
Fig. 1.
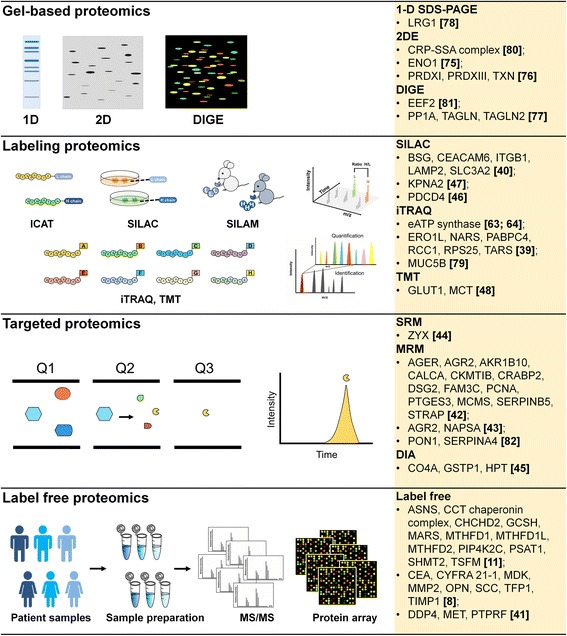
The applications of quantitative proteomics for discovery of biomarkers in lung cancer study. Quantitative proteomics not only provides a list of identified proteins, it also quantifies the changes between normal and disease sample profiles which enables to generate classification models or biomarkers. Biomarkers are measurable biological indicators found in tissue, cells, blood or other body fluids that may be used for detection, diagnosis treatment and monitoring in cancer research by the means of advanced quantitative proteomic approaches: gel-based, stable isotope labeling, targeted proteomics, and label free. In gel-based proteomics, one-dimensional (1D) gel electrophoresis, two-dimensional (2D) polyacrylamide gel electrophoresis, and difference gel electrophoresis (DIGE) approaches have been developed and utilized to separate protein from protein mixtures and identification. In vitro labeling, the peptides are modified by stable isotope labeling (ICAT, iTRAQ, TMT) prior to MS analysis. In vivo labeling, isotope labeling (SILAC and SILAM), specific supplements containing distinct forms of amino acid are given to living cells or mammals prior to MS analysis. The resulting spectrum is able to generate peptide intensity for both identification and quantitation. Targeted proteomics (SRM, MRM, and DIA) using triple quadrupole mass spectrometers systems where the mass of the intact targeted analyte is selected in the first quadrupole (Q1), and then the fragmentation of the Q1 mass-selected precursor ion by collision-induced dissociation in the second quadrupole (Q2), finally a desired product ion is selected in the third quadrupole (Q3), which is then transmitted to the detector. This method of absolute quantitation in targeted proteomics analyses is suitable for identification and quantitation of target peptides within complex mixtures independent on peptide-specific manner. Label-free quantification is an alternative method for samples that cannot directly label and enables the comparison of protein expression across different samples or treatment regardless the number of samples. Protein microarray is another label-free method which is a high-density and high-throughput microarray containing thousands of unique proteins to identify the interactions on a large scale