

Abstract
The transcription factor JUN is frequently overexpressed in multiple genetic sub-types of acute myeloid leukemia (AML), however, the functional role of JUN in AML is not well defined. Here we report that shRNA-mediated inhibition of JUN decreases AML cell survival and propagation in vivo. By performing RNA-seq analysis, we discovered that JUN inhibition reduces the transcriptional output of the Unfolded Protein Response (UPR), an intracellular signaling transduction network activated by endoplasmic reticulum (ER) stress. Specifically, we found that JUN is activated by MEK signaling in response to ER stress and that JUN binds to the promoters of several key UPR effectors, such as XBP1 and ATF4, to activate their transcription and allow AML cells to properly negotiate ER stress. Additionally, we observed that shRNA-mediated inhibition of XBP1 or ATF4 induces AML cell apoptosis and significantly extends disease latency in vivo tying the reduced survival mediated by JUN inhibition to loss of pro-survival UPR signaling. These data uncover a previously unrecognized role of JUN as a regulator of the UPR as well as provide key new insights into the how ER stress responses contribute to AML and identify JUN and the UPR as promising therapeutic targets in this disease.
INTRODUCTION
Although numerous genetic mutations and chromosomal aberrations that drive the development of AML have been identified,1,2 the molecular components that are not mutated, but whose altered expression and function contribute to the etiology and pathophysiology of AML remain largely unknown. The transcription factor JUN is highly expressed and activated in a variety of human cancers, including AML.3–9 In comparison with healthy controls, the mean expression of JUN is substantially higher in AML patient samples bearing t(8;21), t(15;17), inv(16), 11q23 translocations as well as in those possessing complex or normal karyotype.5,6 Elevated JUN expression has also been linked to both AML recurrence and therapy-related myelodysplastic syndrome (MDS)/AML (t-MDS/AML).7,8 However, the functional and molecular roles of JUN in AML cell biology and progression are largely unknown.
The Unfolded Protein Response (UPR) is a signal transduction network comprised of three integrated signaling pathways, PERK, IRE1α and ATF6, that are collectively activated in cells experiencing endoplasmic reticulum (ER) stress.10–12 Under conditions of acute or reparable ER stress, PERK, IRE1 α and ATF6 coordinate the activation of ER-stress correcting transcriptional programs.12,13 Specifically, PERK indirectly activates the transcription factor ATF4, which then stimulates the expression of genes that regulate amino acid import and redox biology to facilitate oxidative protein folding.13–16 The endoribonuclease IRE1α drives the processing of XBP1 mRNA to generate transcripts (called XBP1s) that code for the transcriptional activator XBP1s.17–19 XBP1s and ATF6 engage transcriptional programs that facilitate cellular processes such as protein folding, ER entry of proteins, ER-associated protein degradation (ERAD) and phospholipid biosynthesis.19–24 However, when ER stress is chronic or cannot be mitigated, the UPR initiates both transcriptional (e.g. induction of CHOP) and non-transcriptional programs to promote cell death.12,13,25–29
Components of the UPR are mutated or aberrantly expressed in several settings of human cancer.11,30,31 In AML, the mean expression of XBP1 is significantly higher in AML patients compared to normal human CD34+ hematopoietic stem and progenitor cells (HSPCs) and this increased expression is associated with hypomethylation of the XBP1 promoter.32 Several studies have also observed that markers of activated UPR signaling, such as the presence of the XBP1s splice variant and increased expression of UPR-activated genes GRP78 (encoded by HSPA5), PDI and CALR, are detectable in a significant number of AML patient samples.33–35 However, the regulators of the UPR and the functional consequences of altered UPR signaling in AML have not been defined.
In this study, we have discovered that inhibition of JUN leads to widespread AML cell apoptosis and diminishes leukemia cell propagation in a genetically engineered mouse model (GEMM) of AML driven by the human leukemogenic fusion protein MLL-AF9 as well as human AML cell xenografts. At the molecular level, we have found that JUN regulates the transcription of numerous genes targeted by the UPR, including XBP1 and ATF4. Specifically, we have found that JUN binds to the promoters of XBP1 and ATF4 and that inhibition of JUN reduces UPR transcriptional output and cell survival in both unstressed and ER-stressed AML cells. Moreover, retrospective analyses of gene expression profiles of patient-derived AML cells revealed that JUN expression correlates with UPR target gene expression in multiple genetic sub-types of AML. We have also discovered that JUN is activated in response to ER stress by MEK signaling. Lastly, we have also observed that inhibition of XBP1 or ATF4 leads to widespread AML cell apoptosis and a significant increase in the time of disease onset in vivo. Collectively, these data show that JUN is a key regulator of UPR signaling and through these effects JUN plays a critical role in facilitating AML cell survival and progression.
MATERIALS & METHODS
Cell culture
Human and murine AML cell lines were described previously9 and were cultured in standard culture conditions and cell line identity and purity were verified using the Multiplex Cell Authentication and Contamination Tests (Multiplexion).
Lentiviral and retroviral Constructs
Human and mouse AML cells were transduced with recombinant lentiviruses or retroviruses as described previously.9 Construct information is described in Supplemental Material & Methods.
RNA-seq analysis
Total RNA was extracted using TRIZOL (ThermoFisher – Invitrogen). The sequencing libraries were constructed from 500 ng of total RNA using the Illumina’s TruSeq RNA Sample pre kit V2 (Illumina) following the manufacturer instructions. The fragment size of RNA-seq libraries was verified using the Agilent 2100 Bioanalyzer (Agilent) and the concentrations were determined using Qubit instrument (ThermoFisher — Life Technologies). The libraries were loaded onto the Illumina HiSeq 2500 at 6–10 pM on the rapid mode for 2×100 bp paired end read sequencing. RNA-seq analysis described in Supplemental Materials & Methods.
Western-blot
Western blot analyses were carried out as previously described9 and primary and secondary antibodies are included in Supplemental Materials & Methods.
RNA analysis
RNA was extracted using the QIAGEN RNAeasy kit and converted to cDNA with High Capacity cDNA reverse transcription kit. cDNA was amplified using SYBR Green and primers (listed Supplemental Materials & Methods) on StepOnePlus.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays was performed in THP-1 cells. Cells were crosslinked with 1% formaldehyde for 15 minutes and sonicated for 30 seconds with 30 seconds interval of rest between sonications for 10 cycles, which yielded DNA fragments of ~150–250bp. The chromatin was precipitated by anti-JUN (Cat#: 9165S, Lot#: 9) or IgG control (Cat#: 2729S, Lot#: 7) overnight at 4 °C. The eluted DNA was subjected to Real Time-PCR analysis with primers (listed in Supplemental Materials & Methods) and SYBR Green (Applied Biosystems).
Cell growth and survival
To assess cell death and proliferation, cells from each condition were stained with Annexin V and Propidium Iodide (PI) or anti-BrdU according to the manufacturer’s instruction (BD Biosciences). To assess cells were incubated with CellTiter Aqueous One solution according to the manufacturer’s instructions.
Animal Studies
All animal studies were conducted under the approval of the Fox Chase Cancer Center IUCAC (Protocol #13-1). Detailed descriptions of mouse studies are described in the Supplemental Materials & Methods.
RESULTS
JUN is required for AML cell survival
To assess the functional importance of JUN in AML, several human AML cell lines were transduced with recombinant lentiviruses expressing shRNAs targeting two distinct regions of JUN mRNA (shJUN-1 & -2) or non-targeting control shRNAs (shNT). Following stable knockdown of JUN (Figure 1A and Supplemental Figure 1A), THP-1, U937 and MOLM14 cells from each shRNA condition were seeded at equal concentrations (Day 0) and analyzed for cell number and viability 2 and 4 days later. Cells expressing either shJUN-1 or shJUN-2 displayed a substantial decrease in cell growth compared to shNT-expressing cells (Figure 1B and Supplemental Figure 1B). Compared to shNT controls, JUN shRNA-expressing cells also displayed a significant reduction in BrdU incorporation (Figure 1C and Supplemental Figure 1C) as well as increased signs of apoptosis such as increased caspase-3 cleavage and significantly more Annexin V-positive/propidium iodide-negative (Annexin V+, PI-) events (Figures 1A & D, Supplemental Figures 1D). Comparable JUN inhibition-associated defects in cell expansion and survival were also observed in the human AML cells lines Kasumi-1, HL-60, Mono-mac-6, NOMO1, MV4-11, SKM-1 and OCI-AML3 (Supplemental Figures 1E & F and data not shown). The effects of JUN shRNA treatment on cell growth and viability are very likely to result from on target knockdown of JUN, as shJUN-1 and shJUN-2 had essentially no effect on the human AML cell line ME-1, which express undetectable levels of JUN protein (Supplemental Figures 1E & F and data not shown). Collectively, these data suggest that elevated JUN expression supports AML cell proliferation and survival.
Figure 1. JUN supports AML cell survival and disease propagation in vivo.
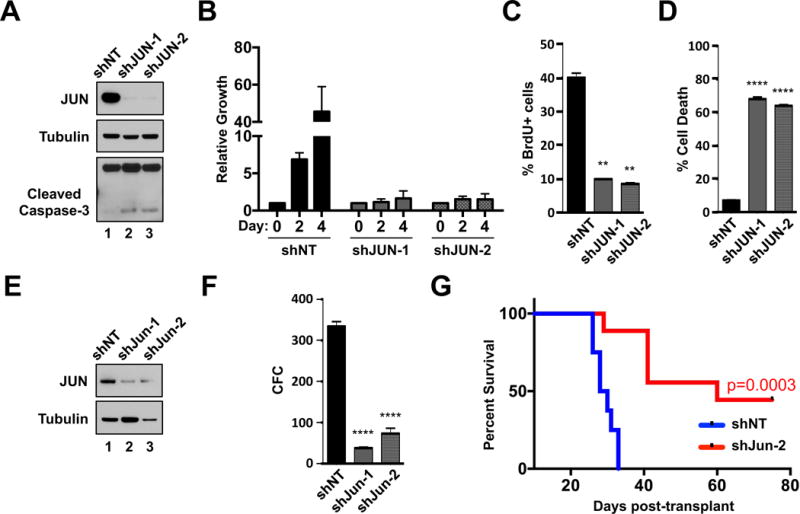
A. THP-1 cells expressing shNT or shJUN-1 & -2 were analyzed by western-blot with the indicated antibodies. B. THP-1 cells expressing shNT or shJUN were seeded at equal densities and then subsequently monitored for cell growth & survival by MTS at days 0, 2 and 4. C. THP-1 cells from each shRNA condition were assessed for BrdU incorporation by flow cytometry. D. Five days after the introduction of shRNAs, THP-1 cells expressing shNT or shJUN-1 or -2 were stained with Annexin V and propidium iodide (PI) and analyzed for cell death by flow cytometry. E & F. FACS-purified GFP+ MLL-AF9 leukemia cells from each shRNA condition were assessed by western blot with the indicated antibodies (E.) as well as colony formation in methylcellulose (F.). G. Kaplan-Meier survival curve analysis of mice transplanted with purified MLL-AF9 cells expressing shNT (blue curve, n = 8) or Jun-2 (red curve, n=9)-targeting shRNAs (P = 0.0003). (** P < 0.01, **** P < 0.0001 vs control).
JUN is required for AML cell expansion in vivo
To determine the role of JUN in leukemia growth ex vivo and in vivo, the GEMM of AML driven by MLL-AF9 was employed36. Specifically, MLL-AF9 leukemia cells recovered from leukemic mice were engineered to co-express GFP in combination with either shNT or shRNAs that target murine Jun expression. Subsequently, cells from each condition were purified by FACS and assessed for protein expression and colony forming capacity (CFC) in methylcellulose (Supplemental Figure 2A). Efficient depletion of JUN (Figure 1E) significantly reduced the CFC of MLL-AF9 leukemia cells (Figure 1F).
To examine whether suppression of JUN impairs leukemia cell expansion in vivo, purified MLL-AF9 leukemia cells expressing either shNT or shJun-2 were transplanted separately into sub-lethally irradiated syngeneic mice (Supplemental Figure 2A). Recipient mice transplanted with shJun-2-expressing leukemia cells displayed a significantly longer disease latency (p=0.0003) than recipients transplanted with shNT-expressing leukemia cells (Figure 1G). As an additional assessment of how JUN inhibition impacts AML growth in vivo, a competitive leukemia assay was performed (Supplemental Figure 2A). Specifically, primary MLL-AF9 leukemia cells were separately transduced with recombinant lentiviruses that co-express GFP and shNT, shJun-1 or shJun-2 at a Multiplicity of infection (MOI) of 0.5. Heterogeneous mixtures of GFP− (untransduced) and GFP+ (transduced) cells from each shRNA-condition were then separately transplanted into sub-lethally irradiated syngeneic recipient mice. Twenty-five days after transplant, all recipient mice from each condition developed frank leukemia with similar disease burdens, characterized by splenomegaly and neutrophilia (Supplemental Figures 2B & C, respectively). Flow cytometric analysis of several hematopoietic organs showed that mice transplanted with MLL-AF9 leukemia cells expressing either shJun-1 or shJun-2 displayed significantly lower percentages of GFP+ cells in the peripheral blood (p=0.0069 and p=0.0059) and spleen (p=0.0344 and p=0.0318) compared to mice transplanted with control (shNT-expressing) MLL-AF9 leukemia cells (Supplemental Figure 1D). The percentage of GFP+ cells in the bone marrow (BM) of mice transplanted with shJun-1 cells was also significantly lower (p=0.043) than mice transplanted with shNT-expressing cells (Supplemental Figure 1D). Although not significant, recipient mice transplanted with shJun-2-expressing MLL-AF9 leukemia cells displayed lower percentages of GFP+ cells in the BM compared to the BM of mice transplanted with shNT-expressing cells (Supplemental Figure 1D).
To ensure that the anti-leukemia effects of JUN inhibition in vivo were not restricted to mouse or MLL-AF9-driven AML, U937 cells were transduced with shNT, shJUN-1 or shJUN-2 and subsequently transplanted into NSG mice (Supplemental Figures 3A–C). Recipient mice transplanted with U937 cells expressing either shJUN-1 or shJUN-2 displayed significantly longer latency (p=0.0014, shNT vs shJUN-1 and p=0.0014, shNT vs shJUN-2) than recipient mice transplanted with shNT-expressing U937 cells (Supplemental Figure 3D). Collectively, these observations suggest that JUN is a critical regulator of AML in vitro and in vivo.
JUN inhibition alters UPR transcriptional output
To identify downstream molecular effectors of JUN, next generation sequencing (RNA-seq) was performed on THP-1 cells expressing shNT, shJUN-1 or shJUN-2. A comparison of the transcriptomes using a statistical cut-off of p<0.001 and fold change (log2) > 1 and < −1 revealed that knockdown of JUN using shRNA-1 significantly altered the transcriptome of THP-1 cells, decreasing the expression of 2514 transcripts while increasing the expression of 1096 (Supplemental Figure 4A). Additionally, 2588 transcripts were decreased and 1083 transcripts were increased in shJUN-2 expressing THP-1 cells compared to shNT-THP-1 cells and a greater than 95% overlap with shJUN-1 (Supplemental Figure 4A).
To identify molecular pathways that are impacted by JUN inhibition, a gene set/pathway enrichment analysis was performed using gene sets from the Molecular Signatures Database.37 Using a Fisher’s exact test (p<0.01), 656 gene sets were significantly altered by JUN inhibition. Organization of these results into a pathway enrichment map revealed that JUN knockdown altered several cellular processes including DNA replication, inflammatory responses, cell cycle regulation, immune processing and regulation as well as transcription factors involved in cancer (Supplemental Figure 4B) consistent with previous published studies.3,4,38–40 Interestingly, gene sets annotated from human B-cells experiencing ER stress41 were also significantly enriched in shJUN-expressing THP-1 cells compared to shNT controls (Figure 2A). Follow-up analysis found that a gene set annotated from promoters bound by the UPR transcriptional effectors ATF4 and CHOP42 was also significantly enriched — more so than any of the curated pathways examined — in THP-1 cells expressing either shJUN compared to shNT controls (Supplemental Figure 4C, shJUN-1, p=2.44 × 10−22 and shJUN-2, p=3.98 × 10−19) indicating that JUN may regulate UPR-related transcriptional programs.
Figure 2. JUN regulates the transcription of key UPR transcriptional effectors.
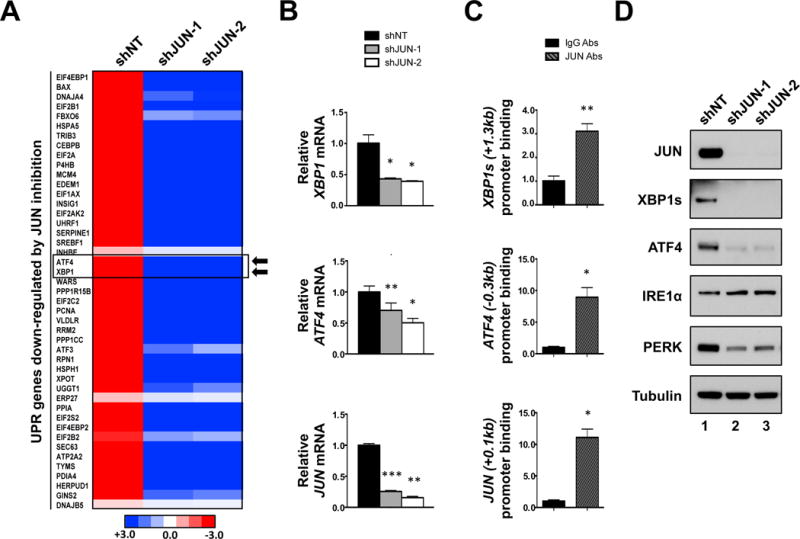
A. Heatmap representing RNA-seq data focusing on UPR transcriptional target genes that are down-regulated in JUN shRNA (shJUN-1 & -2) expressing THP-1 cells compared to shNT controls. B. Real-time qPCR analysis of XBP1, ATF4, and JUN expression in shNT, shJUN-1 and shJUN-2 expressing THP-1 cells. C. ChIP analysis using antibodies against JUN (JUN abs) or rabbit IgG (IgG abs) in THP-1 cells. Precipitated DNA was subjected to qPCR analysis using primers that amplify the indicated regions of the XBP1, ATF4, and JUN promoters. (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 vs control.) D. Lysates of shNT, shJUN-1 and shJUN-2 expressing THP-1 were subjected to western blot analysis with the indicated antibodies.
JUN binds to the promoters of UPR target genes
Numerous UPR-regulated genes were significantly altered by JUN inhibition including the UPR transcriptional effectors ATF4 and XBP1 (Figure 2A). Quantitative real time PCR (qPCR) analysis confirmed that shRNA-mediated inhibition of JUN resulted in decreased levels of XBP1 and ATF4 transcripts in THP-1 (Figure 2B). Additionally, JUN inhibition resulted in significant decreases in the steady-state levels of other UPR-target genes such as CHOP (Supplemental Figure 5A), HSPA5 and HSP90B1 (Figure 4A & Supplemental Figures 12A).
Figure 4. JUN inhibition blunts UPR activation and the ability of AML cells to negotiate ER stress.
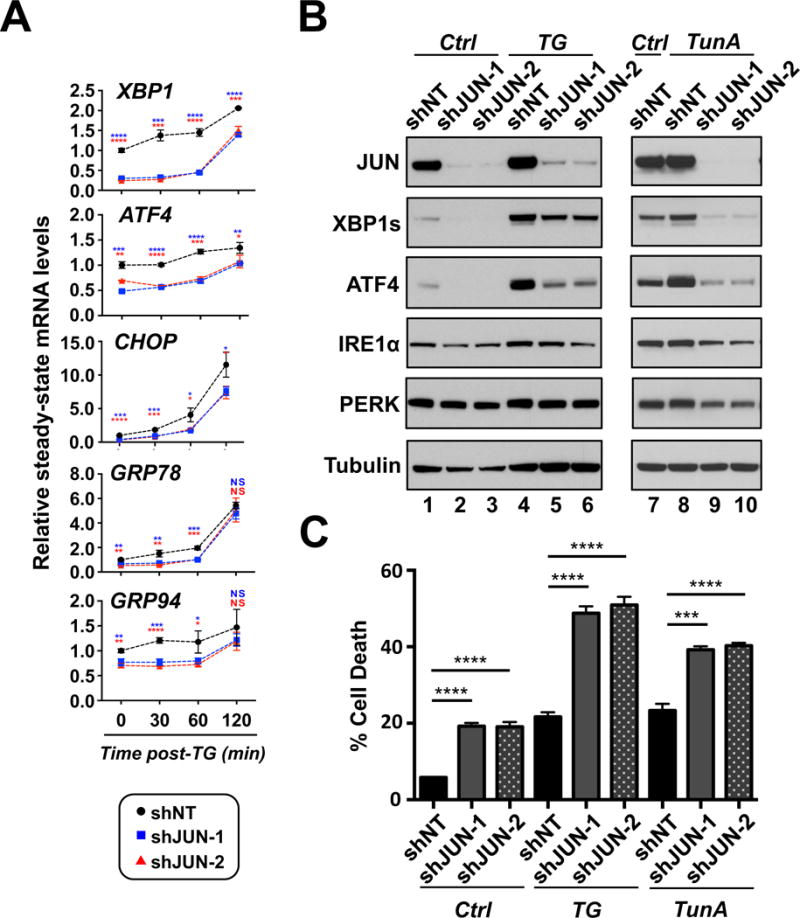
A. THP-1 cells expressing shNT or shJUN-1 or -2 were treated with TG for the indicated times and then subsequently analyzed for XBP1, ATF4, CHOP, GRP78, and GRP94 expression by real-time qPCR. B. THP-1 cells expressing shNT or shJUN-1 or -2 were treated with TG or TunA for 3 hours and then analyzed by western-blot with indicated antibodies. C. Two days after the introduction of shRNAs, THP-1 cells from each shRNA condition were treated with vehicle, 50nM TG or 1 μg/ml TunA for 24 hours followed by flow cytometry analysis for cell death. (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
Retrospective analysis of chromatin-immunoprecipitation sequencing (ChIP-seq) data deposited on the University of California Santa Cruz (UCSC) browser43–45 showed that, in several human cell lines with distinct developmental origins, JUN binding is enriched at positions +1.3, −0.3 and −0.25kb relative to the transcription start site (TSS) of the XBP1, ATF4 and CHOP promoters, respectively (Supplemental Figures 5B). ChIP assays carried out on THP-1 cells using JUN and control IgG antibodies revealed that JUN binding is enriched at the TSS of the JUN promoter (Figure 2C, first panel), consistent with its reported ability to regulate its own expression.3,4 It was also observed that JUN also binds to the +1.3, −0.3 and −0.25kb sites of the XBP1, ATF4 and CHOP promoters, respectively, in THP-1 cells (Figure 2C and Supplemental Figure 5C). Collectively, these results suggest that JUN binds to the ATF4, XBP1s and CHOP promoter regions to drive their transcription.
JUN maintains basal protein levels of XBP1s and ATF4
To assess the impact of JUN inhibition on protein levels of XBP1s and ATF4, western blot analyses performed on lysates from several human AML cell lines expressing shNT, shJUN-1 or shJUN-2. JUN inhibition resulted in decreased protein expression of both ATF4 and XBP1s in THP-1, MOLM14, SKM1 and U937 cells (Figure 2D and Supplemental Figure 6). Depletion of JUN resulted in decreased expression of ATF4 and XBP1s but did not impact the expression or phosphorylation of their UPR upstream sensors, PERK and IRE1α, respectively (Figure 2D and Supplemental Figures 6A & 6B). JUN inhibition in NOMO1 cells resulted in reduced levels of nuclear ATF6f (Supplemental Figure 6C), however, ATF6f was not detectable in U937 or THP-1 cells (data not shown). Although JUN depletion reduced CHOP transcript levels, protein levels of CHOP increased in MOLM14, U937, NOMO1 and THP-1 expressing shJUN-1 or shJUN-2 compared to controls (Supplemental Figures 6B & D).
To confirm that JUN also regulates ATF4 and XBP1s expression in MLL-AF9-expressing mouse leukemia cells, shNT, shJun-1 or shJun-2 expressing MLL-AF9 leukemia cells were isolated by FACS and then subjected to western blot analysis (Supplemental Figure 7A). Inhibition of JUN in mouse MLL-AF9 leukemia cells also resulted in decreased expression of ATF4 and XBP1s (Supplemental Figure 7B). To assess whether increasing JUN levels would yield a concomitant increase in ATF4 and XBP1s expression, MLL-AF9 leukemia cells were infected with recombinant retroviruses expressing GFP alone (MIEG) or in combination with human JUN (MIEG-JUN) (Supplemental Figure 7A). Western blot analysis showed that increased expression of JUN leads to increased expression of ATF4 and XBP1s (Supplemental Figure 7C). Collectively, these observations indicate that JUN positively regulates the expression of ATF4 and XBP1s in human and mouse AML cells.
The expression of JUN and UPR target genes correlate in multiple genetic sub-types of AML
Given that JUN expression is indicative of JUN transcriptional activity3,4 and that JUN expression is increased in certain sub-types of AML such as inv(16), t(8;21), 11q23 and t(15;17) (5,6), the possibility that the expression of JUN-regulated UPR targets may also be increased in those AML sub-types was investigated. Linear regression analyses of gene expression data retrieved from the Hemaexplorer46 databases — a curated database of gene expression profiles from normal and malignant hematopoietic cells — showed that JUN expression positively correlated with the expression of each ATF3, ATF4, CHOP, HSPA5 and PPP1R15B in the AML genetic sub-types inv(16), t(8;21) and 11q23 (Supplemental Table 1 and Supplemental Figure 8). The strongest correlations found occurred between JUN and ATF4 in the inv(16) sub-type (R2=0.85) as well as JUN and ATF3 in several sub-types (inv(16), R2=0.70 and t(8;21), R2=0.62 and 11q23, R2=0.66). Relatively strong correlations were also observed between JUN and each CHOP, HSPA5 and PPP1R15B in inv(16) and 11q23-positive AML samples (Supplemental Table 1 and Supplemental Figure 8). Correlations between JUN and XBP1 were among the weakest, however, this could be the result of XBP1 being largely regulated post-transcriptionally. Using data extracted from the AML TCGA2 dataset, similar correlations between JUN and each of these 6 genes were observed in Cytogenetically normal (CN) and Complex Karotype (CK) sub-types of AML (Supplemental Table 1). Collectively, these data show that JUN correlates with UPR signaling activity in AML patient-derived cells.
JUN expression increases in cells experiencing ER stress
To assess whether JUN expression is altered in cells experiencing ER stress, THP-1 cells were treated with vehicle, Thapsigargin (TG) or Tunicamycin A (TunA) for various times and then subjected to protein and mRNA analyses. As expected, TG or TunA treatment led to a gradual increase in the mRNA and protein levels of XBP1, ATF4, CHOP and GRP78. In THP-1 cells, JUN protein levels began to increase at 30–45 minutes post-TG and — TunA treatment and then continued to gradually increase up to 4-hours post-treatment (Figure 3A and Supplemental Figure 9B & C). JUN mRNA levels began to increase at 30 minutes post-TG or TunA treatment, maxing out at 90 minutes and then steadily declining from 90–240 minutes (Figure 3B and Supplemental Figure 9D). To determine whether ER stress-induced JUN expression is conserved in human and mouse AML cells, MLL-AF9-expressing murine AML cells recovered from leukemia mice were treated with TG and assessed for changes in UPR signaling and transcriptional output at various time points post-treatment (Supplemental Figure 10A). Analysis of protein lysates showed that JUN, ATF4, XBP1s protein levels were all increased by 45 minutes of TG-treatment, whereas CHOP and GRP78 induction required 90 and 120 minutes, respectively (Supplemental Figure 10B). Similar to human cells, JUN transcripts began to increase by 30 minutes of treatment, peaked at 120 minutes and then eventually declined after 4 hours of TG-treatment (Supplemental Figure 10C), confirming that JUN is activated in response to ER-stress in both human and mouse AML.
Figure 3. JUN is activated by MEK signaling in response to ER stress.
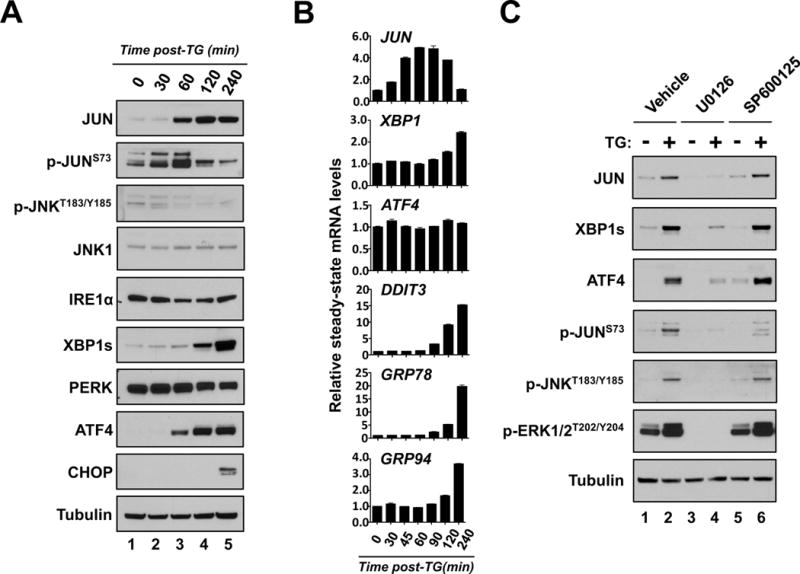
A & B. THP-1 cells were treated with TG and subsequently harvested for protein expression (A.) or (B.) mRNA analysis at the indicated times. A. Protein lysates from each of the indicated time points were subjected to western blot analysis using the indicated antibodies. B. Real-time qPCR analysis of JUN, XBP1, ATF4, CHOP, GRP78 and GRP94 expression at the indicated times. C. THP-1 cells were treated without (−) and with TG (+) in combination with vehicle, 10uM U0126 or 10uM SP600125 for 3 hours. Lysates from each condition were then analyzed by western-blot with indicated antibodies.
MEK signaling activates JUN in response to ER stress
Increased JUN expression is often paralleled by phosphorylation at multiple sites including serine 63 (JUNS63) and 73 (JUNS73) in the amino terminal region of JUN. The phosphorylation of JUN has been shown to be catalyzed by both ERK1/2 as well as the stress-activated kinase JNK.47,48 In response to TG treatment, JUNS73 phosphorylation (p-JUNS73) sharply increases at 30 and 60 minutes but then subsides by 120 minutes post-TG treatment (Figure 3A). Similar to the kinetics of p-JUNS73, phosphorylation of JNKT183/Y185 (p-JNKT183/Y185), which is indicative of JNK activation,49 also increases by 30 minutes and subsides by 120 minutes post-TG treatment (Figure 3A). Both p-JUNS73 and p-JNKT183/Y185 precede the increase in JUN protein levels, however, both decline rapidly as JUN protein levels continue to increase (Figure 3A).
Given that JNK phosphorylation of JUNS73 promotes stabilization and activation of JUN, the impact of JNK inhibition on JUNS73 phosphorylation and JUN protein levels following ER stress was evaluated. Though the JNK inhibitor, SP60012550 partially blocked JUNS73 phosphorylation, it was unable to blunt the increase in JUN protein following TG treatment (Figure 3C). To ensure that these observations are not due to the pleiotropic effects of SP600125, total and phosphorylated levels of JUN in response to ER stress were also examined upon shRNA-mediated inhibition of JNK1 or JNK2. Similar to SP600125 treatment, neither shRNA-mediated inhibition of JNK1 nor JNK2 inhibition obstructed TG-mediated stabilization of JUN (Supplemental Figure 11A & B).
Alternatively, administration of U0126 — a MEK inhibitor that effectively blocks the phosphorylation and activation of ERK1/2 (p-ERK1/2T202/Y204) — obstructed the induction of JUN protein levels, p-JUNS73 and p-JNKT183/Y185 following TG treatment (Figure 3C) suggesting that MEK signaling, possibly through ERK regulates the activation of JUN in response to ER stress.
JUN inhibition disrupts UPR signaling in response to ER stress
To evaluate whether JUN depletion impairs the activation of the UPR following acute pharmacological induction of ER stress, AML cell lines expressing shNT, shJUN-1 or shJUN-2 were treated with TG or TunA and subsequently analyzed for changes in the mRNA and protein expression of key UPR signaling components. Diminished JUN expression in vehicle-treated THP-1, U937 or NOMO1 cells resulted in significantly reduced basal mRNA levels of XBP1, CHOP, GRP78, GRP94 and ATF4 (except for U937 cells) (Figure 4A & Supplemental Figures 12A). The expression of XBP1, CHOP, GRP78, GRP94 and ATF4 were also significantly reduced at 30 and 60 minutes following TG treatment in shJUN-1 & -2 expressing cells compared to similarly treated shNT cells (Figure 4A & Supplemental Figures 12A). Additionally, it was observed that JUN depletion obstructed the induction of ATF4 and XBP1s protein expression in several human AML cell lines treated with TG or TunA (Figure 4B and Supplemental Figure 12B) indicating that JUN inhibition consistently alters UPR signaling in human AML cells exposed to ER stress.
ER stress exacerbates AML cell death induced by JUN depletion
To assess whether AML cells depleted of JUN are more sensitive to ER stress, AML cells expressing shNT, shJUN-1 or shJUN-2 were challenged with either TG or TunA at a time after efficient JUN depletion but prior to maximal cell death. The combination of JUN depletion and either TunA or TG resulted in a significant increase in the percentage of Annexin V+, PI- cells compared to JUN depletion, TunA or TG-treatment alone (Figure 4C and Supplemental Figure 12C). The observed increase in the percentage of Annexin V+/PI- cells in TunA or -TG treated JUN depleted cells was made 24-hours post-treatment. However, at longer times post-stress, the increase in cell death mediated by JUN inhibition alone became so high that the amount of cell death between vehicle- and ER stress-interrogated JUN depleted cells was nearly indistinguishable (data not shown).
Based on the observations that U0126 blocks JUN induction (Figure 3C) and JUN inhibition sensitizes AML cells to TG (Figure 4C and Supplemental Figure 12C), the effects of U0126 and TG treatment on AML cell survival was assessed. Similar to JUN inhibition, U0126 enhanced cell death mediated by moderately-lethal levels of TG (Supplemental Figure 12D). Collectively, these data suggest that JUN depletion acutely sensitizes AML cells to ER stress induced death.
JUN-regulated UPR signaling is activated by conventional AML chemotherapies
The UPR signaling pathway has been implicated in chemotherapy activity and resistance in several solid tumor settings.11 To examine how JUN and UPR components respond to chemotherapy treatment in AML, THP1 cells were treated with a combination of 100nM cytarabine (Ara-C) and 40nM Doxorubicin (Doxo) and subsequently assessed for changes in the expression of JUN and other UPR components over time. Similar to AML cells treated with TG or TunA, JUN expression increased at 60 minutes post-Ara-C/Doxo treatment followed by increased XBP1s and ATF4 expression at 120 and 240 minutes, respectively (Supplemental Figure 13A). Also analogous to TG- or TunA-treated AML cells, inhibition of JUN nullified the increases in XBP1s and ATF4 levels in response to increasing concentrations of Ara-C/Doxo (Supplemental Figure 13B) raising the possibility that JUN and the UPR may play a role chemotherapy efficacy.
XBP1 or ATF4 inhibition induces AML cell apoptosis
To assess the respective functional roles of ATF4 and XBP1 in mediating the effects of JUN depletion on AML cell survival, THP-1 and U937 cells were engineered to express shRNAs targeting either ATF4 or XBP1 (Figure 5A and Supplemental Figure 14A). Similar to JUN depletion, shRNA-mediated inhibition of either XBP1 or ATF4 resulted in increased caspase-3 cleavage (Figure 5A) and the number of Annexin-V+ cells compared to shNT controls (Figure 5B and Supplemental Figure 14B). Also consistent with JUN inhibition, shRNA-mediated inhibition of either ATF4 or XBP1 sensitized THP1 and U937 cells to AML cells to TG- or TunA treatment (Supplemental Figure 15A & B, respectively). Collectively these results indicate that XBP1 or ATF4 are needed to support AML cell survival.
Figure 5. ATF4 and XBP1 support human AML cell survival and disease propagation in vivo.
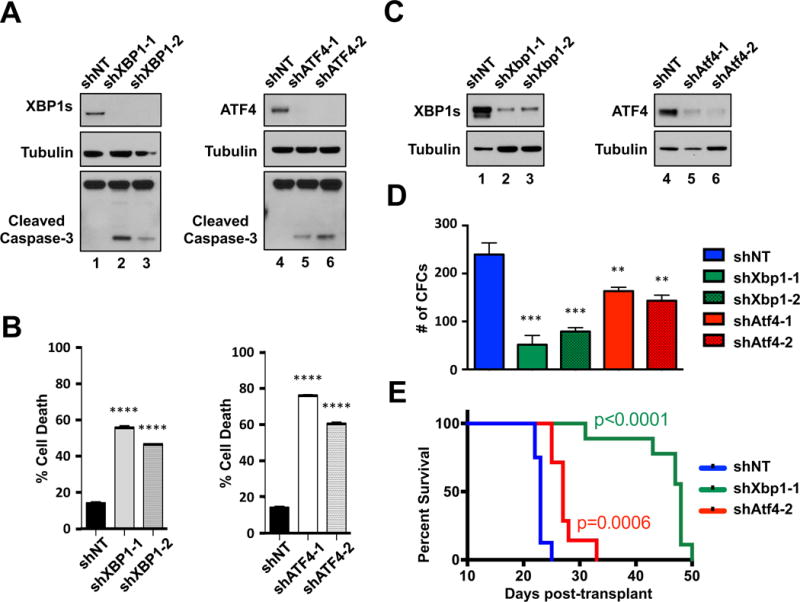
A. THP-1 cells expressing shNT or shRNAs targeting XBP-1 (shXBP1-1 & -2) or ATF4 (shATF4-1 & -2) were analyzed by western blot with the indicated antibodies. B. Five days after the introduction of shRNAs, THP-1 cells expressing shNT, shXBP1-1 or shXBP1-2 or shATF4-1 or shATF4-2 were analyzed for cell death by Annexin V/PI (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001). (C. & D.). Purified GFP+ MLL-AF9 leukemia cells from each shRNA condition were assessed for corresponding protein knockdown (C.) or colony formation in methylcellulose (D.). E. Kaplan-Meier survival curve analysis of mice transplanted with purified MLL-AF9 cells expressing shNT (blue curve, n=8), shXbp1-1 (green curve, n=9) or Atf4-2 (red curve, n=7) -targeting shRNAs (P = 0.0006, shNT vs shAtf4-2 and P < 0.0001, shNT vs shXbp1-1).
Inhibition of XBP1s or ATF4 blocks leukemia expansion in vivo
To evaluate the roles of XBP1 and ATF4 expression in AML growth ex vivo and in vivo, MLL-AF9 leukemia cells were engineered to co-express GFP and shRNAs that efficiently reduce either murine XBP1s or ATF4 expression. Transduced cells were then subjected to FACS to isolate GFP+ cells, which were then assessed for target protein knockdown, colony formation and AML induction in syngeneic recipients (Supplemental Figure 14C). Efficient depletion of either XBP1s or ATF4 (Figure 5C) significantly reduced the colony forming abilities of MLL-AF9 leukemia cells in methylcellulose (Figure 5D). Syngeneic recipient mice transplanted with FACS-purified cells expressing shATF-2 — the more efficient shRNA at reducing ATF4 protein expression — displayed a significantly longer time of disease onset (p=0.0006) compared to recipient mice transplanted with shNT-expressing cells (Figure 5E). Depletion of XBP1s expression also significantly delayed disease onset (p>0.0001) compared to shNT control mice (Figure 5E). Collectively, these observations indicate that suppressing XBP1s or ATF4 expression in this GEMM of AML reduces the ability of leukemia cells to propagate in vivo and suggest that XBP1s and ATF4 are key effectors of the pro-leukemia function of JUN.
DISCUSSION
Here we have identified that the transcription factor JUN, which is elevated in various genetic sub-types of human AML5–9, supports AML cell growth and survival. We also demonstrate that JUN positively regulates basal and ER-stress transcriptional output of the UPR in AML. Moreover, we have found that AML cells also require the UPR transcriptional effectors, XBP1 and ATF4 to maintain AML cell survival and expansion both in vitro and in vivo.
We have also observed that JUN expression positively correlates with the expression of the UPR target genes ATF3, ATF4, CHOP, HSPA5, PPP1R15B and XBP1 in several genetic sub-types of AML. However, the varying correlations between JUN and UPR target gene expression amongst AML sub-type and UPR target gene suggest that the contribution of JUN to UPR signaling may be AML sub-type specific. In 11q23 AMLs, we have found that JUN strongly correlates with UPR target gene expression and that inhibition of JUN diminishes UPR signaling in AML cells expressing MLL-AF9 — the product of the 11q23 translocation t(9;11) — indicating that JUN is a prime regulator of UPR signaling in this AML sub-type. In some AML sub-types, such as t(15;17), where correlations between JUN and UPR signaling are weak it is possible that UPR signaling is regulated by JUN-independent factors in addition to or in replacement of JUN.
It has been proposed that UPR activation proceeds in a time-ordered set of responses, where the initial stages aim to restore ER homeostasis by first shutting down protein synthesis and then engaging damage correcting transcriptional programs, whereas the latter steps function to transition cells towards apoptosis when ER stress persists or the accompanying damage is irreparable.13 Collectively, our data suggest that JUN contributes to the pro-survival, corrective functions of the UPR in AML cells. First, JUN levels increase shortly after chemical induction of ER stress in parallel with the expression of XBP1s and ATF4 but prior to the induction of GRP78 and the pro-death transcription factor CHOP. Second, JUN regulates the expression of XBP1 and ATF4, both of which engage transcriptional programs aimed at restoring ER homeostasis. Third, inhibition of JUN leads to widespread AML cell apoptosis, which can be phenocopied by either XBP1 or ATF4 inhibition. Fourth, although inhibition of JUN resulted in decreased transcription of CHOP, we did observe that CHOP protein levels increase in several AML cell lines following JUN inhibition. This is consistent with previous studies that have shown that perturbations in pro-survival UPR signaling or non-conventional ER stresses result in increased translation of CHOP and subsequent apoptosis.51,52 Such mechanisms could potentially contribute to the pro-apoptotic effects of JUN inhibition. Fifth, chemical ER stressors, such as TG or TunA accelerate AML cell death mediated by JUN, ATF4 or XBP1 inhibition.
JUN is purported to be primarily regulated by two upstream kinase-mediated pathways, JNK and ERK.47,48 JNK activates JUN in response to stresses such as ultraviolet irradiation or inflammation whereas ERK activates JUN in response to growth factor stimulation53. JNK is a well-established target of IRE1α and promotes both survival and death in response to ER stress54. In response to chemically induced ER stress, we have observed that JUN is phosphorylated at Ser73, which is a known substrate of JNK.47,48 However, we have also observed that pharmacological inhibition of MEK signaling — upstream activators of ERK1/2 —blocks ER-stress induced activation of JUN, whereas a chemical inhibitor of JNK does not. Additionally, shRNA-mediated inhibition of JNK1 or JNK2 does not obstruct ER-stress mediated stabilization of JUN. However, JNK1 and JNK2 are able to compensate for the loss of one another and while U0126 effectively blocked TG-mediated activation of ERK1/2 and JUN, it also blocked the activating phosphorylation events on JNK as well as p-JUNSer73. Collectively, these results suggest that ERK1/2 kinases are the primary activators of JUN in response to ER stress, but they do not completely eliminate the possibility that JNK signaling may play a role in ER-stress induced activation of JUN.
The observation that JUN promotes the transcription of key UPR components in AML cells was previously unrecognized. Although previous studies have implicated roles for JUN in regulating ER stress responses, no consistent picture of whether JUN influences ER stress responses has emerged. For example, in murine fibroblasts, JUN inhibition protects cells from lethal doses of the ER stressor TG55 while, in contrast, deletion of JUN in murine hepatocytes results in sustained UPR signaling and subsequent cell damage and death.56 In the hepatoma cell line Huh-7, JUN cooperates with CHOP to induce cell death in response to ER stress,57 however, CHOP has also been described to suppress JUN expression to induce apoptosis in response to chronic ER stress.58 Collectively, our data show that JUN positively regulates the UPR signal transduction network to promote AML cell survival. JUN is a basic leucine zipper (bZIP)-containing transcription factor that regulates the transcription of numerous genes by forming heterodimers with other bZIP-containing transcription factors.3,4 Moreover, the promoter selectivity of JUN is often dictated by its dimerization partner. Interestingly, ATF4 and XBP1s are also bZIP containing proteins raising the possibility they may influence the ability of JUN to regulate UPR transcriptional output. Though future studies will be needed to address this postulate, studies have shown that JUN binds to ATF4 to regulate certain transcriptional programs.59,60
The UPR possesses both pro-survival and pro-death capabilities and therefore it is not surprising that the UPR has both tumor-promoting and –suppressive effects in human cancer.11 The bifurcated impact of the UPR on cell fate insinuates that therapeutically targeting components of this network requires a thorough understanding of the precise events that dictate whether the UPR promotes cell survival or death. The results here describe a previously unrecognized role of JUN as a key UPR regulator thus providing new insights into the roles of both JUN and the UPR in AML. They also suggest that targeting JUN, possibly resulting in the blockage of UPR signaling, will have therapeutic benefit in AML. Despite immense efforts, very few JUN-targeting small molecules have entered early clinical trials and the results of most have failed or remain unknown. Therefore, additional insights of how JUN and the UPR are regulated will be central to developing highly effective therapies that capitalize on the targeting potential of these pathways in AML and possibly other human cancers.
Supplementary Material
Acknowledgments
C.Z. received support from Fox Chase Cancer Center Greenwald Fellowship.
D.D.M. received support from the Rotary Foundation, Grant GG1414529 and the Board of Directors of Fox Chase Cancer Center Fellowship.
S.M.S. received support from Bob and Jeanne Brennan, the W.W. Smith Foundation, the American Society of Hematology Junior Scholar Award and NCI grant R00CA158461.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–74. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20(19):2365–77. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- 4.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131–6. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 5.Rangatia J, Vangala RK, Singh SM, Peer Zada AA, Elsässer A, Kohlmann A, et al. Elevated c-Jun expression in acute myeloid leukemias inhibits C/EBPalpha DNA binding via leucine zipper domain interaction. Oncogene. 2003;22(30):4760–4. doi: 10.1038/sj.onc.1206664. [DOI] [PubMed] [Google Scholar]
- 6.Elsässer A, Franzen M, Kohlmann A, Weisser M, Schnittger S, Schoch C, et al. The fusion protein AML1-ETO in acute myeloid leukemia with translocation t(8;21) induces c-jun protein expression via the proximal AP-1 site of the c-jun promoter in an indirect, JNK-dependent manner. Oncogene. 2003;22(36):5646–57. doi: 10.1038/sj.onc.1206673. [DOI] [PubMed] [Google Scholar]
- 7.Staber PB, Linkesch W, Zauner D, Beham-Schmid C, Guelly C, Schauer S, et al. Common alterations in gene expression and increased proliferation in recurrent acute myeloid leukemia. Oncogene. 2004;23(4):894–904. doi: 10.1038/sj.onc.1207192. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Li M, Sun C, Francisco L, Chakraborty S, Sabado M, et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell. 2011;20(5):591–605. doi: 10.1016/j.ccr.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 11.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 12.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–90. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36(6):329–37. doi: 10.1016/j.tibs.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 15.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 16.Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29(12):2082–96. doi: 10.1038/emboj.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 19.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16(4):452–66. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 21.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003 Nov;23(21):7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, et al. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. 2007;282(10):7024–34. doi: 10.1074/jbc.M609490200. [DOI] [PubMed] [Google Scholar]
- 24.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167(1):35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345(6192):98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015;5(6):586–97. doi: 10.1158/2159-8290.CD-14-1490. [DOI] [PubMed] [Google Scholar]
- 31.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12(9):703–19. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 32.Sun H, Lin DC, Guo X, Masouleh BK, Gery S, Cao Q, et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget. 2016 doi: 10.18632/oncotarget.7702. e-pub ahead of print 25 February 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schardt JA, Weber D, Eyholzer M, Mueller BU, Pabst T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res. 2009;15(11):3834–41. doi: 10.1158/1078-0432.CCR-08-2870. [DOI] [PubMed] [Google Scholar]
- 34.Schardt JA, Eyholzer M, Timchenko NA, Mueller BU, Pabst T. Unfolded protein response suppresses CEBPA by induction of calreticulin in acute myeloid leukaemia. J Cell Mol Med. 2010;14(6B):1509–19. doi: 10.1111/j.1582-4934.2009.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haefliger S, Klebig C, Schaubitzer K, Schardt J, Timchenko N, Mueller BU, et al. Protein disulfide isomerase blocks CEBPA translation and is up-regulated during the unfolded protein response in AML. Blood. 2011;117(22):5931–40. doi: 10.1182/blood-2010-08-304485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442(7104):818–22. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 37.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 39.Uluçkan Ö, Guinea-Viniegra J, Jimenez M, Wagner EF. Signalling in inflammatory skin disease by AP-1 (Fos/Jun) Clin Exp Rheumatol. 2015;33(4 Suppl 92):S44–9. [PubMed] [Google Scholar]
- 40.Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis. 2011;70(Suppl 1):i109–12. doi: 10.1136/ard.2010.140533. [DOI] [PubMed] [Google Scholar]
- 41.Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet. 2010;86(5):719–29. doi: 10.1016/j.ajhg.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–90. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.ENCODE annotation data:; Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63. doi: 10.1093/nar/gks1172. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, et al. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci USA. 2010;107(8):3639–44. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagger FO, Rapin N, Theilgaard-Mönch K, Kaczkowski B, Thoren LA, Jendholm J, et al. HemaExplorer: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucleic Acids Res. 2013;41:D1034–9. doi: 10.1093/nar/gks1021. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991 Oct 17;353(6345):670–4. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 48.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993 Nov;7(11):2135–48. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 49.Kyriakis JM, Avruch J. pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-L-lysine. J Biol Chem. 1990 Oct 5;265(28):17355–63. [PubMed] [Google Scholar]
- 50.Chen YJ, Tan BC, Cheng YY, Chen JS, Lee SC. Differential regulation of CHOP translation by phosphorylated eIF4E under stress conditions. Nucleic Acids Res. 2010 Jan;38(3):764–77. doi: 10.1093/nar/gkp1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hess DA, Humphrey SE, Ishibashi J, Damsz B, Lee AH, Glimcher LH, Konieczny SF. Extensive pancreas regeneration following acinar-specific disruption of Xbp1 in mice. Gastroenterology. 2011 Oct;141(4):1463–72. doi: 10.1053/j.gastro.2011.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001 Nov 20;98(24):13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995 Jul 14;270(28):16483–6. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 54.Brown M, Strudwick N, Suwara M, Sutcliffe LK, Mihai AD, Ali AA, Watson JN, Schröder M. An initial phase of JNK activation inhibits cell death early in the endoplasmic reticulum stress response. J Cell Sci. 2016 Jun 15;129(12):2317–28. doi: 10.1242/jcs.179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao P, Xiao X, Kim AS, Leite MF, Xu J, Zhu X, et al. c-Jun inhibits thapsigargin-induced ER stress through up-regulation of DSCR1/Adapt78. Exp Biol Med (Maywood) 2008;233(10):1289–300. doi: 10.3181/0803-RM-84. [DOI] [PubMed] [Google Scholar]
- 56.Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE, et al. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology. 2012;55(2):408–18. doi: 10.1002/hep.24699. [DOI] [PubMed] [Google Scholar]
- 57.Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G236–43. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Z, Bu Y, Chitnis N, Koumenis C, Fuchs SY, Diehl JA. miR-216b regulation of c-Jun mediates GADD153/CHOP-dependent apoptosis. Nat Commun. 2016;7:11422. doi: 10.1038/ncomms11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001 Apr 30;20(19):2438–52. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- 60.Zhang C, Bai N, Chang A, Zhang Z, Yin J, Shen W, Tian Y, Xiang R, Liu C. ATF4 is directly recruited by TLR4 signaling and positively regulates TLR4-trigged cytokine production in human monocytes. Cell Mol Immunol. 2013;10(1):84–94. doi: 10.1038/cmi.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.