

Abstract
Background & Aims
The liver is an immunologically-privileged organ. Breakdown of liver immune privilege has been reported in chronic liver disease; however, the role of adaptive immunity in liver injury is poorly defined. NIK is known to regulate immune tissue development, but its role in maintaining liver homeostasis remains unknown. This study aimed to assess the role of NIK, particularly thymic NIK, in regulating liver adaptive immunity.
Methods
NIK was deleted systemically or conditionally using the cre/loxp system. CD4+ or CD8+ T cells were depleted using anti-CD4 or anti-CD8 antibody. Donor bone marrows or thymi were transferred into recipient mice. Immune cells were assessed by immunohistochemistry and flow cytometry.
Results
Global, but not liver-specific or hematopoietic lineage cell-specific, deletion of NIK induced fatal liver injury, inflammation, and fibrosis. Likewise, adoptive transfer of NIK-null, but not wild type, thymi into immune-deficient mice induced liver inflammation, injury, and fibrosis in recipients. Liver inflammation was characterized by a massive expansion of T cells, particularly the CD4+ T cell subpopulation. Depletion of CD4+, but not CD8+, T cells fully protected against liver injury, inflammation, and fibrosis in NIK-null mice. NIK deficiency also resulted in inflammation in the lung, kidney, and pancreas, but to a lesser degree relative to the liver.
Conclusions
Thymic NIK suppresses development of autoreactive T cells against liver antigens, and NIK deficiency in the thymus results in CD4+ T cell-orchestrated autoimmune hepatitis and liver fibrosis. Thus, thymic NIK is indispensable for the maintenance of liver immune privilege and liver homeostasis.
Keywords: Liver injury, liver inflammation, liver fibrosis, CD4+ T cells, autoimmune liver disease
Graphical abstract
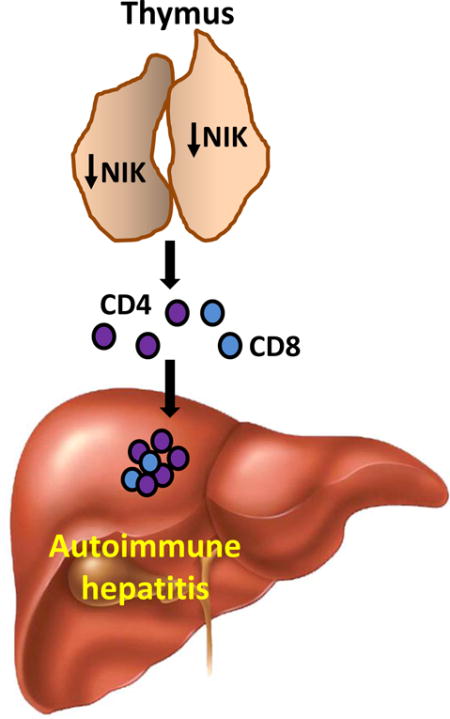
Introduction
The liver is an immunologically-privileged organ. However, liver self-tolerance is compromised in autoimmune liver diseases, including primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), and autoimmune hepatitis (AIH) (1, 2). Hepatic infiltration by T cells, particularly the CD4+ subpopulation, is reported to be associated with alcoholic liver disease, nonalcoholic steatohepatitis (NASH), and hepatotoxin-induced chronic liver injury in rodents and humans (3–7); however, the underlying mechanism responsible for the breakdown of liver immunological privilege is largely unknown. The contribution of adaptive immunity to liver disease progression is also poorly understood.
NF-κB-inducing kinase (NIK), also called MAP3K14, mediates activation of the noncanonical NF-κB2 pathway in response to a subset of cytokines (8, 9). NIK phosphorylates and activates IKKα that in turn phosphorylates NF-κB2 p100 precursors (10, 11). Phosphorylation of p100 induces ubiquitination of p100, resulting in proteolytic cleavage of p100 precursors to generate transcriptionally-active NF-κB2 p52 isoform (11). Functionally, NIK regulates lymphoid tissue development and adaptive immunity in mice (12–14). We recently reported that NIK also regulates hepatocyte-Kupffer cell (KC) crosstalk in the liver (15). Of notice, a homozygous loss of function NIK mutation in humans is associated with abnormal immunity as well as liver dysfunction (16), raising the possibility that NIK regulation of immunity and liver function may be conserved in humans.
In this study, we characterized global as well as tissue-specific NIK knockout (KO) mice. We found that whole body, but not liver-specific or hematopoietic lineage cell-specific, NIK KO mice develop fatal liver inflammation, injury, and fibrosis. Likewise, NIK deficiency in the thymus also results in autoimmune liver disease. We further demonstrated that in NIK KO mice, CD4+ T cells orchestrate immune attacks against liver.
Materials and methods
Generation of NIK KO mice
Animal experiments were conducted following the protocols approved by the University of Michigan Institutional Animal Care and Use Committee (IACUC). Two loxp sites were inserted into 2 NIK introns (NIKflox). To generate whole body NIK KO mice (NIK−/−), NIKflox mice were crossed with EIIA-Cre drives, in which Cre was expressed in germlines (17), to generate NIK+/− mice (NIKflox/+;EIIA-Cre). NIK+/− mice were backcrossed with C57BL/6 WT mice for >6 generations to eliminate EIIA-Cre. To generate hepatocyte-specific or myeloid cell-specific NIK KO mice, NIKflox/flox mice were crossed with albumin-cre or lysM-cre drivers, respectively. Mice were housed on a 12-h light-dark cycle and fed a normal chow diet (9% fat; Lab Diet, St. Louis, MO) ad libitum with free access to water.
Adoptive transfer of bone marrow cells
WT or KO recipient males (5 weeks) were pretreated with GdCl3 (i.p. 10 mg/kg body weight two times at a 4-day interval) and lethal irradiation (2×6 Gy, 3 h apart), and then received donor bone marrow cells (2×106 cells/mouse) via tail vein injection (6 h after irradiation). Donor bone marrow cells were harvested from the femurs and tibias of WT or KO mice (5 weeks) and depleted of red blood cells (RBCs) using a RBC lysis buffer (NH4Cl 155 mM, KHCO3 10 mM, EDTA 0.1 mM, pH 7.3). Recipients drank acidic water (pH 2.6) during GdCl3 treatments and for additional 2 weeks (supplemented with 0.1 mg/ml neomycin) after bone marrow transplantation.
Thymus transplantation
Donor thymi were isolated from WT or NIK KO male littermates (5 weeks). Foxn1nu male recipients (5 weeks) (Stock No: 002019, Jackson laboratory) were anesthetized with isoflurane. A midline incision was made to expose kidney on the left side, and donor thymus (25 mg) was placed under renal capsules. The incision was sutured, and health conditions were monitored daily.
Anti-CD4 or anti-CD8 antibody treatment
Mice (3 weeks) were intraperitoneally injected with anti-CD4 (GK1.5; BioXCell, BE0003-1) or anti-CD8 (YTS169.4; BioXCell, BE0117) antibody (100 μg/mouse) weekly for three consecutive weeks.
Blood analysis
Blood glucose and ALT activity were measured using glucometers (Bayer Corp., Pittsburgh, PA) and an ALT reagent set (Pointe Scientific Inc., Canton, MI), respectively.
Hepatocyte and leukocyte isolation
Primary hepatocytes were prepared from mouse liver using type II collagenase (Worthington Biochem, Lakewood, NJ) (18). To isolate leukocytes, blood samples were collected from tail vein using heparin-coated capillaries and centrifuged at 2000 rpm for 10 min at room temperature. Leukocyte pellets were washed 3 times with RBC lysis buffer.
Real-time quantitative PCR (qPCR)
Total RNAs were extracted using TRIzol reagents (Life technologies). Relative mRNA abundance of different genes was measured using SYBR Green PCR Master Mix (Life Technologies, 4367659).
Immunoblotting
Tissue samples were homogenized in lysis buffer (50 mM Tris, pH 7.5, 1% Nonidet P-40, 150 mM NaCl, 2 mM EGTA, 1 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, 1 mM benzamidine, 10 μg/ml aprotinin, 10 μg/ml leupeptin; 1 mM phenylmethylsulfonyl fluoride). Proteins were separated by SDS-PAGE and immunoblotted with the indicated antibodies.
Hydroxyproline assays
Liver samples were homogenized in 6 N HCl, hydrolyzed at 100 °C for 18 h and centrifuged at 10000 rpm for 5 min. Supernatant was dried in speed-vacuum, dissolved in H2O, and neutralized with 10 N NaOH. Samples were incubated in a chloramine-T solution (60 mM chloramines-T (Sigma, 857319), 20 mM citrate, 50 mM acetate, pH 6.5) for 25 min at room temperature, and then in Ehrlich’s solution (Sigma, 038910) at 65 °C for additional 20 min. Hydroxyproline content was measured using a Beckman Coulter AD 340 Plate Reader (570 nm) and normalized to liver weight.
ROS assays
Liver lysates were mixed with a dichlorofluorescein diacetate fluorescent (DCF, Sigma, D6883) probe (5 μM) for 1 h at 37 °C. DCF fluorescence was measured using a BioTek Synergy 2 Multi-Mode Microplate Reader (485 nm excitation and 527 nm emission).
Immunostaining
Liver frozen sections were prepared using a Leica cryostat (Leica Biosystems Nussloch GmbH, Nussloch, Germany), fixed in 4% paraformaldehyde for 30 min, blocked for 3 h with 5% normal goat serum (Life Technologies) supplemented with 1% BSA, and incubated with the indicated antibodies at 4°C overnight. The sections were incubated with Cy2 or Cy3-conjugated secondary antibodies.
Hematoxylin and eosin (H&E) and Sirius red staining
Liver paraffin sections were stained with H & E or 0.1% Sirius-red (Sigma, 365548) and 0.1% Fast-green (Sigma, F7252) (dissolved in saturated picric acid).
TUNEL assays
Liver frozen sections were fixed with 4% paraformaldehyde and subjected to TUNEL assays using an In Situ Cell Death Detection Kit (Roche Diagnostics, Indianapolis, IN, 11684817910), following manufacturer’s recommended procedures.
Flow cytometry
The liver was perfused with PBS via the portal vein to wash out red blood cells, dissected, and mechanically disrupted between two frosted microscope sides. Liver homogenates were filtered through a 100 μm cell strainer, resuspended in 40% isotonic Percoll solution, loaded on the top of a 70% Percoll solution, and centrifuged at 970×g at room temperatures with no brakes for 30 min. Nonparenchymal cells were collected at the interface of the two Percoll layers, washed, and resuspended in PBS supplemented with 2% FBS. Splenic RBCs were lysed by incubation of cells in 1.66% NH4Cl solution at room temperature for 10 min. Cells (~2×106 in 100 μl) were stained with the indicated antibodies in the presence of anti-FcγR antibody 2.4 G2 (blocking nonspecific binding sites). Cells were also stained with either propidium iodide (PI) or live-dead fixable dye to exclude dead cells from live cells. Surface-stained cells were fixed, permeabilized, and then stained with antibodies against cytokines using Cytofix/Cytoperm kit (BD Bioscience) or antibody against FoxP3 using the Foxp3/Transcription Factor Staining Buffer Kit (eBioscience). Cell fluorescence was assessed using FACSCanto II (BD Biosciences), and data were analyzed with FlowJo software (version 9.7; Tree Star, Ashland, OR). For analysis, forward and side scatter parameters were used for exclusion of doublets. Antibody information was listed in supplementary table 2C.
Statistical Analysis
Data were presented as means ± sem. Differences between groups were analyzed with two-tailed Student’s t test. P < 0.05 was considered statistically significant.
Results
Deletion of NIK results in growth retardation, hypoglycemia, and premature death
To conditionally delete NIK, two loxP sites were inserted into the NIK loci (Fig. 1A). Cre-mediated deletion is expected to excise 5 exons. Global NIK KO mice were generated by crossing NIKflox/flox mice with EIIA-cre drivers (17). EIIA-cre was subsequently removed by backcross with C57BL/6 mice (>6 generations). We confirmed that NIK was disrupted in KO but not wild type (WT) littermates (Fig. 1B).
Fig. 1. Deletion of NIK results in hypoglycemia and premature death.
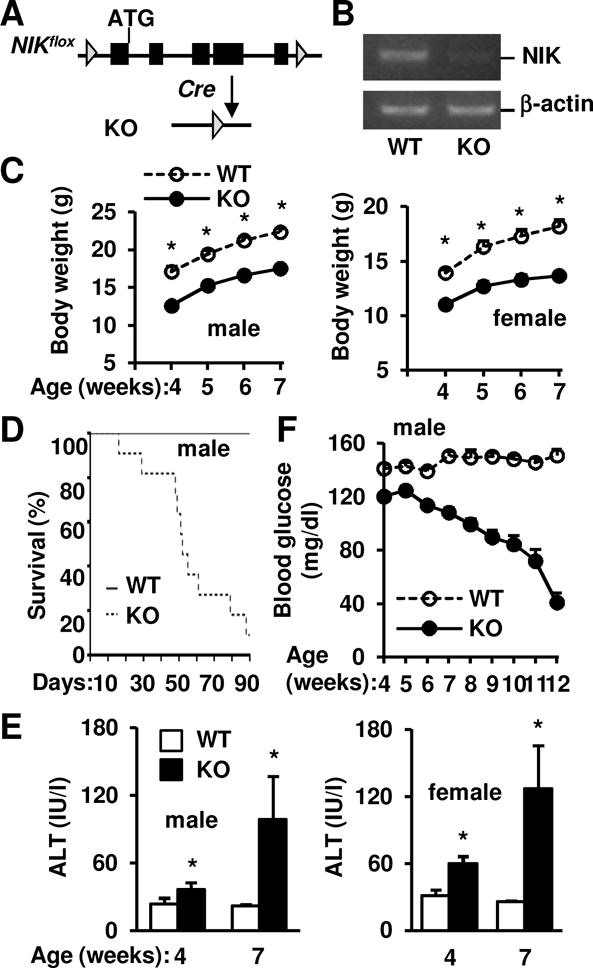
(A) A schematic representation of conditional NIK knockout. (B) Hepatic NIK and β-actin mRNA levels were measured by RT-PCR. (C) Male (WT: n=11, KO: n=12) and female (WT: n=7, KO: n=8) mouse growth curves. (D) Survival curves. WT: n=11, KO: n=12. (E) Plasma ALT activities in males (WT: n=11, KO: n=8) and females (WT: n=7, KO: n=6). (F) Randomly-fed blood glucose. WT: n=6–17, KO: n=4–16. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
Body weight was similar between KO (6.5 ± 0.3 g, n=3) and WT (7.0 ± 0.3 g, n=4, p=0.38) male littermates at 14 days of age; thereafter, both KO males and females progressively gained lesser weight relative to WT littermates (Fig. 1C). Most KO mice died before 13 weeks of age (Fig. 1D). Since hepatocyte-specific overexpression of NIK was shown to induce liver damage (15), we assessed liver injury in KO mice. Plasma alanine aminotransferase (ALT) activity, a liver injury marker, was significantly higher in KO males and females (Fig. 1E). We measured blood glucose because the liver is the primary organ responsible for endogenous glucose production (19). KO mice progressively developed severe hypoglycemia (Fig. 1F). These findings suggest that deletion of NIK impairs liver function, leading to hypoglycemia, growth retardation, and premature death.
NIK KO mice develop severe liver inflammation and injury
We next examined liver integrity by H&E staining of liver sections. Liver structure was disrupted in NIK KO males and females, accompanied by severe immune cell infiltration (Fig. 2A). F4/80+ Kupffer cell (KC)/macrophage (Fig. 2A) and Gr-1+ granulocyte (Supplementary Fig. 1) numbers were substantially higher in KO than in WT littermates. In line with these findings, expression of proinflammatory mediators (TNFα, MCP1, iNOS, and IL-1, -4, -5, -6 and -10) in the liver was also significantly higher in KO mice (Fig. 2B).
Fig. 2. Deletion of NIK results in liver inflammation, oxidative stress, and cell death.
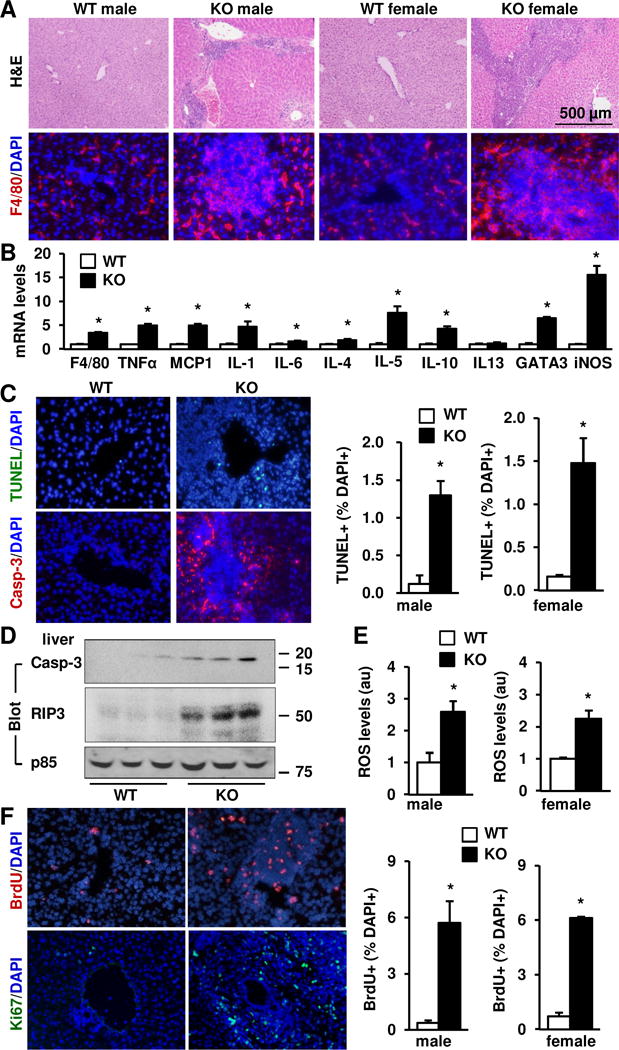
(A) Liver sections from males (7 weeks) and females (7 weeks) were stained with H&E or antibody against F4/80. (B) Gene expression was measured in the livers of WT (n=8–9) and KO (n=7) males at 7 weeks of age by qPCR and normalized to 36B4 levels. (C) Liver apoptotic cells were detected by TUNEL assays or immunostaining liver sections with antibody against activated caspase 3 (Casp-3). TUNEL+ cells were counted and normalized to DAPI+ cells. Male (7 weeks): WT: n=4, KO: n=4; female (7 weeks): WT: n=3, KO: n=3. (D) Liver extracts were prepared from males (7 weeks) and immunoblotted with antibodies against activated caspase-3, RIP3 or the regulatory subunit of p85 (loading control). (E) Liver ROS levels (normalized to liver protein levels) in males at 7 weeks of age (WT: n=5, KO: n=6) and females at 7 weeks of age (WT: n=4, KO: n=4). (F) Liver proliferating cells were detected by immunostaining liver sections with antibodies against BrdU or Ki67. BrdU+ cells were counted and normalized to DAPI+ cells. Males (7 weeks): WT: n=5, KO: n=5; females (7 weeks): WT: n=3, KO: n=3. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
Liver inflammation is associated with hepatocyte damage and death. Consistently, TUNEL+ or activated caspase-3+ (apoptosis marker) cell numbers were significantly higher in KO males and females relative to WT littermates (Fig. 2C). Keratin-8+ hepatocytes and keratin-19+ cholangiocytes accounted for 58% and 22% of total apoptotic cells, respectively (Supplementary Fig. 2). Activated caspase-3 and RIP3 (necrosis marker) protein levels in liver extracts were also substantially higher in KO mice (Fig. 2D). Liver injury is associated with oxidative stress (20, 21); indeed, liver reactive oxygen species (ROS) levels were significantly higher in KO mice (Fig. 2E).
Loss of liver cells is known to promote compensatory liver regeneration (22, 23). Number of proliferating liver cells (BrdU+ or Ki67+ cells) was significantly higher in KO males and females relative to WT littermates (Fig. 2F). F4/80+ KCs/macrophages, CD3+ T cells, α-smooth muscle actin (αSMA+) myofibroblasts, and keratin-8+ hepatocytes accounted for 49%, 14%, 10%, and 9% of total proliferating cells, respectively (Supplementary Fig. 3). Together, these results indicate that endogenous NIK is indispensable for the maintenance of liver integrity and hemostasis in mice.
NIK KO mice develop severe liver fibrosis
Liver inflammation and injury are known to promote liver fibrosis (24–28), prompting us to examine liver αSMA+ myofibroblasts and fibrillar collagens. NIK KO male and female mice developed severe liver fibrosis, as revealed by substantially higher levels of Sirius red/fast green staining and hydroxyproline content in KO mice relative to WT littermates (Fig. 3A–C). Liver αSMA+ myofibroblast number (Fig. 3D) and αSMA protein levels (Fig. 3E) were much higher in KO males and females relative to WT littermates. Consistently, expression of profibrotic genes (e.g. procollagen 1a1, TGFβ1, TIMP1, vimentin, and MMP-9) was significantly higher in KO mouse livers (Fig. 3F). We observed that severity of liver injury, inflammation, and fibrosis progressively increased in KO mice (Supplementary Fig. 4).
Fig. 3. NIK KO mice develop severe liver fibrosis.
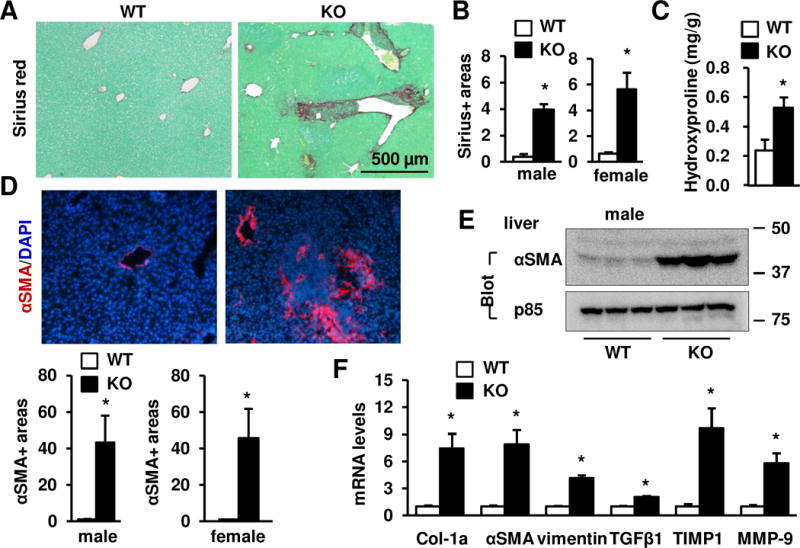
(A) Sirius red/fast green staining of liver sections from 7 week-old males. (B) Sirius red+ areas were quantified using the ImageJ software and normalized to microscopic view areas. Males (7 weeks): WT: n=7; KO: n=5; females (7 weeks): WT: n=4; KO: n=4. (C) Hydroxyproline content (normalized to liver weight) in males at 7 weeks of age. WT: n=4, KO: n=3. (D) Liver sections were immunostained with antibody against αSMA. αSMA+ areas were quantified and normalized to view areas. Male (7 weeks): WT: n=4, KO: n=3; females (7 weeks): WT: n=4, KO: n=4. (E) Liver extracts were immunoblotted with antibodies against αSMA or p85. (F) Gene expression was measured by qPCR and normalized to 36B4 levels. WT: n=8–9, KO: n=7. E-F: Males at 7 weeks of age. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
Deletion of NIK results in a robust CD4+ T cell expansion in the liver
Liver inflammation prompted us to examine adaptive immunity in KO mice. In WT mice, we detected very few CD3+, CD4+, CD8+, or CD25+ T cells in the liver (Fig. 4A), confirming liver being an immunologically-privileged organ. In stark contrast, KO mice had substantially higher levels of CD3+ T cells, particularly CD4+ T cells, in the liver (Fig. 4A). Liver CD4+ T cell expansions were detected at 4 weeks of age when liver cell death and fibrosis were barely detectable (Supplementary Fig. 4). Numbers of liver CD8+ T cells, NK cells, and NKT cells were higher while B220+ B cell number was lower in KO than in WT littermates (Fig. 4A and Supplementary Fig. 1). Consistently, flow cytometry analysis revealed that liver TCRβ+ T cells, CD4+ T cells, and CD8+ T cells were significantly higher in KO than in WT mice (Fig. 4B–C). Surprisingly, spleen TCRβ+, CD4+, and CD8+ T cells were lower in KO mice (Fig. 4B–C).
Fig. 4. Heavy hepatic infiltration of Th1 and Th2 T cells in NIK KO mice.
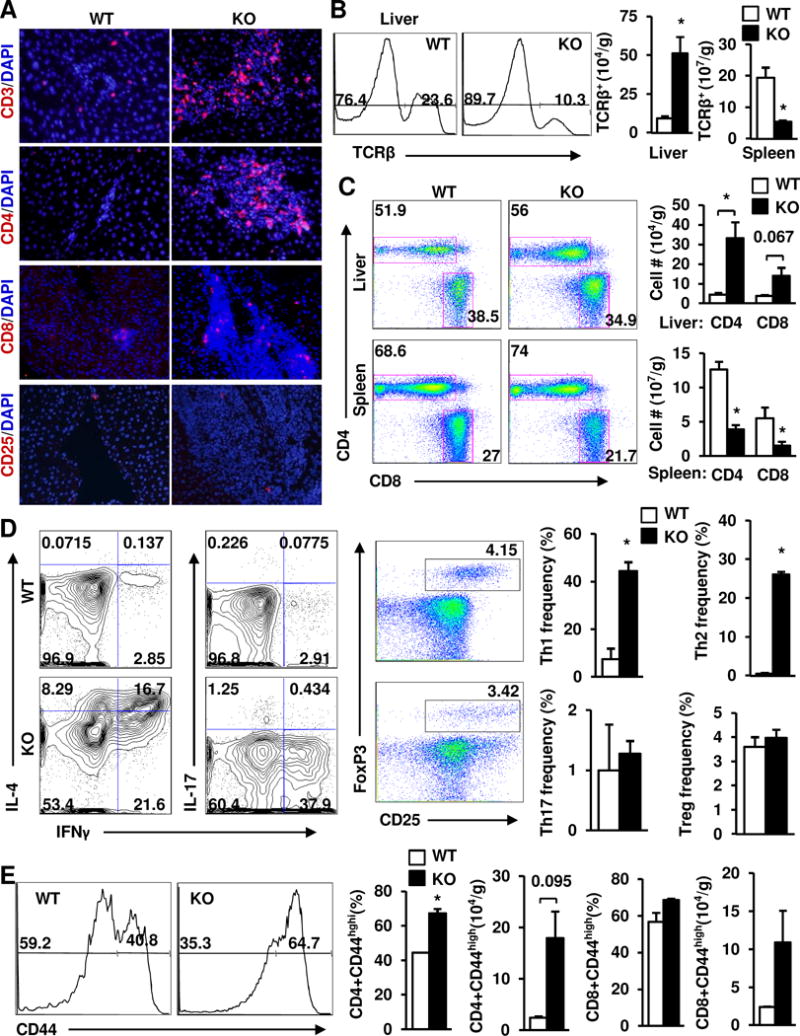
(A) Liver sections were prepared from males (7 weeks) and immunostained with the indicated antibodies (representatives of 4 pairs). (B-E) Livers and spleens were harvested from female mice at 6 weeks of age (n=3) and used to analyze T cell subpopulations by flow cytometry. (B) Total TCRβ+ T cells (normalized to tissue weight). (C) The CD4+ and CD8+ T cell subpopulations (normalized to tissue weight). (D) The Th1, Th2, Th17, and Treg subpopulations. (E) The effector/memory T cell subpopulations (CD44high). Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
CD4+ T cells are predominant adaptive immune cells in KO mouse livers; hence, we further assessed their polarizations and activation. Both Th1 (TCRβ+CD4+IFNγ+IL-4low) and Th2 (TCRβ+CD4+IL-4+IFNγlow) cells were significantly higher in KO than in WT mice; in contrast, Th17 (TCRβ+CD4+IL-17+) and Treg (TCRβ+CD4+CD25+Foxp3+) cells were similar between these two groups (Fig. 4D). Effector/memory CD4+CD44high T cell frequency and number were higher in KO mice (Fig. 4E). These data indicate that liver adaptive immune responses, particularly CD4+ T cell-launched immune responses, are aberrantly activated in KO mice; thus, endogenous NIK is indispensable for the maintenance of liver immune privilege.
Depletion of CD4+ but not CD8+ T cells blocks liver inflammation, injury, and fibrosis in NIK KO mice
We next sought to determine the contributions of CD4+ and CD8+ T cells to autoimmune attacks against liver in KO mice. CD4+ or CD8+ T cells were depleted using neutralizing anti-CD4 or anti-CD8 antibody, respectively. We confirmed that anti-CD4 antibody depleted CD4+ but not CD8+ T cells (Fig. 5A), whereas anti-CD8 antibody depleted CD8+ but not CD4+ T cells (Supplementary Fig. 5A). Depletion of CD4+, but not CD8+, T cells prevented KO mice from weight loss and hypoglycemia (Fig. 5B). Plasma ALT levels were normalized after 4 weeks of anti-CD4, but not anti-CD8, antibody treatment (Supplementary Fig. 5B). Depletion of CD4+ T cells completely blocked liver inflammation and fibrosis in KO mice (Fig. 5C), and substantially lowered liver F4/80+ macrophages/KCs, αSMA+ myofibroblasts, and TUNEL+ apoptotic cells (Fig. 5D). In line with these findings, liver RIP3, activated caspase-3, and αSMA proteins in KO mice were reduced to normal levels after anti-CD4 antibody treatment (Fig. 5E). Depletion of CD4+ T cells decreased expression of proinflammatory mediators to normal levels in KO mice (Fig. 5F). In contrast, depletion of CD8+ T cells did not attenuate liver inflammation, cell death, and fibrosis in KO mice (Supplementary Fig. 5C–D). These results suggest that autoreactive CD4+ T cells are responsible for autoimmune liver disease in KO mice.
Fig. 5. Depletion of CD4+ T cells reverses liver inflammation, injury, and fibrosis in NIK KO mice.
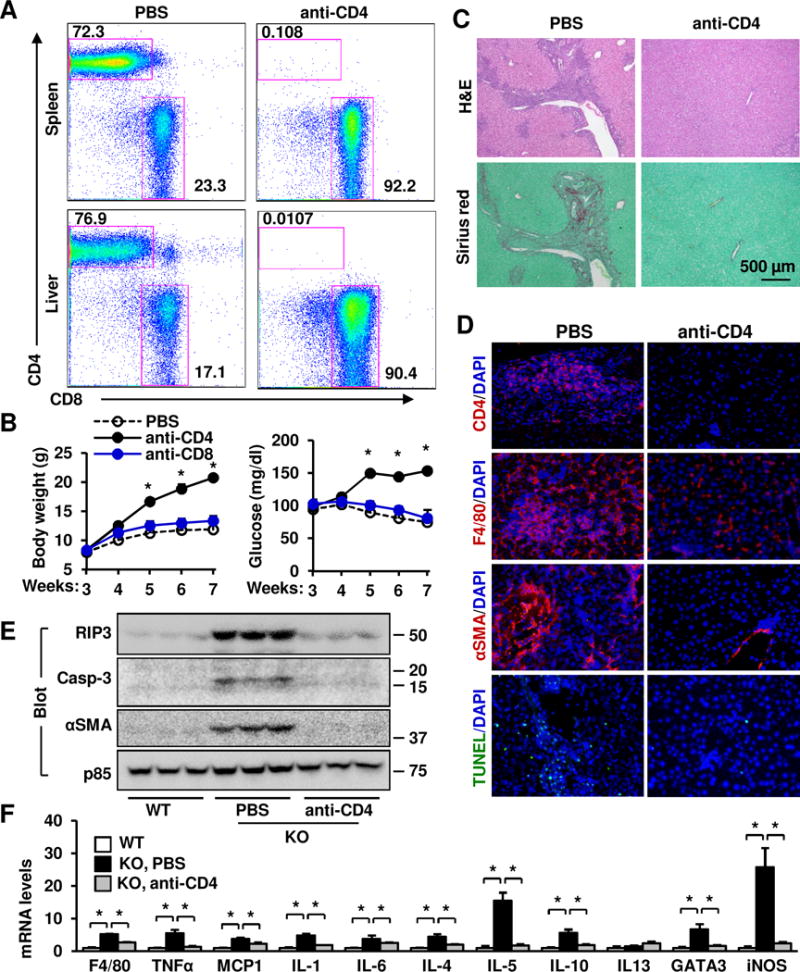
KO male mice were treated with anti-CD4 (or anti-CD8) antibody or PBS (control) at 3 weeks of age. n=3. (A) Flow cytometric analysis of the CD4+ and CD8+ T cell subsets (gated on TCRβ+). The number represents percentages. (B) Growth curves and randomly-fed blood glucose. (C–F) Livers were harvested 4 weeks after treatments. (C–D) Liver sections were stained with the indicated dyes or antibodies (representatives of 3 pairs). (E) Liver extracts were immunoblotted with the indicated antibodies. (F) Gene expression was measured by qPCR and normalized to 36B4 levels. The same WT was used in Fig. 2B and Fig. 5F. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
To determine whether KO mice develop autoimmune inflammation in other tissues, we examined the lung, kidney, and pancreas. We detected a CD3+ T cells expansion in the liver, but not in the lung, kidney, and pancreas, of young KO mice at 3 weeks of age (Supplementary Fig. 6A). However, lung, kidney, and pancreas CD3+ T cell number was higher in adult KO mice relative to WT littermates (>7 weeks) (Supplementary Fig. 6B). Depletion of CD4+ T cells also blocked autoimmune attacks against lung, kidney, and pancreas (Supplementary Fig. 6C–D). These data suggest that NIK deficiency induces CD4+ T cell-mediated autoimmune inflammation in multiple tissues in an age-dependent manner.
Deletion of NIK in the liver or hematopoietic lineage cells does not induce liver inflammation, injury, and fibrosis
To determine whether hepatocyte NIK protects against autoimmune liver injury, we generated liver-specific NIK knockout (NIKΔliver) mice (NIKflox/flox;albumin-cre) by crossing NIKflox/flox mice with albumin-cre drivers. We verified that NIK was deleted in the hepatocytes of NIKΔliver mice (Fig. 6A). Body weight and blood glucose were similar between NIKΔliver and NIKflox/flox mice (data not shown), and the liver was normal even in old NIKΔliver mice at 20 weeks of age (Fig. 6B).
Fig. 6. NIK deficiency in hepatocytes or hematopoietic lineage cells does not induce liver inflammation, injury, and fibrosis.
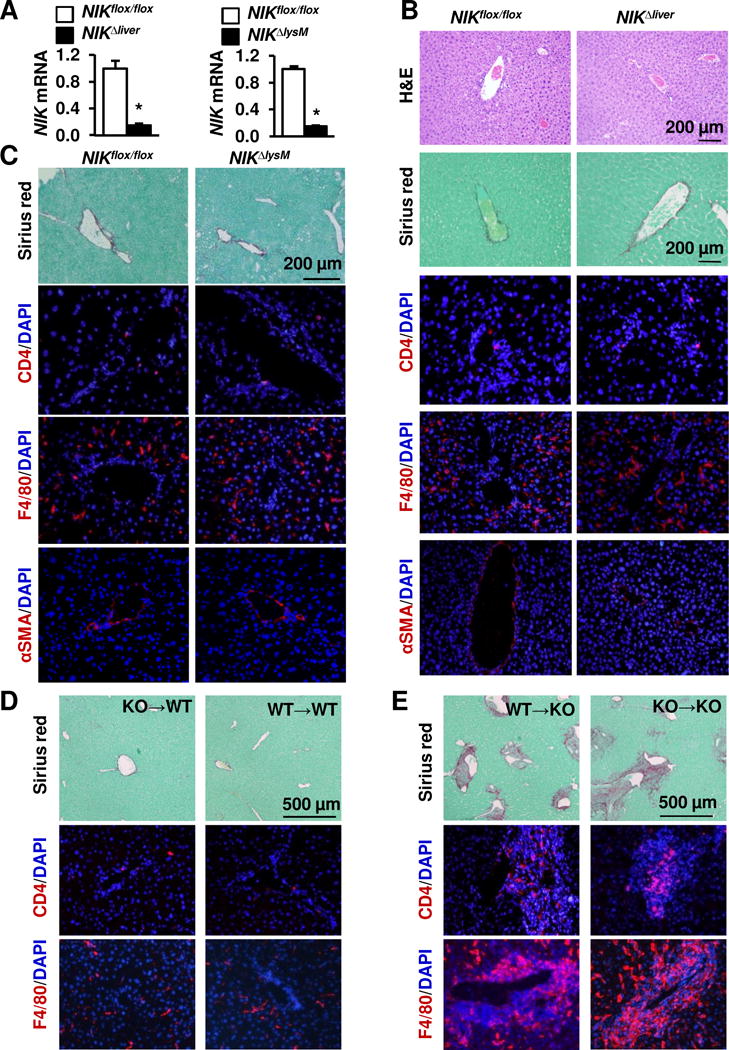
(A) Primary hepatocytes and leukocytes were isolated from NIKflox/flox (n=3) and NIKΔliver (n=3) males (7 weeks). NIK expression was measured by qPCR and normalized to 36B4 levels. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. (B) Liver sections were prepared from males (24 weeks) and stained with H&E, Sirius red/fast green, or the indicated antibodies (representatives of 3–4 pairs). (C) NIKΔlysM and NIKflox/flox males were fed a HFD for 17 weeks. Liver sections were stained with the indicated dyes and antibodies (representatives of 3–4 pairs). (D–E) WT and KO recipients (5 weeks) were treated with GdCl3 and lethal irradiation. Donor bone marrow was prepared from KO or WT males (5 weeks) and adoptively transferred into WT or KO recipients (2×106 cells per mouse). Liver sections were prepared 7 weeks later, and stained with Sirius red or the indicated antibodies.
Next, we assessed the role of NIK in innate immune cells. We generated myeloid cell-specific NIK knockout (NIKΔlysM) mice (NIKflox/flox;lysM-cre) by crossing NIKflox/flox mice with lysM-cre drivers. NIK expression in blood leukocytes was substantially lower in NIKΔlysM mice as expected (Fig. 6A). Body weight and blood glucose were similar between NIKΔlysM and NIKflox/flox mice (data not shown), and the numbers of CD4+ T cells, F4/80+ macrophages/KCs, and αSMA+ myofibroblasts were comparable between NIKΔlysM and NIKflox/flox mice (Fig. 6C). To further assess the role of NIK in hematopoietic lineage cells, including both innate and adaptive immune cells, we conducted adoptive transfers of bone marrows. Recipient mice were pretreated with GdCl3 and lethal irradiation to destroy endogenous KCs and hematopoietic cells, and then reconstituted with either WT or KO donor bone marrows. WT recipients reconstituted with either KO (KO→WT) or WT (WT→WT) donor bone marrows did not develop liver inflammation and fibrosis (Fig. 6D), suggesting that NIK-delicacy in innate and adaptive immune cells is unable to induce liver inflammation and fibrosis. KO recipients reconstituted with either WT (WT→KO) or KO (KO→KO) bone marrows continued to display severe liver inflammation and fibrosis (Fig. 6E), suggesting that NIK deficiency in non-immune cells is likely to be responsible for liver inflammation, injury, and fibrosis.
NIK deficiency in the thymus induces autoimmune hepatitis
We postulated that NIK in the thymus is likely to suppress autoreactive CD4+ T cells against liver antigens. In normal mice, pathogenic autoreactive T cells are removed in the thymic medulla through medullary thymic epithelial cell (mTEC)-mediated negative selection. NIK deficiency dramatically reduced thymic medullary areas in KO mice (Fig. 7A), confirming previous reports (29). AIRE+ mTECs were abundant in WT mice, but barely detectable in KO mice (Fig. 7B). We detected co-localization of AIRE with Phospho-IKKα/β or NF-κB2 in WT but not KO thymi (Fig. 7B), suggesting that NIK is activated predominantly in mTECs.
Fig. 7. Deficiency of thymic NIK results in liver inflammation, injury, and fibrosis.
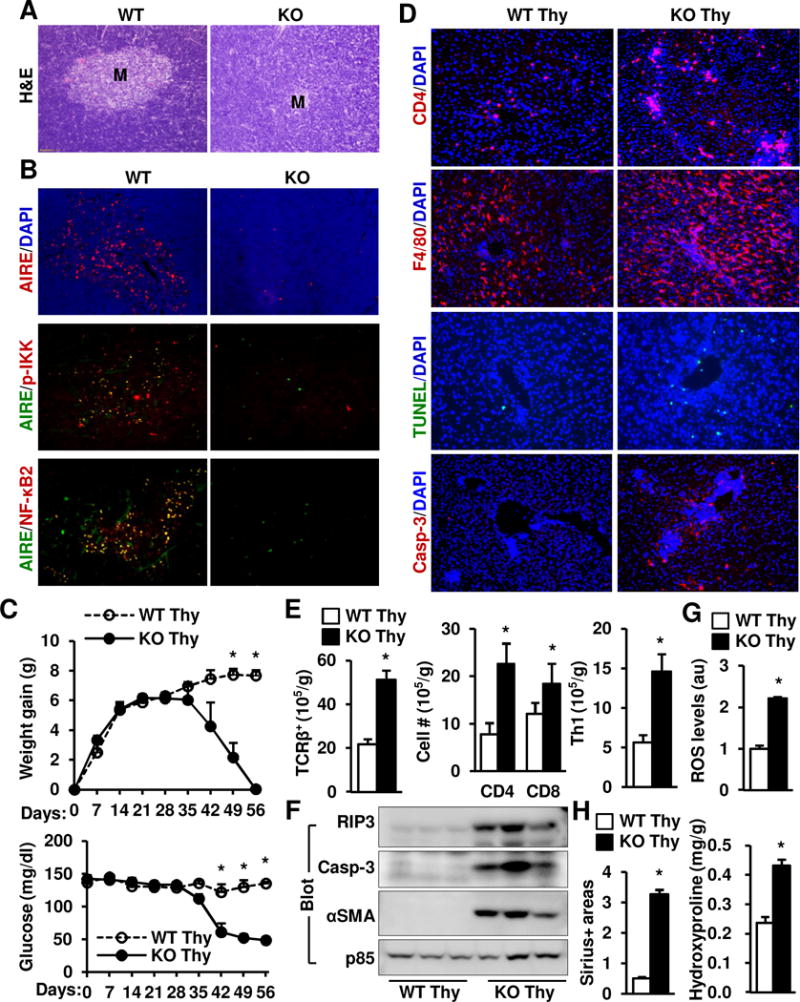
(A–B) Thymus sections were prepared from WT and KO males (5 weeks) and stained with H&E or the indicated antibodies (representatives of 3 pairs). (C–H). Thymi (Thy) were isolated from WT or NIK KO littermates and transplanted under renal capsules of Foxn1nu male recipients. WT Thy: n=4, KO Thy: n=3. (C) Body weight gains and non-fasted blood glucose levels were monitored after transplantation. (D–H) Mice were euthanized 8 weeks after transplantation. (D) Liver sections were prepared 8 weeks after transplantation and stained with the indicated antibodies. Representative images of 3 pairs. (E) Liver T cells were quantified by flow cytometry and normalized to liver weight. (F) Liver extracts were immunoblotted with the indicated antibodies. (G) Liver ROS levels were measured using DCF dye and normalized to liver protein. au: arbitrary units. (H) Liver paraffin sections were stained with Sirius red, and Sirius+ areas were quantified and normalized to view areas. Liver hydroxyproline levels were quantified and normalized to liver weight. Data were statistically analyzed with two-tailed Student’s t test, and presented as mean ± SEM. *p<0.05.
We next sought to determine whether deleting NIK in the thymus induces autoimmune hepatitis. Donor thymi were isolated from WT and KO mice and adoptively transferred into immune-deficient Foxn1−/− recipient mice under renal capsules (Supplementary Fig. 7A). TCRβ+ T cells and the CD4+ and CD8+ T cell subpopulations in the blood and spleen were comparable between WT and KO thymus-graft recipients (Supplementary Fig. 7B), indicating that survival and growth of WT and KO thymic grafts are similar. Body weight and blood glucose were similar between the WT and KO groups within 5 weeks after transplantation; thereafter, the KO but not the WT group progressively lost weight and developed severe hypoglycemia (Fig. 7C). Blood ALT activity was significantly higher in the KO than in the WT groups 8 weeks after transplantation (Supplementary Fig. 7C), and liver TCRβ+ T cells, CD4+ T cells, CD8+ T cells, and Th1 T cells were significantly higher in the KO group (Fig. 7D–E). Liver TUNEL+ and activated caspase-3+ apoptotic cell numbers (Fig. 7D), RIP3 and activated caspase-3 protein levels (Fig. 7F), and ROS levels (Fig. 7G) were substantially higher in the KO group. Furthermore, the KO but not the WT groups developed liver fibrosis (Fig. 7H and Supplementary Fig. 7D). Expression of proinflammatory or profibotic genes was significantly higher in the KO than in the WT group (Supplementary Fig. 7E). These data indicate that NIK deficiency in the thymus is sufficient to induce autoimmune hepatitis.
Discussion
In this study, we found that NIK KO mice developed severe liver inflammation, injury, and fibrosis, leading to premature death. We detected a massive T cell expansion, predominantly Th1 and Th2 CD4+ T cells, in the livers of KO mice. Importantly, depletion of CD4+, but not CD8+, T cells blocked liver inflammation, injury, and fibrosis in KO mice. These results indicate that in NIK KO mice, CD4+ T cells orchestrate autoimmune attack against liver.
To identify NIK target tissues, we deleted NIK in different cell types. Unlike global NIK KO mice, mice with liver-specific, myeloid cell-specific, or hematopoietic lineage cell-specific deletion of NIK did not develop liver inflammation, injury, or fibrosis. Thus, NIK deficiency in hepatocytes or immune cells is unlikely to be responsible for liver destruction. In line with this notion, reconstitution of NIK KO recipients with WT donor hematopoietic lineage cells was unable to correct liver diseases. Together, these data indicate that NIK deficiency in extrahepatic, non-hematopoietic lineage cells causes autoimmune liver disease. We found that NIK KO mice had disrupted thymi with medullary atrophy, raising the possibility that thymic NIK may control thymus growth and central T cell development. Indeed, reconstitution of immune-deficient recipient mice with NIK-null but not WT thymus grafts induced profound liver inflammation, injury, and fibrosis, indicating that NIK deficiency in the thymus causes fatal autoimmune hepatitis.
To gain insight into NIK regulation of T cell development in the thymus, we examined NIK pathways in thymic cells. NIK was activated predominantly in AIRE+ mTECs, which are known to mediate central negative selection to deplete autoreactive T cells (36, 37). Deletion of NIK largely eliminated mTECs in NIK KO mice, suggesting that NIK is required for mTEC growth and maturation. Of notice, inactivation of lymphotoxin β (LTβ), CD40L, or RANKL, cytokines that activate NIK (30), similarly blocks mTEC development (31, 32). Likewise, deletion of IKKα, NF-κB2, or RelB also blocks mTEC development (33–35). Thus, the NIK/IKKα/NF-κB2 pathway is likely to act downstream of LTβ, CD40L, and RANKL to promote mTEC growth and maturation. Therefore, NIK deficiency in the thymus, particularly in mTECs, is likely to impairs mTEC development and mTEC-mediated negative selection. In chimeric mice receiving NIK-null donor thymi, deficiency of thymic NIK induces defects in mTEC development and negative selection, resulting in generation of autoreactive CD4+ T that triger and orchestrate autoimmune liver diseases.
Our data indicate that CD8+ T cells and B cells are unlikely to mediate immune attack against liver in KO mice. Noticeably, F4/80+ KC/macrophage number was substantially higher in NIK KO mice and markedly reduced by depletion of CD4+ T cells. These data suggest that KCs/macrophages may act downstream of autoreactive CD4+ T cells and execute liver destruction. Additional studies are warranted to explore this possibility in the future.
Supplementary Material
Lay Summary.
We found that global or thymus-specific ablation of the NIK gene results in fatal autoimmune liver disease in mice. NIK-deficient mice develop liver inflammation, injury, and fibrosis in a CD4+ T cell-dependent manner. Our findings indicate that thymic NIK is indispensable for the maintenance of liver integrity and homeostasis.
Acknowledgments
We thank Drs. Haoran Su, Yan Liu, Mark J. Canet, Bijie Jiang, Chengxin Sun, Yatrik Shah, Bishr Omary, and Yin Lei for assistance and discussion.
Financial support: This study was supported by grants DK091591, DK094014 (to LR) and AI121156 (to CC) from the National Institutes of Health and by grant 81420108006 (to YL) from National Natural Science Foundation of China. This work utilized the cores supported by the Michigan Diabetes Research and Training Center (NIH DK20572), the University of Michigan’s Cancer Center (NIH CA46592), the University of Michigan Nathan Shock Center (NIH P30AG013283), and the University of Michigan Gut Peptide Research Center (NIH DK34933).
Abbreviations
- NIK
NF-κB-inducing Kinase
- mTEC
medullary thymic epithelial cell
- KC
Kupffer cell
- KO
knockout
- WT
wild type
- ALT
alanine aminotransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Author contributions: HS, LR: Study concept and design; HS, LS, YX, YK, LJ, ZC, KP: acquisition of data; HS, LR: drafting of the manuscript; CC: critical revision of the manuscript for important intellectual content.
References
- 1.Bogdanos DP, Gao B, Gershwin ME. Liver immunology. Compr Physiol. 2013;3:567–598. doi: 10.1002/cphy.c120011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lapierre P, Lamarre A. Regulatory T Cells in Autoimmune and Viral Chronic Hepatitis. J Immunol Res. 2015;2015:479703. doi: 10.1155/2015/479703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsuneyama K, Baba H, Kikuchi K, Nishida T, Nomoto K, Hayashi S, Miwa S, et al. Autoimmune features in metabolic liver disease: a single-center experience and review of the literature. Clin Rev Allergy Immunol. 2013;45:143–148. doi: 10.1007/s12016-013-8383-x. [DOI] [PubMed] [Google Scholar]
- 4.Lemmers A, Moreno C, Gustot T, Marechal R, Degre D, Demetter P, de Nadai P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology. 2009;49:646–657. doi: 10.1002/hep.22680. [DOI] [PubMed] [Google Scholar]
- 5.Cao Q, Batey R, Pang G, Clancy R. Altered T-lymphocyte responsiveness to polyclonal cell activators is responsible for liver cell necrosis in alcohol-fed rats. Alcohol Clin Exp Res. 1998;22:723–729. [PubMed] [Google Scholar]
- 6.Song K, Coleman RA, Zhu X, Alber C, Ballas ZK, Waldschmidt TJ, Cook RT. Chronic ethanol consumption by mice results in activated splenic T cells. J Leukoc Biol. 2002;72:1109–1116. [PubMed] [Google Scholar]
- 7.Inzaugarat ME, Ferreyra Solari NE, Billordo LA, Abecasis R, Gadano AC, Chernavsky AC. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol. 2011;31:1120–1130. doi: 10.1007/s10875-011-9571-1. [DOI] [PubMed] [Google Scholar]
- 8.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 9.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 10.Ling L, Cao Z, Goeddel DV. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc Natl Acad Sci U S A. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 12.Miyawaki S, Nakamura Y, Suzuka H, Koba M, Yasumizu R, Ikehara S, Shibata Y. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur J Immunol. 1994;24:429–434. doi: 10.1002/eji.1830240224. [DOI] [PubMed] [Google Scholar]
- 13.Koike R, Nishimura T, Yasumizu R, Tanaka H, Hataba Y, Watanabe T, Miyawaki S, et al. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur J Immunol. 1996;26:669–675. doi: 10.1002/eji.1830260324. [DOI] [PubMed] [Google Scholar]
- 14.Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 15.Shen H, Sheng L, Chen Z, Jiang L, Su H, Yin L, Omary MB, et al. Mouse hepatocyte overexpression of NF-kappaB-inducing kinase (NIK) triggers fatal macrophage-dependent liver injury and fibrosis. Hepatology. 2014;60:2065–2076. doi: 10.1002/hep.27348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willmann KL, Klaver S, Dogu F, Santos-Valente E, Garncarz W, Bilic I, Mace E, et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun. 2014;5:5360. doi: 10.1038/ncomms6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho KW, Zhou Y, Sheng L, Rui L. Lipocalin-13 regulates glucose metabolism by both insulin-dependent and insulin-independent mechanisms. Mol Cell Biol. 2011;31:450–457. doi: 10.1128/MCB.00459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Jiang L, Rui L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J Biol Chem. 2009;284:11152–11159. doi: 10.1074/jbc.M900754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177–197. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Dig Dis Sci. 2010;55:560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 21.Morris EM, Rector RS, Thyfault JP, Ibdah JA. Mitochondria and redox signaling in steatohepatitis. Antioxid Redox Signal. 2011;15:485–504. doi: 10.1089/ars.2010.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol. 2013;3:485–513. doi: 10.1002/cphy.c120014. [DOI] [PubMed] [Google Scholar]
- 23.Miyajima A, Tanaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell. 2014;14:561–574. doi: 10.1016/j.stem.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarantino G, Saldalamacchia G, Conca P, Arena A. Non-alcoholic fatty liver disease: further expression of the metabolic syndrome. J Gastroenterol Hepatol. 2007;22:293–303. doi: 10.1111/j.1440-1746.2007.04824.x. [DOI] [PubMed] [Google Scholar]
- 27.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 28.Novo E, di Bonzo LV, Cannito S, Colombatto S, Parola M. Hepatic myofibroblasts: a heterogeneous population of multifunctional cells in liver fibrogenesis. Int J Biochem Cell Biol. 2009;41:2089–2093. doi: 10.1016/j.biocel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y, Kuroda N, et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol. 2004;172:2067–2075. doi: 10.4049/jimmunol.172.4.2067. [DOI] [PubMed] [Google Scholar]
- 30.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, Asaumi Y, et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. 2008;29:423–437. doi: 10.1016/j.immuni.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Boehm T, Scheu S, Pfeffer K, Bleul CC. Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J Exp Med. 2003;198:757–769. doi: 10.1084/jem.20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomada D, Liu B, Coghlan L, Hu Y, Richie ER. Thymus medulla formation and central tolerance are restored in IKKalpha−/− mice that express an IKKalpha transgene in keratin 5+ thymic epithelial cells. J Immunol. 2007;178:829–837. doi: 10.4049/jimmunol.178.2.829. [DOI] [PubMed] [Google Scholar]
- 34.Zhu M, Chin RK, Christiansen PA, Lo JC, Liu X, Ware C, Siebenlist U, et al. NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest. 2006;116:2964–2971. doi: 10.1172/JCI28326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 36.Laan M, Peterson P. The many faces of aire in central tolerance. Front Immunol. 2013;4:326. doi: 10.3389/fimmu.2013.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonito AJ, Aloman C, Fiel MI, Danzl NM, Cha S, Weinstein EG, Jeong S, et al. Medullary thymic epithelial cell depletion leads to autoimmune hepatitis. J Clin Invest. 2013;123:3510–3524. doi: 10.1172/JCI65414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.