
Figure 1.
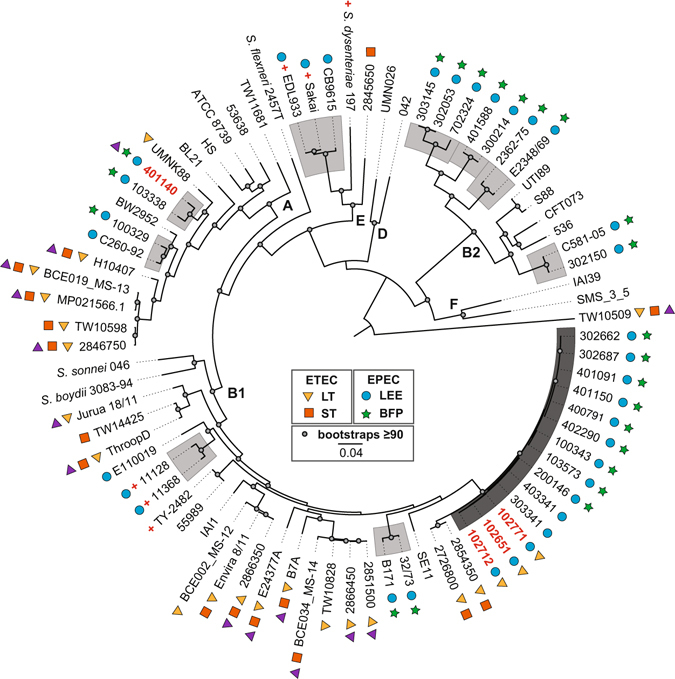
Phylogenomic analysis of the EPEC/ETEC hybrid isolates. The whole-genome sequences of the EPEC/ETEC hybrid isolates were compared with previously sequenced E. coli and Shigella genomes listed in Table S1 using a single nucleotide polymorphism (SNP)-based approach as previously described17, 43. SNPs were detected relative to the completed genome sequence of the laboratory isolate E. coli IAI39 using the In Silico Genotyper (ISG)43. A total of 159,709 conserved SNP sites, which were present in all of the genomes analyzed, were concatenated into a representative sequence for each genome. A maximum-likelihood phylogeny with 100 bootstrap replicates was inferred using RAxML v.7.2.856. The presence of E. coli virulence genes in each of the genomes is indicated by symbols as follows: LT (yellow triangle), ST (orange square), LEE (blue circle), BFP (green star), EatA (purple triangle), and Shiga-toxin (red plus sign). The letters (A, B1, B2, D, E, and F) designate the E. coli and Shigella phylogroups that were previously defined35, 36. The EPEC/ETEC hybrid isolates are indicated in bold red. The phylogenomic lineages of the LEE-containing E. coli are indicated in light grey, while the EPEC7 lineage is in dark grey. Bootstrap values ≥90 are designated by a grey circle. The scale bar represents the distance of 0.05 nucleotide substitutions per site.