deionized water, dehydrated through an ethanol series, and then
placed in 100% ethanol. After two propylene oxide rinses, samples were infiltrated with a mixture of propylene oxide and Eponate 12 resin (Ted Pella, Redding, CA) at a 1:1 ratio overnight and then 1:2 ratio for 6 h. After overnight infiltration in Eponate 12 resin, multiple nematodes were placed in parallel in a small drop of Eponate 12 resin and polymerized in a 60°C oven. On polymerization, the worm halves were reembedded for cross-sectioning. Ultrathin sections were cut 70–80 nm thick with a Leica UltraCut S ultramicrotome (Leica Microsystems, Buffalo Grove, IL). Grids with ultrathin sections were stained with 5% uranyl acetate and 2% lead citrate. The ultrathin sections were imaged on a JEM-1400 transmission electron microscope (JEOL Tokyo, Japan) equipped with a US1000 charge-coupled device camera (Gatan, Pleasanton, CA). Brightness and contrast were adjusted by using Photoshop (Adobe, San Jose, CA).
Swimming assays
For the swimming assays, synchronized young adult animals were transferred into M9 buffer (85 mM NaCl, 42 mM Na2HPO4, 22 mM KH2PO4 and 1 mM MgSO4). Body bends of individual young adult animals were counted for 1 min. A body bend was scored as a complete deflection of the anterior portion of the nematode from the midline. At least 15 animals of each genotype were analyzed. Values are reported as means and standard errors and tested for significance by Dunnett’s test.
Crawling on a plate and optogenetic assays
Crawling on an agar surface (induced backward locomotion) was performed using the methods as described (Hwang et al., 2016; Barnes et al., 2016). Young adult animals were transferred onto a fresh 5.5-cm nematode growth medium (NGM) plate (without bacterial seeding). After a 2-min acclimation period, the front of the animal was gently prodded with a platinum wire to induce backward locomotion, and a movie was recorded. The movie was postprocessed to extract the worm skeleton using custom software written in MATLAB. The velocity of backward crawling was measured and normalized by the length of each worm. For optogenetics assays of muscle kinetics, animals carrying transgene zxIs6 were grown on NGM plates with OP50 E. coli mixed with all-trans-retinal for 3.5 d before experiments. Synchronized young adult animals were loaded into multiple parallel microchannels made of an optically transparent elastic polymer, polydimethylsiloxane (PDMS), and trapped by pneumatically controllable PDMS membrane valves (Stirman et al., 2010; Hwang et al., 2016). To induce ChR2 photoactivation, the trapped animals were simultaneously illuminated with blue light (450–590 nm; 0.3 mW/mm2) for 15 s on the device. For quantitative measurements of a temporal change in projected body area, movies were recorded and postprocessed using custom software written in MATLAB. All curve fitting and statistical analysis were performed using Prism 5 (GraphPad Software). A Wilcoxon rank sum test was conducted for comparison of the data when appropriate.
Competitive fitness assays
Experimental C. elegans strains were competed against a GFP-labeled tester strain with an N2 background (JK2735) that was obtained from the Caenorhabditis Genetics Center. Ten replicate competition assays were run for the experimental strains, wild-type N2, and unc-22(sf21) 3X oc. Competition assays were run for four passages on a 100-mm Petri dish containing 30 ml of NGM-lite (US Biological), seeded with 200 μl of E. coli (OP50), and stored at 20°C during competition. Before the assay, nematode lines were bleach-synchronized (Lewis and Fleming, 1995). Competition assays were initially set up at a ratio of 100 experimental L4 larvae to 100 GFP L4 larvae. The larvae were permitted to develop to adults, exhaust the bacterial lawn, and reproduce. Every 4 d (approximately one nematode generation), the entire dish, containing parents and offspring, was washed with 1 ml of M9 buffer, and the nematodes were pelleted. Approximately 1000 nematodes were transferred to a new dish containing OP50, using methods described in Morran et al. (2009). After the fourth passage, the frequency of the experimental strain relative to the GFP strain was measured in 200 individuals from a cross-section of the Petri dish to determine the relative fitness of the experimental strain to the GFP strain (Morran et al., 2009). An increase in the frequency of the experimental strain relative to the GFP indicates greater competitive fitness for the experimental strain. In addition, 10 replicates of only JK2735 individuals (no experimental strains) were run to test for the loss of GFP as a control. We observed no loss of GFP expression across all 10 replicates over four passages. Values are reported as means and standard errors. A Kruskal–Wallis nonparametric test was performed in JMP 12 (SAS Institute) on the proportion of experimental strain values measured after four generations to compare the competitive fitness of the experimental strains (N2 vs. unc-22(sf21)).
Nicotine sensitivity assays using a WMicrotracker device
The method of nicotine assay was described previously (Matsunaga et al., 2015). Synchronized young adult animals of each genotype were collected in M9 buffer. After a final wash with M9 buffer containing 0.01% Triton X-100 (M9T), a worm slurry (worm pellet: M9T = 1: 5) was prepared. For the assays, 96-well plates were used. To each well was added 40 μl of M9T followed by 10 μl of slurry, delivering ∼50–100 worms/well. The plate was placed in the dark at room temperature for 1 h. Then locomotive activities under no-nicotine conditions were measured for 1 h using a WMicrotracker (Phylumtech) with the program “C. elegans (>L4)” supplied by the manufacturer. After the measurement, 50 μl of M9T containing 0.2 or 0.1% nicotine solution was added to each well, and locomotive activities were measured using the WMicrotracker. For each strain and nicotine concentration, eight independent wells were assayed. On the graphs of locomotive activity versus time, each point represents the mean and standard error.
Western blotting
Equal amounts of total protein from wild-type N2 and unc-22(sf21) nematodes were separated by 5% polyacrylamide–SDS gels (50 V for 40 min, 100 V for 10 min, and 200 V for 60 min), transferred to nitrocellulose membrane (Bio-Rad Laboratories) using transfer buffer (25 mM Tris-base, 192 mM glycine. and 20% methanol [vol/vol]) at 100 V for 2 h, blocked overnight in 5% skim milk (wt/vol)–TBST buffer (0.1% Tween-20 [vol/vol], 1× TBS, pH 7.6), and reacted with anti-twitchin (Benian et al., 1996) at 1:4000 in the same buffer for 1 h at room temperature. After washing, the blot was incubated with anti-rabbit horseradish peroxidase–conjugated secondary antibody (NA9340; GE Healthcare). The signal from the membrane was obtained using a Pierce ECL Western Blotting Substrate (Thermo Scientific) and HyBlot CL film (Denville Scientific).
Brood-size measurements
Synchronized L4-stage animals were transferred onto 3-cm NGM plates (one worm/plate) with fresh OP50 bacteria. Adult worms were transferred every day (24 h) onto new plates for 6 d. The number of viable progeny from each of these adults was totaled. For each strain, the number of progeny from 15 animals was determined. Values are reported as means and standard errors and tested for significance by Dunnett’s test.
Transgenic animals for determining tissue expression of unc-22 promoters
Four plasmids were created, representing each of the four possible unc-22 promoters: unc-22p (ABFGHI)::gfp, unc-22p (D)::gfp, unc-22p (C)::gfp, and unc-22p (E)::gfp. PCR was used to amplify, from genomic DNA, 3.0 kb including the putative promoter sequence upstream of unc-22, a putative 5′-untranslated region, initiator ATG, and the first 24 nucleotides of coding sequence of unc-22. These fragments were then cloned into the pPD_venus vector, which contains a multiple cloning site followed by venus coding sequence and unc-54 3′-untranslated region, using PstI and SmaI sites or PstI and MscI sites. The plasmids were injected at 20 ng/ml into the gonads of wild-type animals along with the pRF4 [rol-6(su1006)] plasmid as coinjection marker (80 ng/ml; Mello and Fire, 1995). At least three independent stable transgenic lines were generated for each plasmid. Fluorescence and differential interference contrast images were obtained with an Axioskop microscope and AxioCam MR camera (Carl Zeiss).
Supplementary Material
Acknowledgments
We thank Dina Greene and Rachel Stanford for their help in making some of the plasmids. G.M.B., H.L., and O.M. gratefully acknowledge support from a Human Frontier Science Program Grant (RGP0044/2012). E.M. was supported in part by the Robert P. Apkarian Integrated Electron Microscopy Core, which is subsidized by the Emory College of Arts and Sciences and the Emory University School of Medicine. Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health (UL1TR000454). EM images were captured with a JEOL JEM-1400 120kV TEM, which is supported by National Institutes of Health Grant S10 RR025679.
Abbreviations used:
- DSF
differential scanning fluorimetry
- DTT
dithiothreitol
- EIGC
Emory Integrated Genomics Core
- EM
electron microscopy
- Fn
fibronectin
- GFP
green fluorescent protein
- Ig
immunoglobulin
- PDMS
polydimethylsiloxane
- SAXS
small-angle x-ray scattering.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-10-0707) on April 20, 2017.
REFERENCES
- Ayme-Southgate A, Southgate R, Saide J, Benian GM, Pardue ML. Both synchronous and asynchronous muscle isoforms of projectin (the Drosophila bent locus product) contain functional kinase domains. J Cell Biol. 1995;128:393–403. doi: 10.1083/jcb.128.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Hwang H, Ono K, Lu H, Ono S. Molecular evolution of troponin I and a role of its N-terminal extension in nematode locomotion. Cytoskeleton. 2016;73:117–130. doi: 10.1002/cm.21281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benian GM, Tinley TL, Tang X, Borodovsky M. The Caenorhabditis elegans gene unc-89, required for muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J Cell Biol. 1996;132:835–848. doi: 10.1083/jcb.132.5.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogomolovas J, Gasch A, Simkovic F, Rigden DJ, Labeit S, Mayans O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 2014;4:140041. doi: 10.1098/rsob.140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogomolovas J, Fleming JR, Anderson BR, Williams R, Lange S, Simon B, Khan MM, Rudolf R, Franke B, Bullard B, et al. Exploration of pathomechanisms triggered by a single-nucleotide polymorphism in titin’s I-band: the cardiomyopathy-linked mutation T2580I. Open Biol. 2016;6:160114. doi: 10.1098/rsob.160114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott JA, Noble MEM, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annu Rev Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Thomson JN. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 1974;250:579–580. doi: 10.1038/250579a0. [DOI] [PubMed] [Google Scholar]
- Francis R, Waterston RH. Muscle cell attachment in Caenorhabditis elegans. J Cell Biol. 1991;114:465–479. doi: 10.1083/jcb.114.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieseler K, Qadota H, Benian GM. Development, structure and maintenance of C. elegans body wall muscle. WormBook. 2016;13:1–59. doi: 10.1895/wormbook.1.81.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene DN, Garcia T, Sutton RB, Gernert KM, Benian GM, Oberhauser AF. Single-molecule force spectroscopy reveals a stepwise unfolding of Caenorhabditis elegans giant protein kinase domains. Biophys J. 2008;95:1360–1370. doi: 10.1529/biophysj.108.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heierhorst J, Probst WC, Kohanski RA, Buku A, Weiss KR. Phosphorylation of myosin regulatory light chains by the molluscan twitchin kinase. Eur J Biochem. 1995;233:426–431. doi: 10.1111/j.1432-1033.1995.426_2.x. [DOI] [PubMed] [Google Scholar]
- Heierhorst J, Tang X, Lei J, Probst WC, Weiss KR, Kemp BE, Benian GM. Substrate specificity and inhibitor sensitivity of Ca2+/S100-dependent twitchin kinases. Eur J Biochem. 1996;242:454–459. doi: 10.1111/j.1432-1033.1996.454rr.x. [DOI] [PubMed] [Google Scholar]
- Hwang H, Barnes DE, Matsunaga Y, Benian GM, Ono S, Lu H. Muscle contraction phenotypic analysis enabled by optogenetics reveals functional relationships of sarcomere components in Caenorhabditis elegans. Sci Rep. 2016;6:19900. doi: 10.1038/srep19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LY, Kontrogianni-Konstantopoulos A. The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J. 2013;27:2001–2012. doi: 10.1096/fj.12-221317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer GH, Garrod S, Woods VL, Taylor SS. Catalytic independent functions of a protein kinase as revealed by a kinase-dead mutant: study of the Lys72His mutant of cAMP-dependent kinase. J Mol Biol. 2005a;351:1110–1122. doi: 10.1016/j.jmb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Iyer GH, Moore MJ, Taylor SS. Consequences of lysine 72 mutation on the phosphorylation and activation state of cAMP-dependent kinase. J Biol Chem. 2005b;280:8800–8807. doi: 10.1074/jbc.M407586200. [DOI] [PubMed] [Google Scholar]
- Katzemich A, West RJ, Fukuzawa A, Sweeney ST, Gautel M, Sparrow J, Bullard B. Binding partners of the kinase domains in Drosophila obscurin and their effect on the structure of the flight muscle. J Cell Sci. 2015;128:3386–3397. doi: 10.1242/jcs.170639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev. 2009;89:1217–1267. doi: 10.1152/physrev.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M, Kötter S Titin, a central mediator for hypertrophic signaling, exercise-induced mechanosignaling and skeletal muscle remodeling. Front Physiol. 2016;7:76. doi: 10.3389/fphys.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger M, Linke WA. The giant protein titin: a regulatory node that integrates myocyte signaling pathways. J Biol Chem. 2011;286:9905–9912. doi: 10.1074/jbc.R110.173260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- Lei J, Tang X, Chambers TC, Pohl J, Benian GM. Protein kinase domain of twitchin has protein kinase activity and an autoinhibitory region. J Biol Chem. 1994;269:21078–21085. [PubMed] [Google Scholar]
- Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- Liewald JF, Brauner M, Stephens GJ, Bouhours M, Schultheis C, Zhen M, Gottschalk A. Optogenetic analysis of synaptic function. Nat Methods. 2008;5:895–902. doi: 10.1038/nmeth.1252. [DOI] [PubMed] [Google Scholar]
- Matsunaga Y, Qadota H, Furukawa M, Choe H, Benian GM. Twitchin kinase interacts with MAPKAP kinase 2 in Caenorhabditis elegans striated muscle. Mol Biol Cell. 2015;26:2096–2111. doi: 10.1091/mbc.E14-05-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayans O, Benian GM, Simkovic F, Rigden DJ. Mechanistic and functional diversity in the mechanosensory kinases of the titin-like family. Biochem Soc Trans. 2013;41:1066–1071. doi: 10.1042/BST20130085. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Miller DM, 3rd, Ortiz I, Berliner GC, Epstein HF. Differential localization of two myosins within nematode thick filaments. Cell. 1983;34:477–490. doi: 10.1016/0092-8674(83)90381-1. [DOI] [PubMed] [Google Scholar]
- Moerman DG, Baillie DL. Genetic organization in C. elegans: fine-structure analysis of the unc-22 gene. Genetics. 1979;91:95–103. doi: 10.1093/genetics/91.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman DG, Benian GM, Barstead RJ, Schriefer LA, Waterston RH. Identification and intracellular localization of the unc-22 gene product of Caenorhabditis elegans. Genes Dev. 1988;2:93–105. doi: 10.1101/gad.2.1.93. [DOI] [PubMed] [Google Scholar]
- Morran LT, Parmenter MD, Phillips PC. Mutation load and rapid adaptation favour outcrossing over self-fertilization. Nature. 2009;462:350–352. doi: 10.1038/nature08496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB. Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HDT, Konarev PV, Svergun DI. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1 – towards automated and web-supported small-angle scattering data analysis. J Appl Cryst. 2007;40:s223–s228. [Google Scholar]
- Qadota H, McGaha LA, Mercer KB, Stark TJ, Ferrara TM, Benian GM. A novel protein phosphatase is a binding partner for the protein kinase domains of UNC-89 (Obscurin) in Caenorhabditis elegans. Mol Biol Cell. 2008;19:2424–2432. doi: 10.1091/mbc.E08-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirman JN, Brauner M, Gottschalk A, Lu H. High-throughput study of synaptic transmission at the neuromuscular junction enabled by optogenetics and microfluidics. J Neurosci Methods. 2010;191:90–93. doi: 10.1016/j.jneumeth.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Castelmur E, Strumpfer J, Franke B, Bogomolovas J, Barbieri S, Qadota H, Konarev PV, Svergun DI, Labeit S, Benian GM, et al. Identification of an N-terminal inhibitory extension as the primary mechanosensory regulator of twitchin kinase. Proc Natl Acad Sci USA. 2012;109:13608–13613. doi: 10.1073/pnas.1200697109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner A, Xiong G, Qadota H, Rogalski T, Vogl AW, Moerman DG, Benian GM. CPNA-1, a copine domain protein, is located at integrin adhesion sites and is required for myofilament stability in Caenorhabditis elegans. Mol Biol Cell. 2013;24:601–616. doi: 10.1091/mbc.E12-06-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KJ, Qadota H, Benian GM. Immunofluorescent localization of proteins in Caenorhabditis elegans muscle. Methods Mol Biol. 2012;798:171–181. doi: 10.1007/978-1-61779-343-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochem J, Gu T, Han M. A new marker for mosaic analysis in Caenorhabditis elegans indicates a fusion between hyp6 and hyp7, two major components of the hypodermis. Genetics. 1998;149:1323–1334. doi: 10.1093/genetics/149.3.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.