

Abstract
Retinal ganglion cells (RGCs) are the sole projecting neurons of the retina and their axons form the optic nerve. Here, we show that embryogenesis‐associated mouse RGC differentiation depends on mitophagy, the programmed autophagic clearance of mitochondria. The elimination of mitochondria during RGC differentiation was coupled to a metabolic shift with increased lactate production and elevated expression of glycolytic enzymes at the mRNA level. Pharmacological and genetic inhibition of either mitophagy or glycolysis consistently inhibited RGC differentiation. Local hypoxia triggered expression of the mitophagy regulator BCL2/adenovirus E1B 19‐kDa‐interacting protein 3‐like (BNIP3L, best known as NIX) at peak RGC differentiation. Retinas from NIX‐deficient mice displayed increased mitochondrial mass, reduced expression of glycolytic enzymes and decreased neuronal differentiation. Similarly, we provide evidence that NIX‐dependent mitophagy contributes to mitochondrial elimination during macrophage polarization towards the proinflammatory and more glycolytic M1 phenotype, but not to M2 macrophage differentiation, which primarily relies on oxidative phosphorylation. In summary, developmentally controlled mitophagy promotes a metabolic switch towards glycolysis, which in turn contributes to cellular differentiation in several distinct developmental contexts.
Keywords: BNIP3L/NIX, hypoxia, macrophages, metabolic reprogramming, retinal ganglion cells
Subject Categories: Autophagy & Cell Death, Metabolism, Neuroscience
Introduction
Autophagy is a catabolic pathway that mediates the degradation and recycling of intracellular components, including whole organelles, to sustain cell homeostasis (Boya et al, 2013). Autophagy is the sole mechanism that allows for the degradation of entire mitochondria, a process commonly known as mitophagy. While mitophagy primarily eliminates damaged or dysfunctional mitochondria (Ashrafi & Schwarz, 2013), this pathway can also mediate the degradation of mitochondria in developmental contexts, a process known as programmed mitophagy (Ney, 2015). BCL2/adenovirus E1B 19‐kDa‐interacting protein 3‐like (BNIP3L), also known as NIX, is an essential regulator of programmed mitophagy during reticulocyte maturation (Aerbajinai et al, 2003; Diwan et al, 2007; Schweers et al, 2007; Sandoval et al, 2008). During programmed mitophagy, NIX is mainly regulated at the transcriptional level and acts as a mitophagy receptor, mediating autophagic sequestration of mitochondria by interacting with LC3 via its LIR domain (Novak et al, 2010).
Metabolic reprogramming is a process by which cells shift their metabolism from oxidative phosphorylation towards glycolysis and convert glucose into lactate even in the presence of oxygen. This phenomenon was first described in cancer cells, but has since been observed in other cell types, including embryonic stem cells, proinflammatory M1 macrophages and cells of the adult retina, and it is thought to be essential to fulfill the metabolic requirements of those cells (Galvan‐Pena & O'Neill, 2014; Ng et al, 2015; Chandel et al, 2016). Hypoxia also triggers metabolic shift towards glycolysis. This type of metabolism is also observed in stem cells and is thought to constitute an adaptation to the hypoxic conditions present in vivo during development, in adult stem cell niches and in the inflamed tissue (Suda et al, 2011; Escribese et al, 2012). Interestingly, hypoxia is a strong inductor of mitophagy (Zhang et al, 2008).
How the complex functionality of the nervous system arises from a pool of undifferentiated neuroepithelial cells is one of the more fascinating aspects of embryonic development (Valenciano et al, 2008). These neuroepithelial cells undergo marked changes to generate differentiated neurons and glial cells. Cell proliferation requires nutrients, energy and biosynthetic activity to duplicate all macromolecular components during each passage of the cell cycle. Differentiation requires profound changes in cellular components and a shift in metabolic activity. Indeed, metabolic pathways may be controlled by the same signals that control cell differentiation (Agathocleous & Harris, 2013). The retina, a model organ for the study of the central nervous system, is a three‐layered structure composed of one glial and six distinct neuronal cell types, which arise from a pool of multipotent retinal progenitor cells (Stenkamp, 2015). In mice, retinal neurogenesis is an orderly process that starts with the differentiation of retinal ganglion cells (RGCs) and is followed by that of other neuronal cell types. During retinal development, cell differentiation follows a central‐to‐peripheral gradient, with the result that the central retina is more developmentally advanced than the peripheral retina (Stenkamp, 2015).
We have previously shown that autophagy genes are essential for the generation of mature neurons from olfactory bulb neural stem cells (Vazquez et al, 2012). Moreover, blockade of autophagy in the embryonic chick retina results in decreased ATP levels and hampers the elimination of apoptotic cells generated during neurogenesis (Mellén et al, 2008, 2009). Interestingly, both these phenotypes are restored by supplying pyruvate as a cell‐permeable precursor (methyl pyruvate), suggesting a connection between autophagy and cell metabolism during neuronal differentiation (Boya et al, 2016). Moreover, cancer cells engage mitophagy to promote metabolic reprogramming towards glycolysis during prolonged mitotic arrest (Domenech et al, 2015). Here, we show that NIX‐ and Atg5‐dependent mitophagy mediates a metabolic shift towards glycolysis that allows RGC generation in the embryonic mouse retina. Furthermore, we describe a mitophagy‐dependent metabolic shift that occurs during the polarization of macrophages towards a proinflammatory and more glycolytic M1 phenotype. Taken together, our data show that programmed mitophagy triggers metabolic reprogramming towards glycolysis during cellular differentiation.
Results
Mitochondrial rarefaction during embryonic RGC differentiation
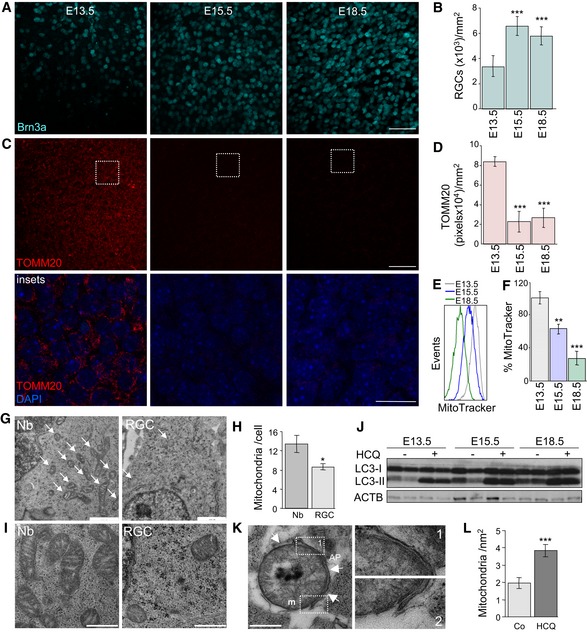
In the mouse retina, neurogenesis starts around day E12.5 with the differentiation of RGCs, the first neurons to be generated. As expected, immunofluorescence analyses revealed the presence of the RGC‐specific transcription factor Brn3a and γ‐synuclein (another RGC marker) from E13.5, with levels peaking at E15.5 in the RGC layer of mouse retinal flatmounts (Fig 1A and B, and Appendix Figs S1 and S2). In the same retinas, immunostaining for the mitochondrial protein TOMM20 decreased after E13.5 (Fig 1C and D, insets below, and Appendix Fig S1C). This decrease in the number of mitochondria during retinal embryonic development was corroborated by labelling dissociated retinas at different embryonic stages with the mitochondrion‐specific dye MitoTracker Deep Red (MTDR), which was quantified by flow cytometry (Fig 1E and F). This decrease in mitochondrial number could not be attributed to decreased expression of genes involved in mitochondrial biogenesis, such as Ppargc1a and Tfam, as their expression remained unchanged during the embryonic stages assessed (Fig EV1A). Moreover, electron microscopy analyses revealed that at E15.5 the number of mitochondria in each RGC was reduced as compared with that of proliferating neuroblasts (Fig 1G and H). Mitochondria from neuroblasts (Nbs) were large and displayed well‐organized cristae, indicative of a high metabolic rate, while those from RGCs were comparatively smaller and contained fewer cristae (Fig 1I). Together these data suggest that programmed elimination of mitochondria occurs during RGC development.
Figure 1. Mitochondrial mass decreases during embryonic retinal development.
-
AImmunostaining of the retinal ganglion cell (RCG)‐specific transcription factor Brn3a (cyan) in the RGC layer of mouse retinal flatmounts at the indicated embryonic days (E). The maximal projection in the RGC layer is shown. Scale bar, 50 μm.
-
BQuantification of RGC density per mm2 at the indicated embryonic stages in retinas stained as in (A) (n = 6 per group). Data are presented as mean ± SEM. ***P < 0.001 (Mann‐Whitney U‐test).
-
CImmunostaining with the mitochondrial marker TOMM20 (red) in the RGC layer of the same retinas as in (A). Scale bar, 50 μm. Insets show TOMM20 staining (red) and nuclei labelled with DAPI (blue) in a confocal plane in the indicated region. Scale bar, 20 μm.
-
DTOMM20‐positive pixel staining at the indicated embryonic stages labelled as in (C) (n = 6 per group). Data are presented as mean ± SEM. ***P < 0.001 (Mann‐Whitney U‐test).
-
E, FFlow cytometry histograms and percentage mean fluorescence intensity (% MitoTracker) in dissociated retinas, corresponding to different embryonic stages, stained with MitoTracker Deep Red (MTDR) (n = 5–12 retinas per group). Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001 (Mann‐Whitney U‐test).
-
GUltrastructural analysis of neuroblast (Nb) and RGC from an E15.5 mouse retina. Scale bar, 1 μm. Arrows indicate mitochondria.
-
HQuantification of the number of mitochondria per cell in G (n = 12–14 cells per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
IMitochondrial morphology (m) in retinal Nb and RGC at E15.5. Scale bars, 500 nm.
-
JImmunoblot of retinas at different embryonic stages incubated for 3 h in the absence (−) or presence (+) of hydroxychloroquine to block autophagic flux.
-
KAutophagosomal membrane (AP) surrounding a mitochondrion (arrows in left‐hand image) in an E15.5 mouse retina in the presence of HCQ to block lysosomal degradation. Scale bar, 500 nm. Insets of the indicated areas show four adjacent membranes (right).
-
LIncrease in the number of mitochondrial per nm2 in RGCs from E15.5 mouse retinas in the presence of HCQ (n = 12 per group). Data are presented as mean ± SEM. ***P < 0.001 (Mann‐Whitney U‐test).
Figure EV1. The increase in mitochondrial number is autophagy‐ and mitophagy dependent.
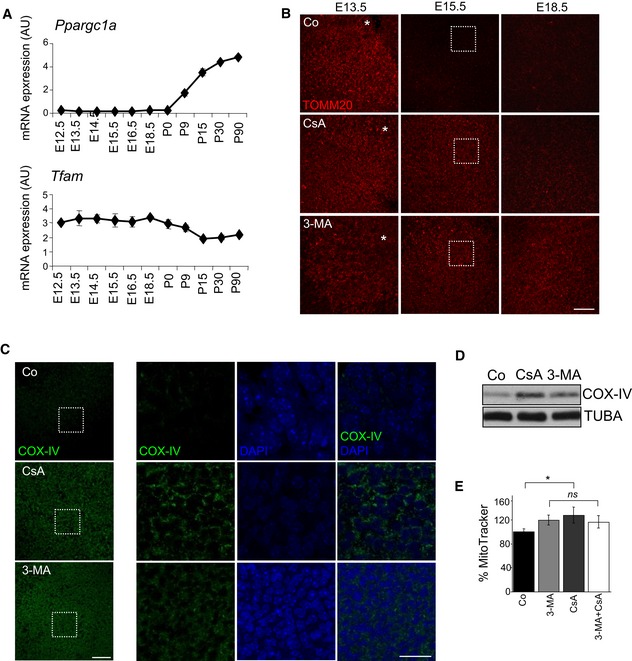
- mRNA expression, determined by qRT–PCR, of the mitochondrial biogenesis regulators Ppargc1a and Tfam (n = 2–3 pools of retinas per group). Data are presented as mean ± SEM.
- TOMM20 immunostaining at the indicated developmental stages in flat‐mounted retinas incubated for 6 h in the presence of 5 μM CsA or 10 mM 3‐MA. The maximal projection of the z‐stack is shown. Scale bar, 50 μm.
- COX‐IV immunostaining in flat‐mounted retinas (E15.5) incubated for 6 h with 3‐MA or CsA. Maximal projections are shown on the left (scale bar, 50 μm) and single confocal planes from the boxed insets are shown on the right. COX‐IV immunostaining is shown in green and DAPI‐stained nuclei in blue. Scale bar in insets, 20 μm.
- COX‐IV immunoblotting of E15.5 retinas incubated for 6 h in 3‐MA or CsA. Tubulin was used as a loading control.
- Percentage of mean fluorescence intensity (% MitoTracker) in E15.5 retinas incubated with 3‐MA, CsA or a combination of both (n = 6–22 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Student's t‐test).
Mitophagy‐mediated elimination of mitochondria during RGC development
Since autophagy is the only known catabolic pathway capable of mediating the degradation of entire mitochondria, we analysed autophagic activity during embryonic development in the mouse retina. The autophagy‐associated lipidation of microtubule‐associated proteins 1A/1B light chain 3 (MAP1LC3, better known as LC3), which gives rise to an increase in the electrophoretic mobility of LC3 (LC3‐II), was increased during retinal development (Fig 1J). Consistently, incubation of retinal explants for 3 h with the lysosomal inhibitor hydroxychloroquine (HCQ) further increased LC3‐II accumulation, indicating that basal autophagic flux is enhanced during the early stages of development of the embryonic mouse retina (Fig 1J). Transmission electron microscopy revealed the presence of mitochondria inside autophagosomes in RGCs at E15.5 in these conditions (Fig 1K) and the number of mitochondria was increased after inhibition of lysosomal degradation (Fig 1L), suggesting that the decrease in mitochondrial mass during retinal development is mediated by autophagy.
Next, we incubated retinas at different embryonic stages with the general autophagy inhibitor 3‐methyladenine (3‐MA) or with cyclosporine A (CsA), an inhibitor of mitochondrial cyclophilin D that suppresses mitophagy in many different cell types and experimental conditions (Kim et al, 2007; Carreira et al, 2010; Domenech et al, 2015; Mauro‐Lizcano et al, 2015). Inhibition of either autophagy or mitophagy for 6 h prevented the decrease in TOMM20 labelling (Figs 2A and B, and EV1B) and resulted in increased MTDR staining, as measured by flow cytometry (Fig 2C and D). Corroborating these results, immunofluorescence and immunoblot analyses revealed increased expression of the mitochondria respiratory chain component COX‐IV (Fig EV1C and D). Incubation of E15.5 retinas with a combination of 3‐MA and CsA failed to further increase mitochondrial number compared with either compound alone, suggesting that both inhibitors act on the same signalling pathway (Fig EV1E). Together, these results indicate that the autophagy‐dependent elimination of mitochondria during retinal development occurs specifically at E15.5.
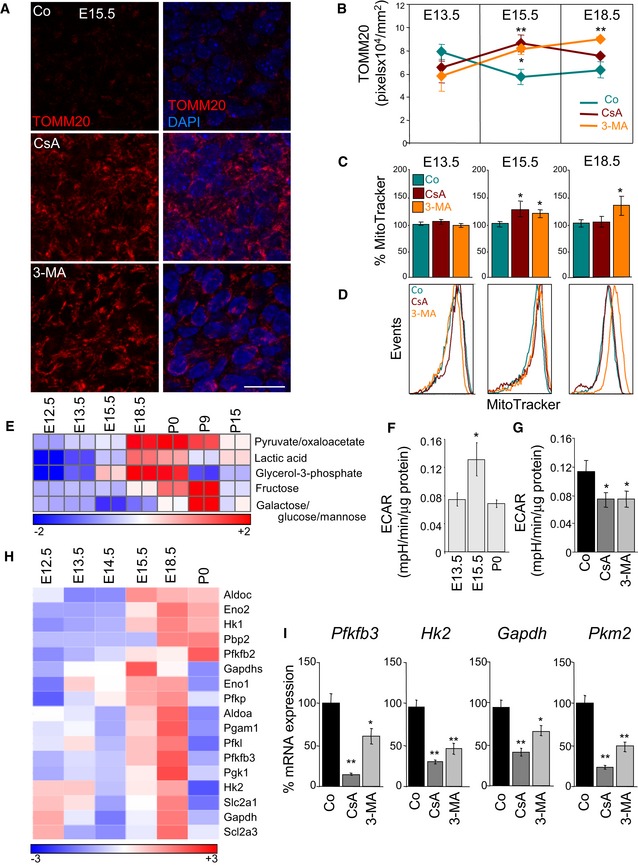
Figure 2. A mitophagy‐dependent metabolic shift towards glycolysis occurs during embryonic retinal development.
-
AMitochondrial labelling by TOMM20 immunostaining in flat‐mounted retinas (E15.5) incubated for 6 h in the presence of 5 μM CsA or 10 mM 3‐MA. TOMM20 immunostaining is shown in red and DAPI‐stained nuclei in blue. A single confocal plane is shown and corresponds to the inset from the lower magnification images displayed in Fig EV1B. Scale bar, 20 μm.
-
BQuantification of TOMM20 immunostaining in retinas cultured as in (A) at the indicated embryonic stages (n = 4–16 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
C, DQuantification of the percentage mean fluorescence intensity (% MitoTracker) and representative flow cytometry histograms at the indicated embryonic stages in retinal explants cultured as in (A) and stained with MTDR (n = 6–57 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test and Student's t‐test).
-
EHeat map indicating expression of glycolysis‐related metabolites in mouse retinas at different developmental stages. P0, Postnatal day 0.
-
F, GExtracellular acidification rate (ECAR) in embryonic retinas at the indicated developmental stages (F) (n = 19–49 pools of retinas per group) and in E15.5 retinas incubated for 6 h in the presence of 3‐MA or CsA (G) (n = 15–19 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test (F) and Student's t‐test (G)).
-
HHeat map showing the relative mRNA expression of glycolysis regulators as determined by transcriptomic analyses in mouse retinas at the indicated developmental stages.
-
ImRNA expression of glycolysis regulators in E15.5 retinas incubated for 6 h with 3‐MA or CsA (n = 3 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
Mitophagy‐dependent metabolic reprogramming in retinal development
Our previous findings in cell lines suggest that high levels of mitophagy may result in a shift from an oxidative to a glycolytic metabolic profile (Domenech et al, 2015). We next used mass spectrometry to measure the levels of selected metabolites during mouse retinal development. Levels of glycolytic metabolites, including lactate, increased from E15.5, but subsequently decreased at birth (Fig 2E). In agreement, cellular metabolic analyses using Seahorse technology revealed that the extracellular acidification rate (ECAR), which is an indirect measurement of glycolysis, increased at E15.5, but decreased at later developmental stages (Fig 2F). This increase in ECAR was prevented by incubation for 6 h with 3‐MA or CsA (Fig 2G). As expected, mitochondrial respiration decreased at E15.5 and increased in retinas treated with 3‐MA or CsA (Appendix Fig S3A and B). RNA transcriptome analyses showed a dramatic increase in mRNA expression of multiple genes encoding glycolytic enzymes (Hk2, Pfkfb3 and Gapdh) and glucose transporters (Scla2a1 and Scla2a3) beginning at E15.5 (Fig 2H). Interestingly, treatment of E15.5 retinal explants with 3‐MA or CsA significantly reduced mRNA expression of these enzymes, implicating mitophagy in the metabolic shift towards glycolysis during embryonic retinal development (Fig 2I).
Mitophagy‐dependent metabolic reprogramming and differentiation of macrophages
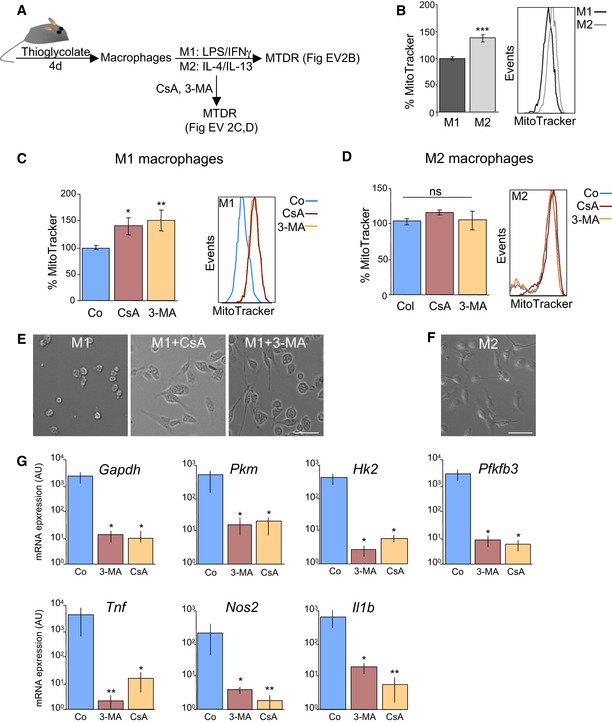
We next investigated whether mitophagy could also play a critical role in other differentiation pathways associated with increased glycolysis. Metabolic reprogramming towards glycolysis has been well described during M1 macrophage activation (O'Neill & Pearce, 2016). Macrophages can be broadly classified into two groups. M1 macrophages, generated in response to proinflammatory conditions such as TLR agonists in combination with IFN‐γ, play important roles in the elimination of bacterial infections and are considered more inflammatory (O'Neill & Pearce, 2016). IL‐4‐activated (M2) macrophages participate in tissue repair and have anti‐inflammatory properties (O'Neill & Pearce, 2016). M1 and M2 macrophages also differ in their metabolic signatures. M1 macrophages display a glycolytic profile, characterized by increased expression of several glycolytic regulators and enhanced lactate production, while M2 macrophages show high levels of oxidative phosphorylation (Rodriguez‐Prados et al, 2010; Izquierdo et al, 2015; Jha et al, 2015). Crucially, macrophages can be reprogrammed by targeting their metabolism (Izquierdo et al, 2015; Mills & O'Neill, 2016). To explore the potential role of mitophagy in M1 macrophage polarization, we induced the differentiation of peritoneal macrophages into either the M1 or M2 phenotype by incubation with LPS/INF‐γ and IL‐4/IL‐13, respectively (Fig EV2A). M1 macrophages showed decreased MTDR staining as compared with M2 macrophages, indicating a reduction in mitochondrial mass (Fig EV2B). MTDR levels dramatically increased when M1, but not M2, macrophages were cultured in the presence of 3‐MA or CsA (Fig EV2C and D). 3‐MA and CsA treatment altered cell morphology from the round shape characteristic of the M1 phenotype to the more elongated shape associated with the M2 activation state (Fig EV2E and F). Moreover, mRNA expression of M1 markers such as Tnf, Nos2 and Il1b, as well as expression of several glycolytic enzymes, was reduced when M1 macrophages were incubated in the presence of autophagy or mitophagy inhibitors (Fig EV2G). Together, these data indicate that mitophagy regulates the glycolytic shift associated with cellular differentiation in several cell types.
Figure EV2. Mitophagy sustains glycolysis in M1 macrophages.
-
ASchematic showing experimental design.
-
BMouse peritoneal macrophages were isolated from adult mice treated for 4 days with thioglycolate and were then incubated in the presence of LPS and IFN‐γ to induce M1 polarization or with IL‐4/IL‐13 to induce the M2 phenotype. Cells were then stained with MitoTracker and assessed by flow cytometry (n = 12 per group). Data are presented as mean ± SEM. ***P < 0.001 (Mann‐Whitney U‐test).
-
C, DM1 (C) or M2 (D) macrophages were incubated for 6 h with 10 mM 3‐MA or 5 μM CsA, and MitoTracker staining was assessed by flow cytometry (C, n = 6–9 per group; D, n = 6–9 per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
E, FRepresentative images of M1 macrophages (E) after incubation with 3‐MA or CsA and M2 macrophages (F). Scale bars, 50 μm.
-
GmRNA expression of the indicated genes in M1 macrophages cultured in the presence of 10 mM 3‐MA or 5 μM CsA (n = 6 per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (ANOVA).
Autophagy‐dependent metabolic reprogramming regulates RGC differentiation
We next investigated whether mitophagy and the concomitant metabolic shift observed during retinal development are implicated in the neuronal differentiation of RGCs. Inhibition of autophagy or mitophagy with CsA or 3‐MA resulted in a decrease in the number of cells positive for the RGC‐specific transcription factor Brn3a at E15.5 (Fig 3A and C). Moreover, blockade of autophagy and mitophagy resulted in alterations in axonal morphology as determined by β‐III‐tubulin labelling in retinal flatmounts (Fig 3B and D), in line with the view that inhibition of autophagy and mitophagy attenuates RGC differentiation. Next, we investigated whether experimental induction of autophagy stimulates RGC differentiation. In the developing retina, neurogenesis follows a central‐to‐peripheral gradient, meaning that the first RGCs are generated close to the optic nerve at E13.5 (Appendix Fig S1A and B). Pharmacological induction of autophagy with rapamycin at E13.5 increased the number of differentiated RGCs in this area (Fig 3E and G) and simultaneously resulted in decreased mitochondrial density, as determined by immunofluorescence (Fig 3F and H) and flow cytometry (Fig 3I–L). All these effects were prevented by simultaneous addition of CsA or 3‐MA, indicating that they involved autophagy‐dependent mitophagy (Fig 3E–L). These findings suggest that RGC differentiation is regulated by autophagy‐dependent mitochondrial degradation.
Figure 3. Retinal ganglion cell (RGC) differentiation is regulated by autophagy‐dependent mitochondrial degradation.
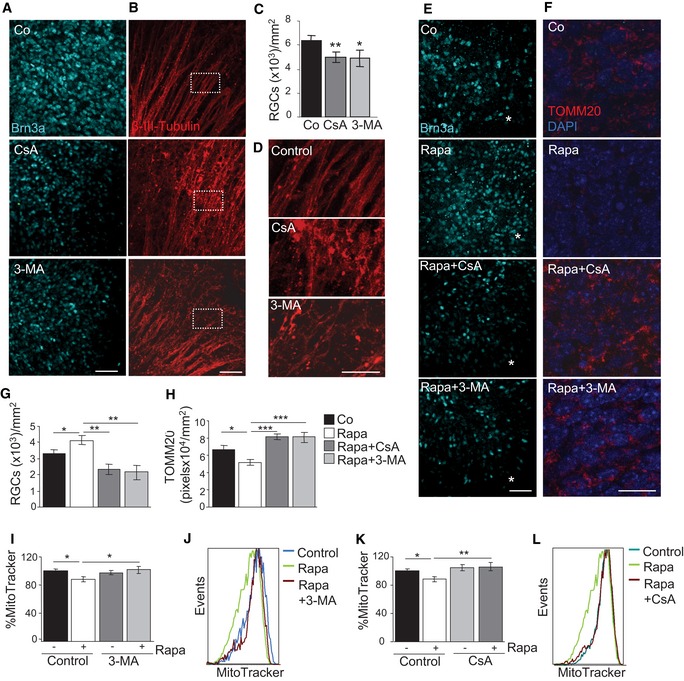
-
A–D(A) Assessment of RGC differentiation by Brn3a immunostaining in E15.5 retinas incubated for 6 h with 10 mM 3‐MA or 5 μM CsA. Maximal projection is displayed. Scale bar, 50 μm. (B) β‐III‐tubulin immunostaining of RGC axons in retinal flatmounts from E15.5 retinas incubated for 6 h with 10 mM 3‐MA or 5 μM CsA. Maximal projection is displayed. Scale bar, 50 μm. Magnified images of boxed regions are shown in (D). (C) Quantification of the number of RGCs in E15.5 retinas incubated as in (A) (n = 8–12 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
EAssessment of RGC differentiation by Brn3a immunostaining in retinal flatmounts from E13.5 retinas incubated with 200 nM rapamycin in the absence or presence of 3‐MA or CsA. Scale bar, 50 μm. Asterisks indicate the optic nerve.
-
FTOMM20 immunostaining in E13.5 retinas incubated as in (E). Scale bar, 20 μm.
-
GQuantification of the number of RGCs in retinas cultured as in (E) (n = 5–18 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
HQuantification of TOMM20 immunostaining in E13.5 retinas incubated as in (F) (n = 9–22 per group). Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001 (Mann‐Whitney U‐test).
-
I, J(I) Percentage mean fluorescence intensity (% MitoTracker) in E13.5 retinas incubated for 6 h with rapamycin in the absence or presence of 3‐MA, and a representative flow cytometry histogram (J) (n = 8–23 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
K, L(K) Percentage mean fluorescence intensity (% MitoTracker) in E13.5 retinas incubated for 6 h with rapamycin in the absence or presence of CsA, and a representative flow cytometry histogram (L) (n = 8–23 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
In support of this interpretation, immunofluorescence revealed more TOMM20‐positive structures in retinas isolated from Atg5‐deficient mice versus autophagy‐competent wild‐type controls (Fig 4A and B, and Appendix Fig S4A). Interestingly, at E13.5 this increase was observed only in the central and more mature area of the retina, where young RGCs (Y) are located (see Appendix Fig S1A and B); no differences were observed between wild‐type and autophagy‐deficient retinas in the area in which the neuroblasts (Nbs) are located (Fig 4C). Similarly, pharmacological inhibition of autophagy and mitophagy increased mitochondrial number in young RGCs, but not in neuroblasts (Appendix Fig S4C). Conversely, MTDR staining throughout the entire retina revealed no differences in mitochondrial mass at E13.5 between wt and autophagy‐deficient retinas. This can be explained by the facts that flow cytometry is performed in all retinal cells, and young RGCs only account for a small percentage of the total number of cells in the retina at this early stage of differentiation (Fig 4D). At E15.5 and E18.5, Atg5‐deficient retinas exhibited an increase in MTDR staining, as detected by flow cytometry (Fig 4D). Incubation of E15.5 Atg5‐deficient retinas with 3‐MA or CsA did not further increase MTDR staining, suggesting that pharmacological and genetic inhibition of autophagy gives rise to comparable phenotypic effects (Appendix Fig S4D and E). Analysis of autophagy‐deficient retinas revealed increased TOMM20 immunofluorescence at E15.5 in retinal sections (Fig 4E) and increased COX‐IV levels in flatmounts (Appendix Fig S4B).
Figure 4. Genetic inhibition of autophagy impairs RGC differentiation.
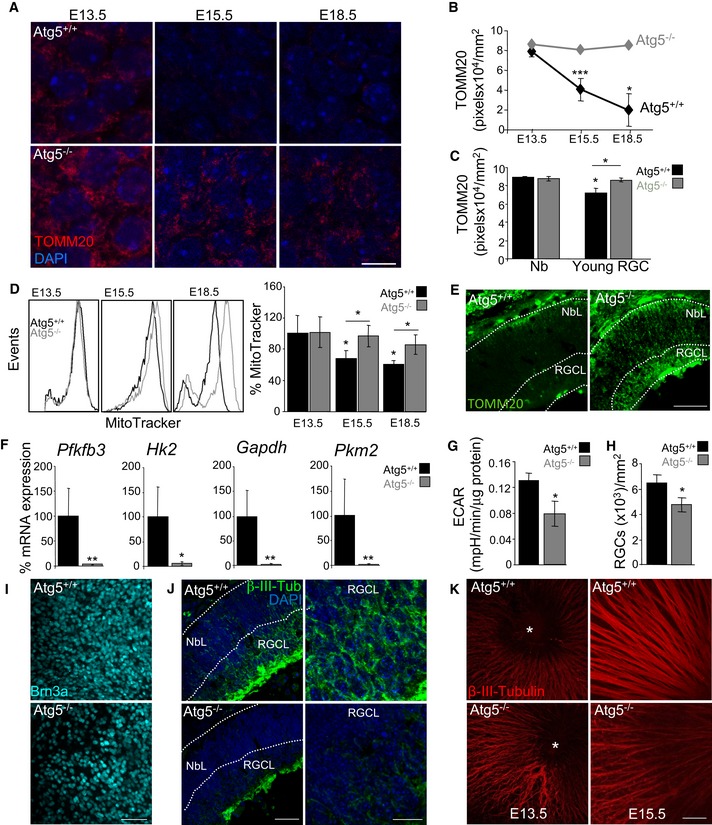
- TOMM20 immunostaining in wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) retinal whole mounts corresponding to the indicated embryonic stages. At E13.5, the central retina is shown where the young RGCs are located. Scale bar, 10 μm.
- Quantification of TOMM20 immunostaining in retinas from (A) (n = 4–15 retinas per group). Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001 (Mann‐Whitney U‐test).
- Quantification of TOMM20 immunostaining in peripheral (Nb) and central (young RGCs) areas of wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) retinas at E13.5 (n = 4–15 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
- Flow cytometry histograms and mean fluorescence intensity in dissociated retinas, stained with MTDR, corresponding to different embryonic stages, and corresponding quantification (n = 7–16 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
- TOMM20 immunostaining in E15.5 retinal sections from wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) retinas. NbL, neuroblast layer; RGCL, retinal ganglion cell layer. Scale bar, 75 μm.
- mRNA expression of the indicated genes in wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) retinas at E15.5 (n = 5–6 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
- Extracellular acidification rate (ECAR) in wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) at E15.5 (n = 11–15 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
- Quantification of the number of RGCs in (I) (n = 11–12 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
- Assessment of RGC differentiation by Brn3a immunostaining in retinal flatmounts from wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) mouse retinas at E15.5. Scale bar, 50 μm.
- β‐III‐tubulin immunostaining (green) of RGCs in retinal sections from E15.5 Atg5‐deficient mice. DAPI‐stained nuclei are shown in blue. Scale bar, 75 μm. Higher magnification images of the RGCL are shown on the right. Scale bar, 20 μm.
- β‐III‐tubulin immunostaining of RGCs axons in retinal flatmounts from E13.5 and E15.5 retinas from wild‐type (Atg5+/+) and Atg5‐deficient (Atg5−/−) animals. Scale bar, 50 μm.
Of note, Atg5‐deficient retinas showed reduced mRNA expression of several regulators of glycolysis with respect to wild‐type retinas (Fig 4F), as well as decreased lactate production as determined by ECAR (Fig 4G), in line with the deficient metabolic reprogramming towards glycolysis observed following pharmacological inhibition of autophagy or mitophagy during retinal development. Expression of the RGC‐specific transcription factor Brn3a was reduced in Atg5‐deficient retinal flatmounts as compared with controls (Fig 4H and I) and in retinal sections stained with Brn3a and γ‐synuclein (Fig EV3). Finally, in autophagy‐deficient animals staining for neuronal‐specific β‐III‐tubulin was decreased in retinal sections and revealed axonal alterations (Fig 4J and K). Together these data indicate an increase in mitochondrial mass, attenuation of the glycolytic shift and altered neuronal differentiation in Atg5‐deficient retinas.
Figure EV3. Atg‐5 deficient retinas display reduced numbers of RGCs.
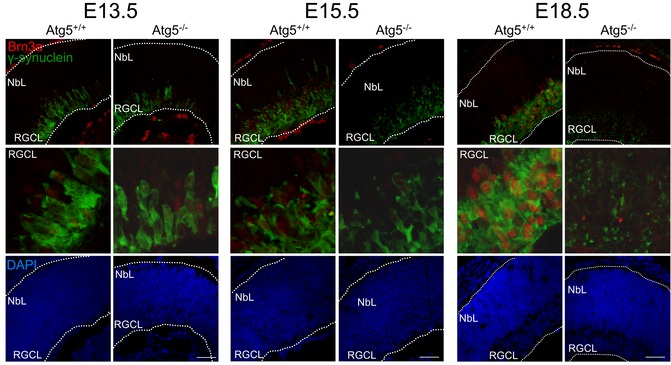
Brn3a (red) and g‐synuclein (green) staining in retinas from wt and Atg5‐deficient animals at the indicated embryonic stages. Higher magnifications on the RGCL are also depicted. DAPI was used to label nuclei. Scale bar Scale bar, 50 μm.
Hypoxia triggers mitophagy‐dependent metabolic reprogramming in RGC differentiation
Metabolic reprogramming towards glycolysis reduces oxygen consumption and is a classical response to tissue hypoxia. We thus investigated whether hypoxia occurred during mouse embryonic retinal development using the Hypoxyprobe method, which detects adducts formed between pimonidazole and proteins in conditions of low oxygen tension and has been previously used to assess hypoxic areas during embryonic development (Lee et al, 2001). We detected an elevated formation of pimonidazole adducts in the embryonic mouse retina, which were greatly reduced in the adult tissue with a faint staining in some RGCs (Fig 5A). More importantly, staining for pimonidazole adducts was observed in the somas of young RGCs as they were still migrating (Fig 5A). Staining for GLUT1 (encoded by SLC2A1, a HIF‐1 target gene) was increased at E15.5 and E18.5, confirming the transcriptomic data and further suggesting that hypoxia‐dependent metabolic reprogramming occurs at these developmental stages. Indeed, the expression of multiple HIF‐1 target genes was upregulated in embryonic mouse retinas between E15.5 and E18.5, subsequently decreasing at birth (Fig 5B). Together these data demonstrate the existence of tissue hypoxia during mouse embryonic retinal development and an increase in the expression of multiple HIF‐1 alpha‐responsive genes.
Figure 5. Hypoxia regulates mitophagy during retinal development.
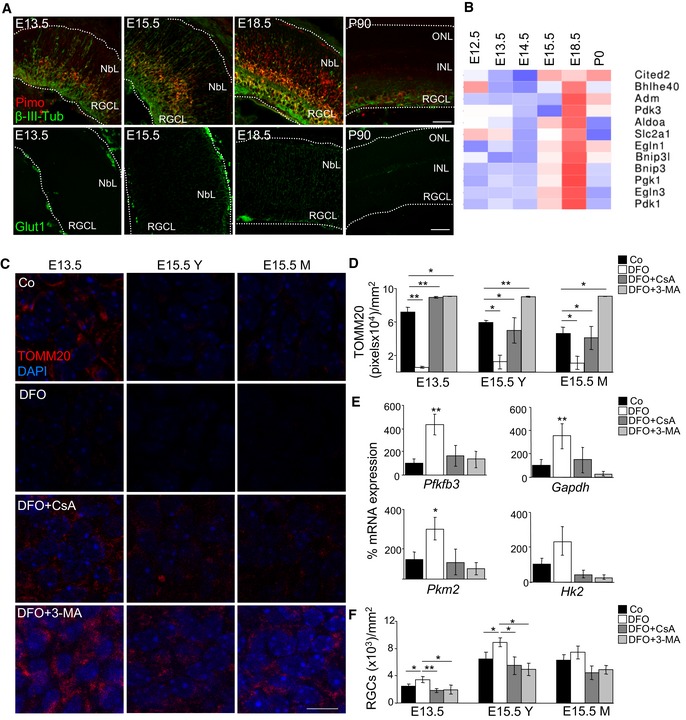
-
AImmunostaining for pimonidazole (red), β‐III‐tubulin (green, upper row) and SLC2A1 (green, bottom row) in mouse eye sections at the indicated stages. NbL, neuroblast layer; RGCL, retinal ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bar, 50 μm.
-
BHeat map showing the relative mRNA expression of HIF‐1 alpha target genes as determined by transcriptomic analyses in mouse retinas at the indicated developmental stages.
-
C, DTOMM20 immunostaining and corresponding quantification in retinal flatmounts corresponding to E13.5 and E15.5 retinas incubated with the hypoxia inducer DFO (1 mM) in the absence or presence of CsA or 3‐MA (n = 3–8 retinas per group). Scale bar, 10 μm. Y (young) indicates the peripheral area of the E15.5 retina containing less mature RGCs, and M (mature) indicates the centre of the retina, containing more mature RGCs. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
EmRNA expression of the indicated genes in E15.5 retinas treated as in C (n = 3–8 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
FQuantification of RGC number after Brn3a immunostaining in retinas treated as in (C) (n = 3–9 per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
To evaluate the relationship between hypoxia and mitophagy during RGC differentiation, E13.5 and E15.5 retinas were cultured with deferoxamine (DFO), an iron chelator that inhibits prolyl hydroxylases leading to HIF‐1 alpha stabilization (Safran & Kaelin, 2003). DFO induced marked TOMM20 degradation (Fig 5C and D), decreased MTDR staining (Fig EV4A), increased mRNA expression of glycolytic genes (Fig 5E) and stimulated RGC differentiation as determined by Brn3a immunostaining (Figs 5F and EV4B). More importantly, all these DFO‐induced effects were completely blocked by CsA or 3‐MA (Figs 5C–F, and EV4A and B), suggesting that they are mitophagy dependent. Furthermore, DFO treatment of Atg5‐deficient retinas had no additional effect on mitochondrial mass or RGC differentiation, supporting the view that the hypoxia occurs upstream of mitophagy (Appendix Fig S5). Finally, DFO treatment resulted in a decrease in mitochondrial membrane potential that was not affected by mitophagy inhibition, suggesting that this mitochondrial alteration occurs upstream, and perhaps as a trigger of mitophagy (Fig EV4C). Next, we investigated whether these phenomena were also observed in vivo during retinal development. In agreement with our ex vivo data, mitochondrial membrane potential was reduced during retinal development (Fig EV4D). Altogether these findings indicate that hypoxia‐induced mitophagy regulates RGC differentiation during retinal development.
Figure EV4. Hypoxia regulates RGC differentiation.
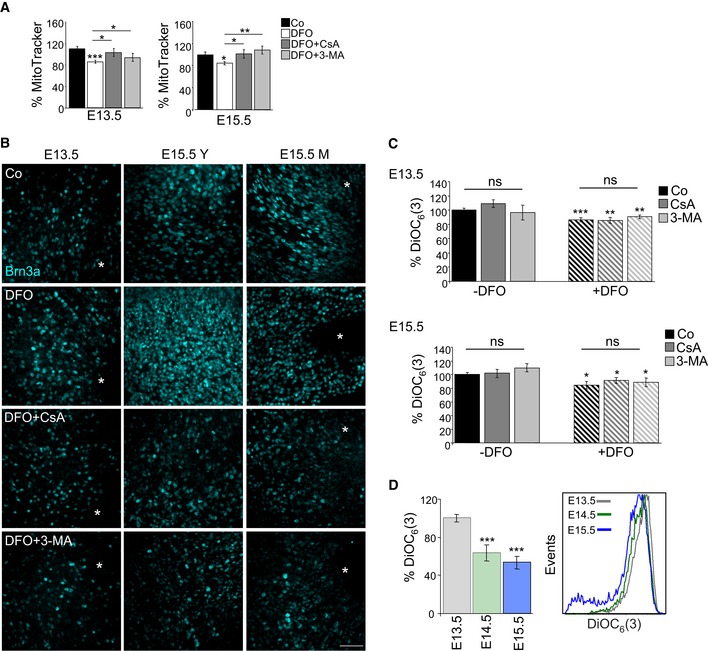
- Percentage mean fluorescence intensity (% MitoTracker) in E13.5 and E15.5 retinas incubated for 6 h with DFO in the absence or presence of 3‐MA or CsA (n = 8–14 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Mann‐Whitney U‐test and Student's t‐test).
- Assessment of RGC differentiation by Brn3a immunostaining in retinal flatmounts. Image shows Brn3a immunostaining in retinal flatmounts corresponding to E13.5 and E15.5 retinas incubated with the hypoxia inducer DFO (1 mM) in the absence or presence of CsA or 3‐MA. Scale bar, 50 μm. Y (young) indicates the peripheral area of the E15.5 retina containing less mature RGCs, and M (mature) indicates the centre of the retina, containing more mature RGCs.
- Chemical hypoxia results in decreased mitochondrial membrane potential as determined by flow cytometry with DiOC6(3) in E13.5 (n = 5–24 retinas per group) and E15.5 (n = 9–21 retinas per group) retinas incubated with the hypoxia inducer DFO (1 mM) in the absence or presence of CsA or 3‐MA. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Mann‐Whitney U‐test).
- Determination of mitochondrial membrane potential by flow cytometry with DiOC6(3) in retinas at the indicated embryonic stages (n = 10–16 retinas per group). Data are presented as mean ± SEM. ***P < 0.001 (Mann‐Whitney U‐test).
Obligatory contribution of NIX to mitophagy and RGC differentiation
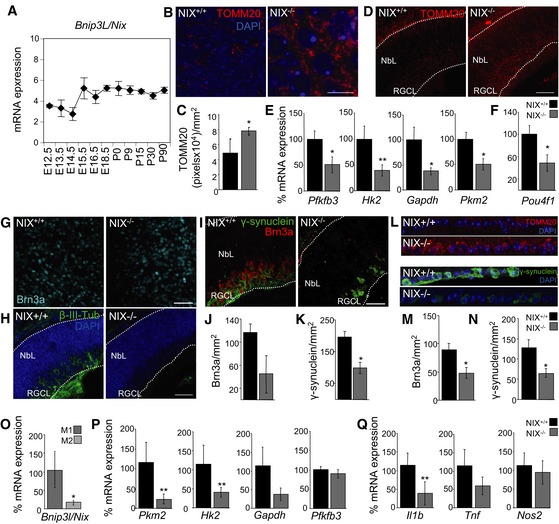
Among HIF‐1‐responsive genes, expression of the mitophagy regulator BNIP3L (NIX) increased rapidly at E15.5 (Figs 5B and 6A). This suggests that NIX upregulation may constitute the molecular link between hypoxia and mitophagy during retinal development. In support of this hypothesis, retinas from NIX‐deficient mice exhibited increased mitochondrial mass as determined by TOMM20 immunofluorescence in retinal flatmounts (Fig 6B and C) and in retinal sections (Fig 6D). Moreover, NIX‐deficient retinas showed reduced mRNA expression of several glycolytic genes including Pfkfb3, Hk2, Gapdh and Pkm2 (Fig 6E), as well as the RGC‐specific transcription factor Brn3a (known as Pou4f1; Fig 6F). These alterations correlated with a decrease in RGC numbers in retinal flatmounts and in sections stained with Brn3a and γ‐synuclein (Fig 6G–K). Moreover, adult NIX mice displayed increased mitochondrial levels and a reduced number of RGCs stained with Brn3a and γ‐synuclein (Fig 6L and N). Further supporting the view that NIX may constitute a molecular link between hypoxia and cell differentiation, NIX mRNA expression was upregulated when retinas were treated with DFO, which recapitulates the observed in vivo RGC differentiation pathway (Appendix Fig S6A and Fig 5C–F). Taken together, our results show that during retinal development activation of NIX‐dependent mitophagy at E15.5 regulates metabolic reprogramming mediating RGC differentiation.
Figure 6. NIX regulates mitophagy during retinal development.
-
AqPCR measurement of BNIP3L/NIX mRNA expression in mouse retinas at indicated developmental stages (n = 2–3 pools of retinas per embryonic stage). Data are presented as mean ± SEM.
-
B, CTOMM20 immunostaining (B) and corresponding quantification (C) (n = 4–8 retinas per group) in retinal flatmounts from wild‐type and NIX‐deficient mice at E15.5. Scale bar, 10 μm. Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
DImmunostaining for TOMM20 (red) in retinal sections from E15.5 wild‐type and NIX‐deficient mice. DAPI‐stained nuclei are shown in blue. NbL, neuroblast layer; RGCL, retinal ganglion cell layer. Scale bar, 75 μm.
-
EmRNA expression of the indicated genes in E15.5 wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mouse retinas (n = 5–8 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
FmRNA expression of Pou4f1 (Brn3a) in E15.5 wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) retinas (n = 5–8 pools of two retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Student's t‐test).
-
GAssessment of RGC differentiation by Brn3a immunostaining in retinal flatmounts from E15.5 wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice. Scale bar, 50 μm.
-
Hβ‐III‐tubulin immunostaining (green) in retinal sections from E15.5 wild‐type and NIX−/−‐deficient mice. DAPI‐stained nuclei are shown in blue. NbL, neuroblast layer; RGCL, retinal ganglion cell layer. Scale bar, 75 μm.
-
IBrn3a (red) and γ‐synuclein (green) staining in retinal sections from E15.5 wild‐type and NIX‐deficient mice. Scale bar, 50 μm.
-
JQuantification of RGC number after Brn3a immunostaining in the RGC layer in eye sections from E15.5 wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice (n = 4 eyes per group). Data are presented as mean ± SEM (Mann‐Whitney U‐test).
-
KQuantification of RGC number after γ‐synuclein immunostaining in the RGC layer in eye sections from E15.5 wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice (n = 4 eyes per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
LImmunostaining of the RGCL for TOMM20 (red), γ‐synuclein (green) and DAPI (blue) in retinal sections from adult wild‐type and NIX‐deficient mice.
-
MQuantification of RGC number after Brn3a immunostaining in the RGC layer in eye sections from adult wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice (n = 3 eyes per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
NQuantification of RGC number after γ‐synuclein immunostaining in the RGC layer in eye sections from adult wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice (n = 3 eyes per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
OqPCR measurement of BNIP3L/NIX mRNA expression in M1 and M2 macrophages (n = 6 per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
P, QqPCR measurement of mRNA expression of glycolytic (P) and proinflammatory markers (Q) in M1 peritoneal macrophages from wild‐type (NIX+/+) and NIX‐deficient (NIX−/−) mice (n = 6–8 per group). Data are presented as mean ± SEM. **P < 0.01 (Mann‐Whitney U‐test).
Obligatory contribution of NIX to M1 differentiation
We next investigated whether M1 macrophage polarization was also NIX dependent. We found that NIX mRNA expression was upregulated in M1 versus M2 macrophages (Fig 6O), in line with the fact that mitophagy is active in M1 but not M2 macrophages. Interestingly, we observed no differences in mRNA levels of other mitophagy regulators between wild‐type and NIX‐deficient macrophages (Appendix Fig S6B). Next, we isolated peritoneal macrophages from wild‐type and NIX‐deficient mice and directed their differentiation to either the M1 or M2 phenotype by incubation with LPS/INF‐γ or IL‐4/IL‐13, respectively. In NIX‐deficient M1‐macrophages, we observed reduced mRNA expression of Pkm2 and Hk2, which encode glycolytic enzymes (Fig 6P), and reduced expression of the proinflammatory cytokine‐encoding gene Il1b (Fig 6Q), indicating a NIX‐dependent metabolic shift during M1 macrophage polarization. No differences in mRNA expression of M1 or M2 markers between NIX‐deficient and wild‐type animals were observed in macrophages differentiated into the M2 macrophages (Appendix Fig S6C and D). Taken together, these data show that a NIX‐dependent metabolic shift is essential for macrophage and RGC differentiation.
Contribution of glycolysis to RGC differentiation
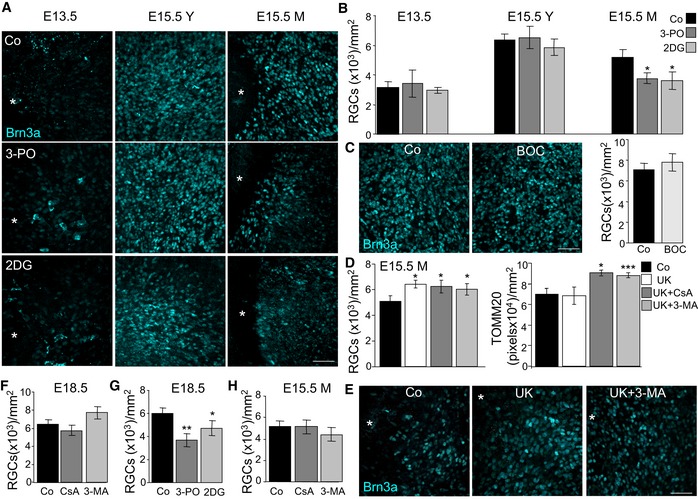
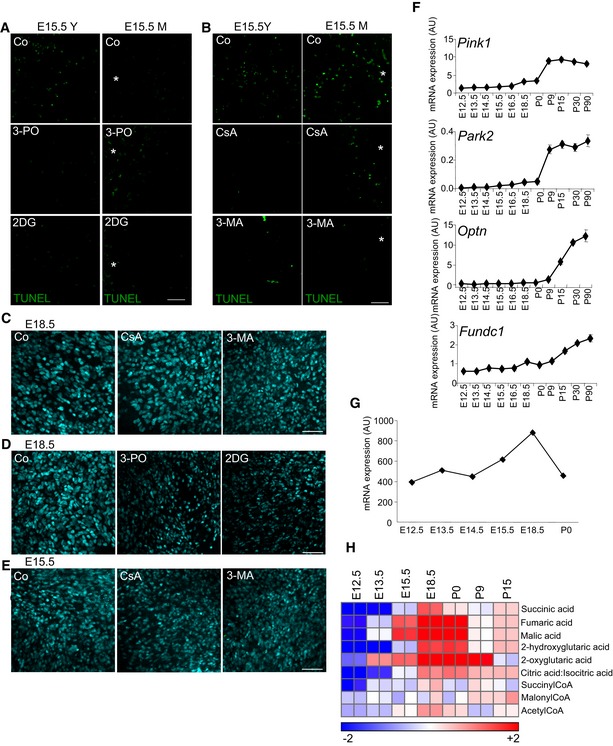
Our analyses revealed a mitophagy‐dependent metabolic shift towards glycolysis in the developing retina during the developmental stages studied. We next sought to determine whether this metabolic shift was required for RGC differentiation. We blocked glycolysis using 2‐deoxyglucose (2DG) or 3‐(3‐pyridinyl)‐1‐(4‐pyridinyl)‐2‐propen‐1‐one (3‐PO), which inhibit hexokinase and 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase, respectively. Inhibition of glycolysis decreased the number of differentiated RGCs in the central and more mature area of the retina at E15.5 (E15.5M), but had no effect on RGCs at E13.5 or on RGCs located in the peripheral retina at E15.5 (E15.5Y; Fig 7A and B, and Appendix Fig S1). This decrease in neuronal differentiation was not due to increased cell death after glycolysis inhibition, as no increase in cell death (as determined by TUNEL) was observed after the different treatments (Fig EV5A and B), and caspase inhibition did not alter RGC differentiation at E15.5 (Fig 7C). By contrast, blocking the entry of pyruvate into the mitochondria with UK5099, a strategy shown to increase glycolysis (Zhong et al, 2015), resulted in increased RGC number and neither 3‐MA nor CsA had any effect on cell differentiation under these conditions (Fig 7D and E). Interestingly, increased glycolysis did not alter mitochondrial number, indicating that the metabolic change essential for cell differentiation occurs downstream of mitophagy (Fig 7D and E).
Figure 7. Glycolysis regulates RGC differentiation.
-
ABrn3a immunostaining of RGCs in flat‐mounted retinas corresponding to the indicated embryonic stages incubated with the glycolysis inhibitors 3‐PO (10 μM) or 2DG (10 mM). Scale bar, 50 μm.
-
BQuantification of the number of RGCs in E13.5 and E15.5 retinas incubated as in (A) (n = 3–12 retinas per group). Data are presented as mean ± SEM. *P < 0.05 (Mann‐Whitney U‐test).
-
CCaspase inhibition does not influence RGC differentiation as determined by Brn3a immunostaining of RGCs in flat‐mounted E15.5 retinas treated with the pan‐caspase inhibitor Bob‐D‐FMK. Scale bar, 50 μm (n = 4 retinas per group). Data are presented as mean ± SEM (Mann‐Whitney U‐test).
-
DQuantification of RGC number and TOMM20 immunostaining in E15.5 retinas treated with 40 μM of UK5099 in the absence or presence of CsA or 3‐MA (n = 6–10 retinas per group). Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001 (Mann‐Whitney U‐test).
-
EBrn3a immunostaining of RGCs in flat‐mounted retinas corresponding to E15.5 retinas incubated with UK in absence or presence of 3‐MA. Scale bar, 50 μm.
-
F, GQuantification of RGC number in E18.5 retinas treated with CsA or 3‐MA (F) (n = 3–7 retinas per group) and with 3‐PO or 2DG (G) (n = 6–12 retinas per group). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 (Mann‐Whitney U‐test).
-
HQuantification of RGC number in E15.5 retinas treated with CsA and 3‐MA (n = 9–12 retinas per group). Data are presented as mean ± SEM (Mann‐Whitney U‐test).
Figure EV5. RGCs differentiation is regulated by a mitophagy‐dependent glycolytic switch.
-
A, BTUNEL staining to assess cell death in E15.5 retinas treated with the indicated treatments. Scale bar, 50 μm.
-
CBrn3a immunostaining of RGCs in flat‐mounted E18.5 retinas incubated with CsA or 3‐MA. Scale bar, 50 μm.
-
DBrn3a immunostaining of RGCs in flat‐mounted E18.5 retinas incubated with 3‐PO or 2DG. Scale bar, 50 μm.
-
EBrn3a immunostaining of RGCs in the mature region of flat‐mounted E15.5 retinas incubated with CsA or 3‐MA. Scale bar, 50 μm.
-
FqRT–PCR analysis of mRNA expression of the mitophagy regulators Pink1, Park2, Optn and Fundc1 (n = 2–3 pools of retinas per group). Data are presented as mean ± SEM.
-
GmRNA expression of LDHA in retinas at the indicated ages.
-
HVAccumulation of TCA cycle intermediates during retinal development.
Finally, to investigate the role of autophagy and mitophagy in the maturation of RGCs at later developmental stages, we cultured E18.5 retinas in the presence of CsA or 3‐MA. No changes in RGC numbers were observed (Figs 7F and EV5C). By contrast, the inhibition of glycolysis reduced RGC numbers at E18.5 (Figs 7G and EV5D). Together these data confirm the involvement of glycolysis in RGC maturation. Again, our data suggest that mitophagy precedes the metabolic switch, as RGC maturation was unaffected by the inhibition of autophagy or mitophagy in the mature region at E15.5 (Figs 7H and EV5E). These findings demonstrate that autophagy‐dependent mitochondrial degradation induces a metabolic shift essential for cell differentiation.
Discussion
Here, we demonstrate that mitochondrial degradation via programmed mitophagy induces a metabolic shift towards glycolysis that is essential for cell differentiation. Specifically, this mitophagy‐dependent metabolic reprogramming is required for the correct differentiation of RGCs, the first neurons generated in the vertebrate retina, and for proinflammatory M1 macrophage polarization. In the developing retina, mitophagy is regulated by hypoxia and the HIF‐1 target gene BNIP3L/NIX, which triggers mitochondrial elimination via autophagy resulting in a metabolic shift towards glycolysis (Fig 8). We observed that neurogenesis was also attenuated in autophagy‐competent retinas following inhibition of glycolysis, indicating that this increase in glycolysis is essential for cell differentiation. Taken together, our findings demonstrate for the first time that mitophagy regulates metabolic reprogramming towards glycolysis during cell differentiation.
Figure 8. Working model.
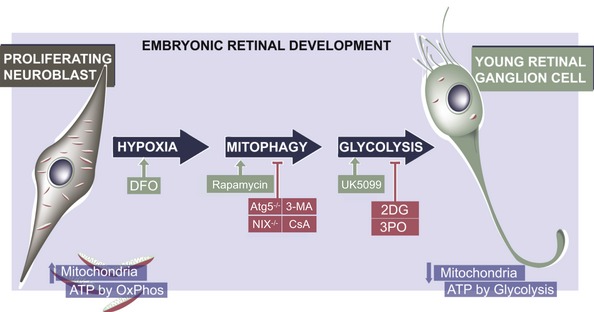
During embryonic retinal development, proliferating neuroblasts display an increase in mitochondrial number and rely on oxidative phosphorylation for ATP production, while recently differentiated retinal ganglion cells (RGCs) rely more on glycolysis and contain fewer mitochondria. The metabolic shift from the former profile to the latter is regulated by a hypoxia‐like response, which results in an increase in BNIP3L/NIX‐dependent mitophagy. This in turn decreases mitochondrial number, increases glycolysis and promotes RGC differentiation. Inhibition of autophagy (in Atg5‐deficient mice or following 3‐MA treatment), mitophagy (in NIX −/− retinas or following CsA treatment) or glycolysis (using the glycolysis inhibitor 2‐deoxyglucose (2DG) or the Pfkfb3 inhibitor 3PO) impairs neuronal differentiation (red boxes). Conversely, RGC differentiation is enhanced (green boxes) in response to deferoxamine mesylate (DFO)‐induced chemical hypoxia, mTOR inhibition with rapamycin, and after augmenting glycolysis with UK5099.
NIX transcription is usually maintained at low levels, but is induced to mediate the elimination of mitochondria during reticulocyte maturation (Diwan et al, 2007; Schweers et al, 2007; Sandoval et al, 2008) and the generation of memory NK cells (O'Sullivan et al, 2015). In the embryonic mouse retina, NIX levels peak shortly after E15.5, while those of other known mitophagy regulators such as PINK1 and Parkin increase postnatally (Fig EV5F). A similar increase in NIX transcription is observed in M1 in comparison with M2 macrophages, suggesting a pivotal role of NIX in programmed mitophagy also during macrophage polarization. Several transcription factors such as TFEB, SP1, FOXO3a, E2F1 and HIF‐1 regulate NIX transcription (Hamacher‐Brady & Brady, 2016). HIF‐1 regulates the expression of NIX in several cancer cell types, tumour‐associated macrophages and endothelial cells (Sowter et al, 2001; Ney, 2015). The accumulation of several HIF‐1‐regulated transcripts and the increased number of pimonidazole adducts observed during RGC differentiation suggests that a hypoxia‐like response may regulate programmed mitophagy during retinal development. In agreement with this view, increased HIF‐1α staining has been observed in the embryonic retina (Kurihara et al, 2010). In addition, we found that artificial induction of chemical hypoxia in E15.5 retinas increased NIX mRNA expression, reduced mitochondrial mass and increased neuronal differentiation. All these effects were attenuated when autophagy or mitophagy was blocked. While tissue hypoxia is a classical trigger for M1 macrophage polarization during inflammation, LPS and other proinflammatory stimuli have been shown to induce hypoxia‐independent HIF‐1 regulation (Escribese et al, 2012). Based on these findings, it is tempting to speculate that a HIF‐1‐dependent pathway acts as a trigger for NIX‐dependent metabolic reprogramming during the macrophage polarization and neuronal differentiation.
Beginning at E15.5, we observed a glycolytic switch in the retina, as indicated by the accumulation of glycerol‐3‐phosphate and lactate, as well as increased transcription of several glycolytic enzymes. Supporting the view that anaerobic glycolysis is upregulated during embryonic retina development, lactate dehydrogenase levels were also increased (Fig EV5G). This switch to a glycolytic profile was transient and was reversed at birth; levels of lactate, as well as mRNA expression of many glycolytic enzymes, began to decrease at birth (postnatal day 0, P0). Furthermore, this shift was mitophagy dependent; lactate accumulation was attenuated in Atg5‐deficient retinas, and both 3‐MA and CsA reduced lactate production and decreased the expression of glycolytic enzymes. These data corroborate our previous findings demonstrating that the regulation of mitochondrial mass by mitophagy can mediate metabolic reprogramming towards glycolysis in cancer cell lines (Domenech et al, 2015).
While previous results by our group and others have demonstrated the key role of autophagy in neuronal differentiation (Vazquez et al, 2012; Wang et al, 2013), the present study demonstrates for the first time that mitophagy is also essential for this process. NIX‐ and Atg5‐deficient mice displayed reduced numbers of differentiated RGCs during retinal development. Furthermore, activation of autophagy using the mTOR inhibitor rapamycin induced mitophagy and increased neuronal differentiation, an effect that was blocked by 3‐MA and CsA. Inhibition of glycolysis interfered with RGC differentiation, indicating that the mitophagy‐dependent metabolic shift is essential for RGC maturation. Interestingly, this effect occurred at E15.5 and at E18.5, while inhibition of autophagy or mitophagy only reduced RGC numbers at earlier developmental stages—where hypoxia takes place—in line with the view that hypoxia‐mediated mitophagy occurs upstream of the metabolic shift towards glycolysis.
The importance of metabolic regulation for stem cell function has been well described, and as a general rule stem cells predominantly rely on glycolytic metabolism (Chandel et al, 2016). On the other hand, the differentiation of most cell types is typically associated with increased mitochondrial activity (Agathocleous et al, 2012; Wagatsuma & Sakuma, 2013; Agostini et al, 2016; Cedikova et al, 2016; Ellen Kreipke et al, 2016). However, in agreement with our observations in RGCs, this view has been recently challenged (Forni et al, 2016). A decrease in mitochondrial activity and increased mitophagy are observed during the differentiation of mouse skin mesenchymal stem cells into chondrocytes (Forni et al, 2016). While those authors did not manipulate autophagy/mitophagy, they did show that manipulation of mitochondrial dynamic could abrogate the metabolic shift and cell differentiation. Interestingly, chondrocyte differentiation is also regulated by hypoxia (Araldi & Schipani, 2010). These observations suggest that in hypoxic conditions, as found in the embryonic retina and in the foetal growth plate during bone formation, mitophagy‐dependent metabolic reprogramming may regulate cell differentiation.
The large numbers of mitochondria found in adult RGCs (Appendix Fig S7A) suggest that mitochondrial biogenesis is increased in these cells during the postnatal period, as cell metabolism shifts to a more oxidative profile (Lathrop & Steketee, 2013; Yu et al, 2013). Furthermore, mitophagy coupled to extensive mitochondrial biogenesis could constitute a mechanism by which substrate preference is changed in order to sustain oxidative metabolism, as recently described during perinatal maturation of the mouse heart (Gong et al, 2015). However, the adult retina is dependent on aerobic glycolysis, as already observed by Warburg (1956) and recently reviewed by Ng et al (2015). This dependency is associated with the high metabolic demands of photoreceptor cells, the predominant cell type in the adult retina. Photoreceptors resemble rapidly proliferating cells in that they lose and regenerate almost 10% of their outer segment each day during disc shedding. Thus, glycolytic metabolism is implicated in two phases of retinal development: during embryonic development, as demonstrated in the present study, and in the mature retina, as previously described (Warburg, 1956; Appendix Fig S7B and C).
Here, we demonstrate the generality of our findings in a second paradigm associated with metabolic reprogramming. During polarization, M1 macrophages shift their metabolism towards glycolysis to fuel the metabolic demands imposed by inflammatory responses (O'Neill & Pearce, 2016). We found that blockade of autophagy and mitophagy during M1 macrophage polarization resulted in an increase in mitochondrial mass, and reduced expression of glycolytic genes and M1‐associated proinflammatory cytokines. This effect was also observed in NIX‐deficient macrophages. Interestingly, this decrease in M1 polarization did not result in increased M2 responses in NIX‐deficient macrophages, suggesting that mitophagy controls M1 responses. Moreover, our data indicate that a decreased M1 phenotype does not result in increased M2 phenotype, in agreement with recent evidence (Van den Bossche et al, 2016). In line with previous findings linking rapamycin treatment with increased M1 polarization (Mercalli et al, 2013), we observed an increase in RGC differentiation during retinal in response to this autophagy/mitophagy inducer. Together with the observed arrest of M1 macrophage polarization in response to blockade of autophagy and mitophagy, our data indicate that a cell differentiation pathway involving mitophagy and glycolysis is active in several cellular contexts.
Why is the mitochondrial mass of RGCs transiently reduced during differentiation? One possible explanation for this metabolic switch is the increased anabolic requirements of these neurons, which at this stage begin to generate long axons to connect the retina to the brain. In support of this hypothesis, we detected profound defects in axon morphology in autophagy‐deficient retinas. Moreover, we observed an accumulation of TCA intermediates including succinate, fumarate and malate in wild‐type mouse retinas between E15.5 and P0 (Fig EV5H). Interestingly, these metabolites also accumulate in M1 macrophages, a phenomenon known as the “broken” TCA cycle, and may actually trigger the inflammatory response (Jha et al, 2015; O'Neill, 2015). It is thus plausible that programmed mitophagy is essential to promote this metabolic reprogramming towards glycolysis to fulfil the anabolic requirements for neuronal differentiation and M1 macrophage polarization. Supporting this hypothesis, glycolysis and activity of the pentose phosphate pathway have been observed during the early stages of in vitro differentiation of cortical neurons (Agostini et al, 2016). The transient increase in glycolysis observed in the embryonic retina thus indicates a special mitophagy‐dependent metabolic requirement during the early stages of RGC differentiation. Alternatively, mitophagy may constitute a drastic mitochondrial quality control mechanism, which eliminates damaged organelles that would otherwise act as a potentially deleterious source of ROS in these long‐lived, postmitotic cells. Near‐perfect mitochondrial function is indeed critical for vision, as evidenced by the close association between blindness and mitochondriopathies (Schrier & Falk, 2011), and the mitochondrial damage that underlies several disease conditions involving RGC dysfunction (e.g. glaucoma; Chrysostomou et al, 2013).
Taken together, our data demonstrate that programmed mitophagy is essential for metabolic reprogramming towards glycolysis associated with cell differentiation. More importantly, we demonstrate that neurogenesis during mammalian neuronal differentiation is dependent on a marked increase in mitophagy.
Materials and Methods
Animal procedures
All animal procedures and study protocols were approved by the local ethics committee for animal experimentation and the ethics committees of the CSIC and the Albert Einstein College of Medicine and were carried out in accordance with US and EU regulations and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Wild‐type C57BL/6J and CD1 mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Atg5 mice (Kuma et al, 2004) were kindly provided by Prof. Noboru Mizushima (Department of Biochemistry and Molecular Biology, Graduate School and Faculty of Medicine, The University of Tokyo). NIX/BNIP3L mice (Diwan et al, 2007) were kindly provided by Prof. Gerald W. Dorn, II, Washington University in St. Louis). Both male and female animals were used in this study. Mice were reared in a barrier‐controlled facility (20°C; 12‐h light/dark cycle) with ad libitum access to food and water. Animals were crossed and the morning on which the vaginal plug was detected was designated embryonic day (E) 0.5. Animals were euthanized by cervical dislocation and embryos removed by caesarean section. The embryos were staged and then placed in a Petri dish in 1× phosphate‐buffered saline (PBS). Eyes were removed under a dissecting microscope and fixed for 1 h in 4% PFA. Alternatively, neuroretinas were dissected by separating the pigment epithelium, lens, vitreous body, vessels and other tissues. For hypoxia staining, we used the Hypoxyprobe kit (Cat. # HP3‐100kit). Pimonidazole at 60 mg/kg body weight was injected intraperitoneally. Three hours later, mice were euthanized by cervical dislocation and embryos removed by caesarean section.
Organotypic retinal cultures
Neuroretinas were cultured for 6 h in DMEM:HAM/F12 (42400, Gibco) supplemented with 10 mM sodium bicarbonate (S7561), 1.1 mg/l pyruvate (P2256), 100 mg/l apotransferrin (T2252), 0.02 mg/ml sodium selenite (S5261), 0.32 mg/l putrescine (P5780), 0.2 mg/l progesterone (P8783), all from Sigma, and 0.05 g/l gentamicin (15710, Gibco), and pH set to 7.14. Before conducting experiments, the medium was supplemented with 2 mM glutamine (25030, Gibco) and 10 nM recombinant human insulin (I2643, Sigma). Retinas were treated by adding the following compounds to the medium, as indicated: 10 mM 3‐methyladenine (3‐MA; M9281, Sigma), 200 nM rapamycin (R‐5000, Euromedex), 5 μM CsA (30024, Sigma), 10 mM 2DG (D8375, Sigma), 10 μM 3‐PO (525330, Millipore), 40 μM UK5099 (PZ0160) 1 mM DFO (D9533, Sigma), 15 μg/ml hydroxychloroquine (HCQ; 880872.4, Laboratorios Rubió S.A.) and 25 μM Boc‐D‐FMK (218575, Calbiochem). After culture, the retinas were washed twice with PBS and analysed as described below.
Flow cytometry
After isolation of culture, retinas were incubated for 5 min at 37°C in 0.5 mg/ml trypsin (3703, Worthington) in HBSS (14170‐088, Gibco) and then dissociated by gentle pipetting. Trypsin‐mediated digestion was arrested by the addition of foetal bovine serum, and cells were centrifuged at 290 g and resuspended in HBSS (14170‐088, Gibco). Cells were then incubated with 10 nM MitoTracker Deep Red (M22426, Invitrogen) or 40 nM DiOC6(3) (D273). Next, 10,000 cells were acquired in the FL4 and FL1 channels of an FC500 flow cytometer (Beckman Coulter), and the mean fluorescence intensity (MFI) of the viable cell population (determined in FSC and SSC channels) was analysed and normalized against that of untreated cells (Esteban‐Martinez et al, 2017).
Immunofluorescence staining of flat‐mounted retinas or cryosections and confocal microscopy analysis
Fixed eyes were cryopreserved in 15% sucrose in 1× PBS overnight, followed by 30% sucrose for 1 week. Eyes were embedded in Tissue‐Tek OCT (Sakura Finetechnical Co. Ltd., Tokyo, Japan) and sectioned at 12 μm on a LEICA CM 1950 cryostat. The sections were then fixed for 15 min with 4% PFA, blocked with BGT (BSA, glycine, Triton), permeated with 1% (w/v) Triton X‐100 and incubated overnight with selected antibodies as described below. Alternatively, freshly dissected or cultured retinas were flat‐mounted onto nitrocellulose membranes, fixed overnight in 4% paraformaldehyde (w/v) in 0.1 M phosphate buffer (pH 7.4) and then permeated with 1% (w/v) Triton X‐100. The retinas or cryosections were subsequently incubated overnight at 4°C with primary antibodies raised against the following antigens: TOMM20 (sc‐11415, Santa Cruz), COXIV (459600, Invitrogen), Brn3a (MAB1585, Millipore), β‐III‐tubulin (MMS‐435P, COVANCE), GLUT1 (AB400084, Abcam) and PAb2627AP (HP3‐100 kit, Hypoxyprobe). The γ‐synuclein antibody (Nguyen et al, 2011) was kindly provided by Prof. Marsh‐Armstrong (University of California at Davis). Next, samples were incubated with Alexa secondary antibodies (Invitrogen) diluted at 1:200 in 10% NGS in PBS. Nuclei were labelled with 1 μg/ml DAPI (D9542, Sigma), and samples were mounted with DABCO (D27802, Sigma). When necessary, the signal was amplified using fluorochrome‐conjugated streptavidin with the TSA System (SAT704A001EA, PerkinElmer). Confocal analysis was performed with an SP5 Leica confocal microscope. The interval between confocal planes was 1 μm for eye sections and 2 μm for flat‐mounted retinas. If not stated otherwise, maximum projections of the all z‐stacks are displayed. The TOMM20‐positive area was automatically quantified in ImageJ. The macro was created by transforming the maximum projections in the RGC layer into 16‐bit images, and a minimum detection threshold common to all images was set to define positive staining. The number of positive pixels was quantified automatically and expressed as number of pixels per 104/mm2. Quantification of RGCs was performed by counting the total number of Brn3a‐positive nuclei in the maximum projections of flat‐mounted retinas. TUNEL staining was performed as described previously (Mellén et al, 2009).
Immunoblotting
Retinas were maintained at −80°C until processing. Proteins were extracted from at least three pooled retinas in lysis buffer consisting of 50 mM Tris–HCl (pH 6.8), 10% glycerol (v/v), 2% SDS (w/v), protease inhibitor cocktail (P8783) and phosphatase inhibitors (1 mM sodium orthovanadate (S6508), 1 mM sodium fluoride (201154) and 5 mM sodium pyrophosphate (221368), all from Sigma. Retinas were homogenized using a plastic pestle until completely disaggregated. Next, samples were heated for 10 min at 95°C and stored at −20°C. Protein concentration was determined using the Pierce BCA Protein Assay Kit (23227, Pierce Thermo Fisher Scientific) and measured at 540 nm. Total protein extract (15 μg) was mixed with 10 mM DTT and bromophenol blue, loaded into Any kD Criterion TGX Precast Gels (567–1124, Bio‐Rad) and transferred onto PVDF membranes (170–4157, Bio‐Rad) for 14 min at 25 V using a Trans‐Blot Turbo Transfer System (Bio‐Rad). After transfer, membranes were activated with 100% methanol for 2 min and blocked for 1 h at room temperature with 5% non‐fat milk in PBS‐T (1× PBS, 0.5% Tween 20 (v/v)). The membranes were subsequently incubated with primary antibodies anti‐LC3 (L7543, Sigma), PCNA, (Delta Biolabs C19), GAPDH (Ab8245, Abcam), TIMM23 (BD 611222), α‐tubulin (sc‐8035, Santa Cruz), β‐actin (A5441, Sigma), TOMM20 (sc‐11415), COX‐IV (459600, Invitrogen) and PFKFB3 (131238, Cell Signaling).
Metabolic determinations
For mass spectrometry, 20 mg of retinal tissue was used obtained from pools of 28 retinas from E12.5, 35 retinas from E13.5, 24 retinas from E15.5, 24 retinas from E18.5, 24 retinas from P0 and four retinas from P9 and P15 per developmental stage. The tissue was first weighted for each condition and then solubilized with 500 μl of cold (−20°C) lysate buffer (MeOH/water/chloroform, 9/1/1, −20°C with internal standards) in 1.5 ml polypropylene Precellys lysis tubes). They were then homogenized three times for 20 s using Precellys 24 tissue homogenizer (Bertin Technologies), followed by centrifugation (10 min at 15,000 × g and 4°C). The upper phase of the supernatant was collected. Another extraction of 500 μl of lysate buffer, followed by another homogenization, was next performed. After centrifugation, the supernatant was collected and added to the first extraction. The supernatant was then evaporated in microcentrifuge tubes at 40°C in a pneumatically assisted concentrator (Techne DB3, Staffordshire, UK). Methanol (300 μl) was added to the dried extract and split into two parts of 150 μl: the first was used for the GC‐MS experiment and the second for LC‐MS determinations. The GC‐MS aliquots were solubilized in methanol and transferred to a glass tube and evaporated again. Fifty microlitres of methoxyamine (20 mg/ml in pyridine) was added to the dried extracts, which were then stored at room temperature in darkness for 16 h. The following day, 80 μl of MSTFA was added and final derivatization performed at 40°C for 30 min. Samples were then transferred to glass vials and directly injected into GC‐MS. After the second evaporation of the LC‐MS aliquots, the LC‐MS‐dried extracts were solubilized with 50 μl of MilliQ water, centrifuged (10 min at 15,000 × g and 4°C), transferred to glass vials and directly injected into UHPLC/MS. Target analysis was performed as previously described (Enot et al, 2015). After manual verifications and QC protocols, 83 metabolites were selected for GC targeted analysis and 17 metabolites for LC targeted analysis. Real‐time analysis of the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) was performed using a Seahorse XF‐24 metabolic extracellular flux analyser (Seahorse Bioscience). After dissection, neuroretinas were flat‐mounted onto nitrocellulose membranes and resuspended in XF assay medium (DMEM/F12, D0547, Sigma) and assessed in a Seahorse XF24 islet capture microplate. Values were normalized per μg of protein.
qPCR and RNA sequencing
RNA was extracted from 2 to 3 pools of six retinas (E12.5–E18.5) and of three retinas at P0. For qPCR, 5 to 6 pools of two retinas were used for ex vivo experiments and three pools of two retinas for the experiments with the Atg5−/− and the NIX−/− mice. Total RNA from retinas was extracted using TRIzol reagent, and reverse transcription performed using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer's instructions. Quantitative real‐time PCR was performed in a Light Cycler® 480 Instrument (Roche, Mannheim, Germany) with Taqman Universal PCR Master Mix using Taqman assays (Life Technologies, Carlsbad, CA, USA). The following probes were used: Ppargc1a, Mm01208835_m1; Tfam, Mm00447485_m1; Pfkfb3, Mm00504650_m1; Hk2, Mm00443385_m1; Pkm2, Mm00834102_gH; Gapdh, Mm99999915_g1; Pink1, Mm00550827_m1; Parkin, Mm00450187_m1; Optineurin, Mm01333245_m1; Fundc1, Mm00511132_m1; Bnip3L/Nix, Mm00786306; Pou4f1, Mm02343791; Il1b, Mm00434228_m1; Nos2, Mm00440502_m1; Tnf, Mm00443258_m1; Igf1, Mm00439560; Arginase1, Mm00475988; Mannose receptor, Mm00485148; 18S, Hs99999901_s1. Assays were performed in duplicate, and results were normalized according to expression levels of 18S RNA. Results are expressed using the (cycle threshold) quantification method.
Transcriptomic data have been deposited in the GEO repository (accession number GSE84299). Total RNA was extracted from a pool of 24 retinas per developmental stage. Total RNA (1 μg) with RIN (RNA integrity number) numbers ranging from 8.6 to 9.9 (Agilent 2100 Bioanalyzer) was used. PolyA+ fractions were processed using the TruSeq Stranded mRNA Sample Preparation kit (Agilent). The resulting directional cDNA libraries were sequenced for 50 bases in a single‐read format (IlluminaHiSeq 2000) and analysed with Nextpresso, a computational pipeline that performs a complete automated analysis using state‐of‐the‐art tools. Nextpresso works as follows: reads are quality‐checked with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned to the mouse genome (GRCm38/mm10) with TopHat‐2.0.10 (Trapnell et al, 2012), using Bowtie 1.0.0 and Samtools 0.1.19, allowing two mismatches and five multihits (Langmead et al, 2009; Li et al, 2009). Transcript assembly and the estimation of abundance and differential expression are determined with Cufflinks 2.2.1 (Trapnell et al, 2012), using the mouse genome annotation data set GRCm38/mm10 from the UCSC Genome Browser (Speir et al, 2016). Heatmap figures were subsequently generated with GENE‐E (http://www.broadinstitute.org/cancer/software/GENE-E/index.html). Hypoxia‐regulated genes were selected from HypoxiaDB as previously described (Khurana et al, 2013).
Primary macrophage isolation and activation
Adult CD1 or NIX mice were intraperitoneally injected with 1 ml of 4% (w/v) thioglycolate broth to elicit peritoneal macrophages (Rodriguez‐Prados et al, 2010). After 4 days, the animals were euthanized and intraperitoneally injected with 10 ml of cold sterile PBS, followed by aspiration and peritoneal lavage. Peritoneal macrophages were washed twice with 10 ml cold PBS, counted and resuspended at a concentration of 0.5 × 106 cells/ml in complete RPMI 1640 medium supplemented with 10% FBS with antibiotics. Next, 0.5 × 106 cells or 1 × 106 cells were cultured for 24 h in 24‐ and 12‐well plates for flow cytometry and RNA isolation, respectively, in the presence of LPS (100 μg/ml, E. coli 055:B5, Sigma‐Aldrich) and mIFN‐γ (20 μg/ml, Immunotools, Germany) or with mIL‐4+ mIL‐13 (20 μg/ml, Immunotools). Cells were subsequently cultured for 6 h with 10 mM 3‐MA and 5 μM CsA and then analysed by microscopy or trypsinized for mRNA extraction or flow cytometry assessment.
Transmission electron microscopy
Retinas were fixed for 4 h at 4°C in Karnovsky buffer (4% paraformaldehyde (w/v) and 2.5% glutaraldehyde (v/v) in cacodylate buffer (Sigma), pH 7.4), washed and fixed again in aqueous 5% (w/v) osmium tetroxide, 1% potassium ferricyanide. Next, the retinas were dehydrated and immersed in propylene oxide for 3 × 10 min and embedded in Epon resin (EMS). The resin was polymerized at 50°C overnight. An ultramicrotome (Vitracut E, Reichert‐Jung) was used to cut ultra‐thin sections (50 nm) for electron microscopy analysis. Sections were stained with uranyl acetate and lead citrate and analysed using a Zeiss EM 902 transmission electron microscope (Germany), set at 90 kV. Mitochondrial number was quantified in ImageJ by analysing at least 14 different images per retinal region (retinal ganglion cell layer and neuroblast layer).
Statistical analyses
Data are presented as the mean ± the standard error of the mean of at least three independent experiments performed in triplicate. Differences between treatments were analysed using a Student's t‐test, ANOVA or in the case of non‐normally distributed data, a nonparametric Mann–Whitney U‐test (SPSS 17.0). Statistical significance was set at P < 0.05. *P < 0.05, **P < 0.01, ***P < 0.001.
Author contributions
LE‐M designed and performed most of the experiments and helped in manuscript writing. ES‐F collaborated in the immunostaining of flat‐mounted retinas, confocal microscopy of flat‐mounted retinas, RNA extraction and qRT–PCR and with macrophage experiments. RSM performed the extraction and polarization of NIX‐deficient macrophages and collaborated in eye isolation of NIX‐deficient mice. ES collaborated in the immunoblotting and mice genotyping. GM discussed the data and together with DE and SD performed and analysed the mass spectrometry assay. OG analysed the transcriptomic data. MS‐R and MM helped with data discussion. AC provided the material and mice needed for experiments with NIX‐deficient animals. AMC helped with discussion of the data. GK collaborated in the mass spectrometry, data discussion and manuscript writing. PB conceived the project, designed and supervised all the experiments and wrote the manuscript with comments from co‐authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Expanded View
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Acknowledgements
Research in P.B. laboratory is supported by grants BFU2015‐65623, BFU2015‐71869‐REDT from Spain's Ministerio de Economía y Competitividad and I‐link 0701 (to P.B. and A.M.C.) from CSIC. L.E.M. was recipient of a JAE‐Pre grant from CSIC. G.M. is funded by the Ramón y Cajal Program (RYC‐2013‐12751), supported by Spain's Ministerio de Economía y Competitividad (BFU2015‐68539) and the BBVA Foundation (BBM_BIO_3105). GK is supported by the Ligue Contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the framework of E‐Rare‐2, the ERA‐Net for Research on Rare Diseases; Association Pour la Recherche sur le Cancer (ARC); Cancéropôle Ile‐de‐France; Institut National du Cancer (INCa); Institut Universitaire de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LeDucq Foundation; the LabEx Immuno‐Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI). A.C. research is supported by NIH grants EY012200 and EY014237. M.S.R. was supported by the Asociación Española contra el Cáncer (AECC, AIOA120833SALA). Work in the M.M. laboratory was supported by grants from the MINECO: (SAF2015‐69920‐R), Consolider‐Ingenio 2010 Programme (SAF2014‐57791‐REDC), Excellence Network CellSYS (BFU2014‐52125‐REDT) and the OncoCycle Programme (S2010/BMD‐2470) from the Comunidad de Madrid. We thank N. Marsh‐Armstrong and S. Martin‐Puig for reagents; O. Howard for English‐language editing; M. Casas for graphical abstract artwork; E. Rial for reagents and Seahorse analysis; and A. Corbi, T. Suárez, H.L. Vieira, A. Zorzano, J.M. Sanchez‐Puelles, M. Morán, I. Lopez‐Sanchez and I. Trounce for discussion.
The EMBO Journal (2017) 36: 1688–1706
See also: A Deczkowska & M Schwartz (June 2017)
References
- Aerbajinai W, Giattina M, Lee YT, Raffeld M, Miller JL (2003) The proapoptotic factor Nix is coexpressed with Bcl‐xL during terminal erythroid differentiation. Blood 102: 712–717 [DOI] [PubMed] [Google Scholar]
- Agathocleous M, Love NK, Randlett O, Harris JJ, Liu J, Murray AJ, Harris WA (2012) Metabolic differentiation in the embryonic retina. Nat Cell Biol 14: 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M, Harris WA (2013) Metabolism in physiological cell proliferation and differentiation. Trends Cell Biol 23: 484–492 [DOI] [PubMed] [Google Scholar]
- Agostini M, Romeo F, Inoue S, Niklison‐Chirou MV, Elia AJ, Dinsdale D, Morone N, Knight RA, Mak TW, Melino G (2016) Metabolic reprogramming during neuronal differentiation. Cell Death Differ 23: 1502–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araldi E, Schipani E (2010) Hypoxia, HIFs and bone development. Bone 47: 190–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20: 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Reggiori F, Codogno P (2013) Emerging regulation and functions of autophagy. Nat Cell Biol 15: 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Esteban‐Martinez L, Serrano‐Puebla A, Gomez‐Sintes R, Villarejo‐Zori B (2016) Autophagy in the eye: development, degeneration, and aging. Prog Retin Eye Res 55: 206–245 [DOI] [PubMed] [Google Scholar]
- Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA (2010) Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 6: 462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedikova M, Kripnerova M, Dvorakova J, Pitule P, Grundmanova M, Babuska V, Mullerova D, Kuncova J (2016) Mitochondria in white, brown, and beige adipocytes. Stem Cells Int 2016: 6067349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Jasper H, Ho TT, Passegue E (2016) Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol 18: 823–832 [DOI] [PubMed] [Google Scholar]
- Chrysostomou V, Rezania F, Trounce IA, Crowston JG (2013) Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol 13: 12–15 [DOI] [PubMed] [Google Scholar]
- Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, Molkentin JD, Macleod KF, Kalfa TA, Dorn GW II (2007) Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA 104: 6794–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech E, Maestre C, Esteban‐Martinez L, Partida D, Pascual R, Fernandez‐Miranda G, Seco E, Campos‐Olivas R, Perez M, Megias D, Allen K, Lopez M, Saha AK, Velasco G, Rial E, Mendez R, Boya P, Salazar‐Roa M, Malumbres M (2015) AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol 17: 1304–1316 [DOI] [PubMed] [Google Scholar]
- Ellen Kreipke R, Wang Y, Miklas JW, Mathieu J, Ruohola‐Baker H (2016) Metabolic remodeling in early development and cardiomyocyte maturation. Semin Cell Dev Biol 52: 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enot DP, Niso‐Santano M, Durand S, Chery A, Pietrocola F, Vacchelli E, Madeo F, Galluzzi L, Kroemer G (2015) Metabolomic analyses reveal that anti‐aging metabolites are depleted by palmitate but increased by oleate in vivo . Cell Cycle 14: 2399–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribese MM, Casas M, Corbi AL (2012) Influence of low oxygen tensions on macrophage polarization. Immunobiology 217: 1233–1240 [DOI] [PubMed] [Google Scholar]
- Esteban‐Martinez L, Villarejo‐Zori B, Boya P (2017) Cytofluorometric assessment of mitophagic flux in mammalian cells and tissues. Methods Enzymol 588: 209–217 [DOI] [PubMed] [Google Scholar]
- Forni MF, Peloggia J, Trudeau K, Shirihai O, Kowaltowski AJ (2016) Murine mesenchymal stem cell commitment to differentiation is regulated by mitochondrial dynamics. Stem Cells 34: 743–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan‐Pena S, O'Neill LA (2014) Metabolic reprograming in macrophage polarization. Front Immunol 5: 420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW II (2015) Parkin‐mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350: aad2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher‐Brady A, Brady NR (2016) Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73: 775–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo E, Cuevas VD, Fernandez‐Arroyo S, Riera‐Borrull M, Orta‐Zavalza E, Joven J, Rial E, Corbi AL, Escribese MM (2015) Reshaping of human macrophage polarization through modulation of glucose catabolic pathways. J Immunol 195: 2442–2451 [DOI] [PubMed] [Google Scholar]
- Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430 [DOI] [PubMed] [Google Scholar]
- Khurana P, Sugadev R, Jain J, Singh SB (2013) HypoxiaDB: a database of hypoxia‐regulated proteins. Database (Oxford) 2013: bat074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez‐Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Kurihara T, Kubota Y, Ozawa Y, Takubo K, Noda K, Simon MC, Johnson RS, Suematsu M, Tsubota K, Ishida S, Goda N, Suda T, Okano H (2010) von Hippel‐Lindau protein regulates transition from the fetal to the adult circulatory system in retina. Development 137: 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop KL, Steketee MB (2013) Mitochondrial dynamics in retinal ganglion cell axon regeneration and growth cone guidance. J Ocul Biol 1: 9 [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Jeong CH, Koo SY, Son MJ, Song HS, Bae SK, Raleigh JA, Chung HY, Yoo MA, Kim KW (2001) Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn 220: 175–186 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro‐Lizcano M, Esteban‐Martinez L, Seco E, Serrano‐Puebla A, Garcia‐Ledo L, Figueiredo‐Pereira C, Vieira HL, Boya P (2015) New method to assess mitophagy flux by flow cytometry. Autophagy 11: 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellén MA, de la Rosa EJ, Boya P (2008) The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ 15: 1279–1290 [DOI] [PubMed] [Google Scholar]
- Mellén MA, de la Rosa EJ, Boya P (2009) Autophagy is not universally required for phosphatidyl‐serine exposure and apoptotic cell engulfment during neural development. Autophagy 5: 964–972 [DOI] [PubMed] [Google Scholar]
- Mercalli A, Calavita I, Dugnani E, Citro A, Cantarelli E, Nano R, Melzi R, Maffi P, Secchi A, Sordi V, Piemonti L (2013) Rapamycin unbalances the polarization of human macrophages to M1. Immunology 140: 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, O'Neill LA (2016) Reprogramming mitochondrial metabolism in macrophages as an anti‐inflammatory signal. Eur J Immunol 46: 13–21 [DOI] [PubMed] [Google Scholar]
- Ney PA (2015) Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 1853: 2775–2783 [DOI] [PubMed] [Google Scholar]
- Ng SK, Wood JP, Chidlow G, Han G, Kittipassorn T, Peet DJ, Casson RJ (2015) Cancer‐like metabolism of the mammalian retina. Clin Exp Ophthalmol 43: 367–376 [DOI] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente‐Soriano FJ, Yang Z, Davis CH, Bedont JL, Son JL, Wei JO, Buchman VL, Zack DJ, Vidal‐Sanz M, Ellisman MH, Marsh‐Armstrong N (2011) Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci USA 108: 1176–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dotsch V, Ney PA, Dikic I (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA (2015) A broken krebs cycle in macrophages. Immunity 42: 393–394 [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213: 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan TE, Johnson LR, Kang HH, Sun JC (2015) BNIP3‐ and BNIP3L‐mediated mitophagy promotes the generation of natural killer cell memory. Immunity 43: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin‐Sanz P, Cascante M, Bosca L (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185: 605–614 [DOI] [PubMed] [Google Scholar]
- Safran M, Kaelin WG Jr (2003) HIF hydroxylation and the mammalian oxygen‐sensing pathway. J Clin Invest 111: 779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier SA, Falk MJ (2011) Mitochondrial disorders and the eye. Curr Opin Ophthalmol 22: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL (2001) HIF‐1‐dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61: 6669–6673 [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Fujita PA, Eisenhart C, Diekhans M, Clawson H, Casper J, Barber GP et al (2016) The UCSC genome browser database: 2016 update. Nucleic Acids Res 44: D717–D725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp DL (2015) Development of the vertebrate eye and retina. Prog Mol Biol Transl Sci 134: 397–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, Semenza GL (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9: 298–310 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenciano AI, Boya P, de la Rosa EJ (2008) Early neural cell death: numbers and cues from the developing neuroretina. Int J Dev Biol 53: 1515–1528 [DOI] [PubMed] [Google Scholar]
- Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, Luque‐Martin R, Chen HJ, Boshuizen MC, Ahmed M, Hoeksema MA, de Vos AF, de Winther MP (2016) Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep 17: 684–696 [DOI] [PubMed] [Google Scholar]
- Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, De Pablo F (2012) Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8: 187–199 [DOI] [PubMed] [Google Scholar]
- Wagatsuma A, Sakuma K (2013) Mitochondria as a potential regulator of myogenesis. ScientificWorldJournal 2013: 593267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liang CC, Bian ZC, Zhu Y, Guan JL (2013) FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci 16: 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956) On the origin of cancer cells. Science 123: 309–314 [DOI] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Balaratnasingam C, Morgan WH, Yu PK, Su EN (2013) Retinal ganglion cells: energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res 36: 217–246 [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhong Y, Li X, Yu D, Li Y, Long Y, Yuan Y, Ji Z, Zhang M, Wen JG, Nesland JM, Suo Z (2015) Application of mitochondrial pyruvate carrier blocker UK5099 creates metabolic reprogram and greater stem‐like properties in LnCap prostate cancer cells in vitro . Oncotarget 6: 37758–37769 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Expanded View
Expanded View Figures PDF
Review Process File
Source Data for Figure 1