

Abstract
Accumulating studies suggest that the epidermal growth factor receptor (EGFR) activation is associated with the physiology and pathophysiology of the cardiovascular system, and inhibition of EGFR activity is emerging as a potential therapeutic strategy to treat diseases, including hypertension, cardiac hypertrophy, renal fibrosis and abdominal aortic aneurysm. The capacity of G protein-coupled receptor (GPCR) agonists, such as angiotensin II (AngII), to promote EGFR signaling is well described – a process termed EGFR “transactivation” – yet delineating the molecular processes and functional relevance of this crosstalk has been challenging. Moreover, these critical findings are dispersed among many different fields. The aim of our review is to highlight the recent advancement of the signaling cascades and downstream consequences of EGFR transactivation within the cardiovascular renal system in vitro and in vivo. We will also focus on linking EGFR transactivation to animal models of the disease as well as the potential therapeutic applications.
INTRODUCTION
EGF Family Ligands and Receptors
The future Nobel laureate, Dr. Stanley Cohen and colleagues discovered by accident what would come to be known as the epidermal growth factor (EGF) more than half a century ago. EGF was identified in a submaxillary gland extract that induced precocious eyelid opening and incisor eruption due to stimulation of epidermal growth and keratinization when injected into newborn mice (1). The receptor for EGF belongs to the ErbB family of receptors. ErbBs are typical receptor tyrosine kinases that were implicated in cancer etiology in the early 1980s when the avian erythroblastosis tumor virus was found to encode an aberrant form of the human EGF receptor (EGFR is also known as ErbB1 or HER1) (2). The ErbB further also includes ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4). There are 11 known ErbB ligands. The binding relationship between the ligands and receptors, and the specific nature of the receptors are illustrated in Figure 1. Upon ligand binding, dimerization (both homo and hetero) of EGFR/ErbBs results in their intracellular tyrosine kinase activation and autophosphorylation at several tyrosine residues (2–4). These phosphorylated sites within the intracellular domain of EGFR/ErbBs act as docking sites for adaptor proteins, facilitating downstream signaling cascades. The most widely studied signaling pathways involve recruitment of Shc/Grb2 to activate the Ras/ERK (extracellular signal-regulated kinase) cascade and of phosphoinositide 3-kinase (PI3K) p85 for the PI3K/Akt/p70 S6 kinase cascade (2; 3).
Figure 1.
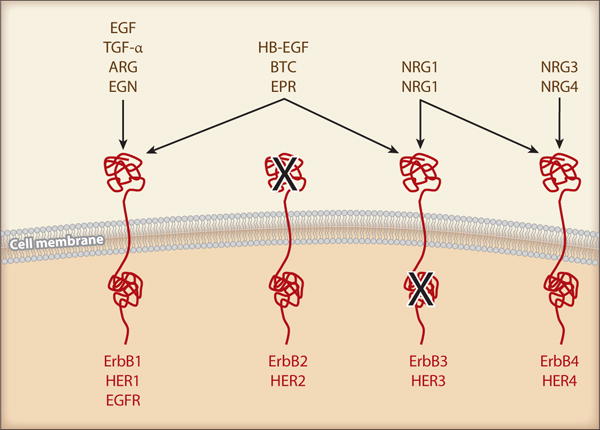
Mechanism of EGFR transactivation by GPCRs and the Aim of review
The EGFR has become one of the most widely studied molecules in cancer biology. EGFR antagonism through antibodies and small molecule inhibitors has had beneficial effects in patients suffering from non-small-cell lung cancer, colorectal cancer and pancreatic cancer (3; 4). Intriguingly, the scope of EGFR research has expanded in recent years to include cardiovascular physiology and pathophysiology (5; 6). Research into cardiovascular-specific EGFR signaling has provided unique upstream signaling cascades that provide necessary components to activate downstream EGFR signaling (7). In conjunction with ligand binding-mediated EGFR activation and downstream signaling, consecutive activation of EGFR upon activation of GPCRs has established a new mechanism for EGFR activation known as EGFR “trans”-activation (8; 9). While mechanistic insights related to EGFR transactivation are still being elucidated, it is now well recognized that GPCR-mediated transactivation of EGFR can occur through initial heterotrimeric G protein dissociation, activation of ligand-specific intermediates including non-receptor tyrosine kinase, intracellular Ca2+, and reactive oxygen species (ROS), followed by a metalloprotease activation and shedding of EGFR ligands (7; 10–12). As well, EGFR activation in some instances has been shown to be G protein-independent and β- arrestin-dependent (13). Among these upstream regulators, specific interest into the role and mechanism of A Disintegrin and Metalloprotease 17 (ADAM17) in mediating EGFR ligand shedding and subsequent EGFR transactivation has been highlighted in recent years (10; 11; 14; 15) (Figure 2).
Figure 2.
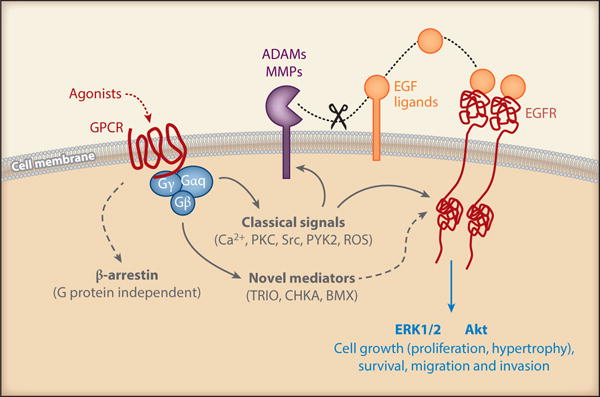
Of all of the GPCR ligands implicated in EGFR transactivation, special emphasis has been placed on angiotensin II (AngII), its EGFR transactivation mechanism via AngII type-1 receptor (AT1R), and the effect AngII plays via EGFR in cardiovascular pathology (16; 17). While key elements of AT1R/EGFR signaling have been elucidated, tissue specific alterations in this signaling cascade make this field an area of growing interest. The aim of this review is to provide an up-to-date understanding of EGFR/ErbB activation, with special emphasis placed on transactivation via GPCRs including AT1R and its potential roles in initiation and progression of cardiovascular and renal diseases. Mechanistic insight will be provided through reviews on cell-based models, animal models, and where applicable, in humans, and the areas of need for continued investigation will be highlighted.
VASCULAR SMOOTH MUSCLE
Pathophysiological vascular remodeling precedes end-organ damage associated with hypertension and is also a key component in the development of atherosclerosis. Prevention of vascular remodeling is therefore considered to be important for prevention of cardiovascular diseases (18). Vascular remodeling involves hypertrophy, hyperplasia, migration and phenotypic change of vascular smooth muscle cells (VSMCs). There is sufficient evidence showing critical involvement of the EGFR in mediating vascular remodeling.
In Vitro Findings
All members of EGFR/ErbB as well as most of their ligands are expressed by VSMCs (5). The EGFR ligands expressed in VSMCs, such as heparin-binding EGF-like growth factor (HB-EGF), epiregulin or betacellulin, are all reported to have strong mitogenic and chemotactic effects (19). One of the well acknowledged inducers of EGFR transactivation, AngII, plays a significant role in the development and aggravation of various cardiovascular diseases such as hypertension and atherosclerosis by inducing vascular remodeling (20). In vitro studies have shown that AngII induces Gq/Ca2+-dependent and/or Src-dependent transactivation of EGFR leading to activation of downstream kinases (ERK, Akt, p38 mitogen-activated protein kinase (MAPK), p70S6K) and subsequent NADPH oxidase activation, ROS generation, and protein synthesis resulting in hypertrophy, hyperplasia, and migration of VSMCs (17; 21–25). Similarly, the activation of Gq-coupled endothelin-A receptor stimulates EGFR transactivation in VSMCs, and specific inhibition of EGFR kinase activity suppresses growth responses induced by endothelin-1 in VSMCs (26). Likewise, VSMCs derived from VSMC-selective EGFR knockout mice show reduced ERK activation in response to several vascular agonists, including endothelin-1 (27).
Recently, studies using viral vectors encoding dominant negative ADAM17 (25) and ADAM17 siRNA-embedded miRNA have shown that AngII-mediated EGFR transactivation and subsequent VSMC hypertrophy requires a tyrosine phosphorylation-dependent ADAM17 activation and subsequent HB-EGF shedding (11). AngII-induced EGFR phosphorylation has also been reported to require cytosolic Ca2+-dependent phospholipase A2 (cPLA2), arachidonic acid, p38 MAPK and phospholipase D in cultured VSMCs, (28). Plasma kallikrein also stimulates G protein-coupled protease activated receptors (PARs) and promotes EGFR transactivation via ADAM17 independent of bradykinin in VSMCs (29). In addition, the matrix metalloproteinases (MMP2 and MMP7) seem to be critical for adrenergic vascular tone by inducing EGFR transactivation and resultant PI3K activation and ATP synthesis (30–32).
Many of the signaling proteins implicated in EGFR transactivation via AT1R are localized to lipid rafts and/or caveolae, which are cholesterol rich membrane micro domains acting as signaling platforms to facilitate temporal and spatial localization of signal transduction (33). The metalloprotease ADAM17 is compartmentalized within caveolae (34). Infection of adenovirus encoding caveolin-1 (Cav1), the major structural protein of caveolae, inhibits AngII-induced HB-EGF shedding, EGFR transactivation, ERK activation, hypertrophy and migration of VSMC, but not intracellular Ca2+ elevation (35). However, silencing Cav1 expression also leads to diminished AngII-induced EGFR transactivation (36). Therefore, tight regulation via Cav1/caveolae might be a critical component to EGFR transactivation.
Aldosterone is known to increase EGFR expression in VSMCs (37). Providing further relevance to AngII-mediated EGFR activation, synergy between AngII/AT1R and aldosterone/mineralocorticoid receptor (MR) has been reported in VSMC EGFR transactivation. (38; 39). The synergistic effect of these molecules in the renin angiotensin aldosterone system (RAAS) highlights the complexity underpinning vascular dysfunction whereby both AngII and aldosterone stimulate simultaneous EGFR activation through the AT1R and the MR, respectively. Aldosterone and AngII interaction also enhances a tyrosine kinase c-Src-independent ERK1/2 signaling and c-Src-dependent signaling through EGFR and platelet-derived growth factor (PDGF) receptor activation, which stimulates VSMC migration (40). It should be noted that there is increasing evidence that MR-mediated EGFR transactivation may be a major mechanism underlying non-genomic functions of MR (41). VSMCs incubated with aldosterone show progression of inflammation through increased 12/15-lipoxygenase expression, which can be suppressed with the EGFR inhibitor AG1478 (42). In aged rat VSMCs, there is increased MR and EGFR expression compared to adult rat VSMCs, and aldosterone treatment in adult VSMCs shifts the proinflammatory expression phenotype towards that of aged VSMCs. Furthermore, AG1478 can attenuate the age-associated proinflammatory profile (43). More in depth understanding of MR signaling mechanisms will be provided in the following in vivo section, and cardiac and kidney section.
Oxidative stress plays a critical role in cardiovascular disease, and ROS such as H2O2 have been shown to transactivate EGFR via metalloprotease–dependent HB-EGF production (44). Likewise, thrombin stimulates EGFR transactivation via NADPH oxidase 1 (Nox1)-derived ROS in VSMCs (45). Other pathophysiological factors implicated in cardiovascular diseases, including oxidized low density lipoprotein (oxLDL) (46) and mechanical stress (47), are also reported to promote EGFR ligand shedding and EGFR transactivation. Moreover, high glucose is reported to enhance AngII signal transduction via EGFR N-glycosylation. The 170 kD N-glycosylated EGFR can be transactivated by EGFR ligands or GPCR ligands including AngII, but the 145 kD non-N-glycosylated EGFR can only be activated by EGF ligands, suggesting a potential mechanism for the pathogenic effects of AngII in diabetic vascular disorders (48). In addition to the involvement in growth responses and ROS production, EGFR transactivation in VSMCs mediates a wide variety of cellular responses including expression of key genes and pathways implicated in inflammation, fibrosis and apoptosis as summarized in Table 1. Taken together, EGFR transactivation may be an important signaling convergence point by which many cardiovascular risk factors promote vascular remodeling.
Table 1.
Vascular epidermal growth factor receptor pathophysiology
| Model | Intervention | Outcome and intermediate | Reference(s) |
|---|---|---|---|
| Hypertension (AngII infusion or SHR) | EGFR antisense | Reduced hypertension ERK1/2 | 55, 56 |
| Hypertension/renal injury (5/6 nephrectomy) | PKI-166 | Reduced progression of hypertension; partially restored contractile response | 59 |
| Hypertension/vascular remodeling (AngII infusion) | Erlotinib | Reduced vascular remodeling; did not reduce hypertension; reduced ADAM17 and ER stress | 61 |
| Hypertension/vascular remodeling (AngII infusion) | AG1478 waved-2 mouse | Reduced cerebral arteriole hypertrophy; did not reduce hypertension | 63 |
| Hypertension/renal dysfunction (L-NAME treatment and removal) | Gefitinib | Restored functional endothelial cells; did not reduce hypertension | 89 |
| Restenosis (balloon angioplasty) | Monoclonal antibody targeting EGFR | Inhibited medial smooth muscle cell proliferation and intimal hyperplasia | 66 |
| Restenosis (balloon angioplasty) | ADAM17 dominant-negative adenovirus | Reduced intimal hyperplasia EGFR inhibition | 68 |
| Vascular remodeling (partial ligation) | HB-EGF−/− mouse | Inhibited medial smooth muscle cell proliferation and intimal hyperplasia | 69 |
| Abdominal aortic aneurysm (AngII with BAPN infusion) | Erllotinib | Reduced abdominal aorta diameter and increased survival; decreased ADAM17, MMP2, IL-6, and ER stress markers | 76 |
| Endothelial dysfunction (aldosterone/salt/nephrectomy) | Waved-2 mouse | Restored endothelial dysfunction and inhibited AngII-induced contraction; did not affect vascular remodeling | 90 |
| Endothelial dysfunction (STZ-induced type 1 diabetes) | AG1478 | Restored endothelial function and eNOS expression; reduced ER stress | 91 |
| Endothelial dysfunction (db−/− mice) | AG1478 | Restored endothelial function and eNOS expression | 92 |
| Neovascularization (oxygen-induced retinopathy) | Erlotinib TIMP-3−/− mouse | Reduced neovascularization | 93 |
| Neovascularization (oxygen-induced retinopathy) | EC ADAM17 KO mouse | Reduced neovascularization and HB-EGF production | 94 |
Abbreviations: ADAM, a disintegrin and metalloprotease; AngII, angiotensin II; BAPN, beta aminopropionitrile; EC, endothelial cell; EGFR, epidermal growth factor receptor; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; HB-EGF, heparin-binding epidermal growth factor–like growth factor; IL-6, interleukin 6; KO, knockout; SHR, spontaneously hypertensive rat; STZ, streptozotocin; TIMP-3, tissue inhibitor of metalloprotease-3.
Cumulatively, in vitro analysis has helped illustrate the complex signaling dynamics of the EGFR transactivation potentially involved in many vascular pathological conditions. These cell studies further help lay the framework for understanding the signaling phenotype observed in animal models of vascular diseases including hypertension. The contribution of VSMC EGFR transactivation in vivo will be discussed in the following section.
Vascular Smooth Muscle EGFR Transactivation In Vivo
Hypertension
It is interesting to observe the multifaceted role of EGFR in non-cancer diseased states such as in cardiovascular diseases. For example, use of an EGFR antisense oligodeoxynucleotide can abolish hypertension in male Sprague-Dawley rats infused with AngII as well as in spontaneously hypertensive rats (49; 50). To support the role of EGFR in hypertension, it has been shown that EGFR inhibitors AG1478 or PD153035 reduce pressure-induced myogenic tone (51) and adrenergic vasoconstriction (30) in rodent mesenteric arteries, which also involves a metalloprotease-dependent HB-EGF production. AG1478 also prevents development of hypertension and enhanced vascular myosin light chain expression in fructose-fed rats (52). In addition, the EGFR inhibitor PKI-166 attenuates the progression of hypertension in 5/6 nephrectomized rats (53). These data highlight the pivotal roles of vascular EGFR activation contributing to the development of hypertension and maintenance of blood pressure, which is supported by the hypotensive phenotype and responsiveness to AngII observed in vascular-selective EGFR deficient mice (54).
In contrast, treatment of mice with erlotinib, a clinically utilized EGFR inhibitor, failed to reduce elevated blood pressure while allowing significant protection from vascular hypertrophy, perivascular fibrosis (55) and renal fibrosis (56) in AngII-infused mice. The erlotinib-mediated tissue protection is associated with attenuation of tissue growth factor-β (TGFβ) induction (56), ADAM17 induction, endoplasmic reticulum (ER) stress and oxidative stress (55). AngII-induced hypertrophy of cerebral arterioles but not hypertension is attenuated in mice treated with AG1478 as well as in waved-2 mice having non-functional EGFR (57). Additionally, ADAM17 induction by AngII was attenuated by ER chaperone 4-phenylbutyrate or inhibition of hypoxia inducible factor 1, suggesting a positive feed back loop for EGFR transactivation induced by AngII potentially contributing to hypertensive end-organ damage (55; 58). In humans, positive correlation of plasma EGF levels to diastolic blood pressure and carotid artery stiffness has been reported (59). These accumulating findings support the notion that EGFR is a critical therapeutic target in hypertension intervention. Nevertheless, further investigation into the EGFR specific mechanisms contributing to hypertension complications is warranted.
Restenosis and Atherosclerosis
In a balloon catheter model of rat carotid intimal hyperplasia/restenosis, the delivery of an EGFR blocking antibody (60) or EGFR inhibitory protein (61) is sufficient to inhibit medial hypertrophy and intimal hyperplasia. In the same model, phosphor- EGFR-positive cells in the neointima is significantly reduced and intimal hyperplasia was attenuated when carotid arteries are treated with an adenovirus encoding dominant-negative ADAM17 (35). In line with these findings, partial ligation induces EGFR activation and carotid artery hypertrophic remodeling which is prevented in HB-EGF deficient mice (62). Moreover, enhanced expression and plasma secretion of HB-EGF and enhanced arterial EGFR activation has been observed in a primate model of diet-induced atherosclerosis (63). The concept of the contribution of EGFR in atherosclerosis is further supported by human data demonstrating elevated expression of EGFR ligands (64) and ADAM17 in patients with atherosclerosis (65).
Abdominal Aortic Aneurysm
Both EGFR and ADAM17 have been identified as highly ranked nexus genes up-regulated in abdominal aortic aneurysms (AAA) of apolipoprotein E (ApoE) −/− mice infused with AngII (66). Additionally, enhanced ADAM17 expression is observed in human AAAs (67). Thus, silencing of ADAM17 prevents CaCl2-induced AAA in mice (68). In agreement with the involvement of Cav1 in ADAM17-dependent EGFR transactivation in VSMCs, AAA development and rupture in mice treated with AngII and β-aminopropionitrile has been shown to be prevented in Cav1 deficient mice (36). Likewise, erlotinib treatment inhibits AAA formation and rupture in this model, associated with reductions in endoplasmic reticulum stress, oxidative stress, expression of ADAM17, interleukin-6, and MMPs. Activation of EGFR is also observed in human AAAs (69). These results indicate that inhibition of EGFR could be a potential treatment for AAA.
Vascular smooth muscle EGFR transactivation is thus a potential therapeutic target to treat cardiovascular diseases. Downstream mediators of EGFR for vascular pathophysiology are summarized in Table 2.
Table 2.
Cardiac epidermal growth factor receptor pathophysiology
| Model | Intervention | Outcome and intermediate | Reference(s) |
|---|---|---|---|
| Cardiac hypertrophy (AngII infusion or SHR) | EGFR antisense | Reduced cardiac hypertrophy and ERK1/2 activation | 55, 56 |
| Cardiac hypertrophy (AngII infusion) | Erlotinib | Reduced cardiac hypertrophy, ADAM17 expression, and ER stress | 61 |
| Cardiac hypertrophy (AngII or ISO infusion) | Cardiac RALT transgenic mice | Reduced cardiac hypertrophy and fibrosis; reduced activation of Akt and ERK and expression of TGF-β1 | 112 |
| Cardiac hypertrophy (AngII infusion or TAC) | KB-R7785 (ADAM inhibitor) | Reduced cardiac hypertrophy and HB-EGF shedding | 98 |
| Cardiac hypertrophy (AngII or cardiac AT1R transgenic) | Cardiac dnEGFR transgenic mice | Reduced cardiac hypertrophy and ANP expression | 113 |
| Cardiac hypertrophy (aldosterone/salt/nephrectomy) | Cardiac dnEGFR transgenic mice | Did not alter cardiac hypertrophy or fibrosis | 114 |
| Cardiac hypertrophy/failure (ISO infusion) | Gefitinib | Reduced hypertrophy and apoptosis; altered βAR-sensitive cytokines including CCL2 and TNF-α | 120 |
| Cardiac hypertrophy | SM22-mediated cardiac EGFR KO | Resulted in cardiac hypertrophy and enhanced expression of ANP/BNP with high mortality in mice | 60 |
| Cardiac hypertrophy/dysfunction | EKB-560 AG1478 | Increased wall thickness and decreased cardiac function with 3-month treatment of the EGFR inhibitor | 122 |
| Aortic valve stenosis | Waved-2 mouse | Resulted in aortic valve stenosis, cardiac hypertrophy, fibrosis, and dysfunction in C57BL6/J background mice | 123 |
| Cardiac dilation/dysfunction (ISO infusion) | Erlotinib | Decreased cardiac function and increased LV dilation; decreased p-ERK expression and increased apoptosis | 102 |
| Diabetic cardiomyopathy (STZ-induced type 1 diabetes) | AG1478 | Reduced cardiac fibrosis, ER stress markers, and expression of PAI-1 and collagen-I | 91 |
| Preconditioning (ischemia/reperfusion) | AG1478 | Attenuated preconditioning by the inhibitor | 124 |
| Arrhythmias (ischemia/reperfusion) | AG556 | Prevented arrhythmias and tyrosine phosphorylation of cardiac Na+ and L-type Ca2+ channels | 127 |
| Myocardial infarction (coronary ligation in TIMP-3−/− mice) | Cetuximab | Attenuated cardiac rupture, TGF-β expression, and collagen synthesis in mice with myocardial infarction | 128 |
Abbreviations: ADAM, a disintegrin and metalloprotease; AngII, angiotensin II; ANP, atrial natriuretic peptide; AT1R, AngII type 1 receptor; βAR, β-adrenergic receptor; BNP, B-type natriuretic peptide; CCL2, chemokine C-C motif ligand 2; dnEGFR, dominant-negative EGFR; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; HB-EGF, heparin-binding epidermal growth factor–like growth factor; ISO, isoproterenol; KO, knockout; LV, left ventricle; PAI-1, plasminogen activator inhibitor-1; RALT, receptor-associated late transducer; SHR, spontaneously hypertensive rat; STZ, streptozotocin; TAC, transverse aortic constriction; TGF, transforming growth factor; TNF-α, tumor necrosis factor alpha.
ENDOTHELIUM
Unlike VSMC EGFR activity, endothelial transactivation of EGFR, either through AngII or other stimuli, is less well studied. However, there have been significant strides in providing clarity to EGFR signaling in cell-based models, and some correlational animal studies.
EGFR Activation in Endothelial Cells
Human umbilical vein endothelial cell (HUVEC) migration stimulated by AngII is mediated through focal adhesion kinase and paxilin phosphorylation and is dependent upon signaling involving EGFR transactivation, as assessed through pharmacological inhibition (70). Similarly, vascular endothelial growth factor (VEGF)- stimulated HUVEC migration is dependent upon HB-EGF shedding and EGFR transactivation (71). AngII-induced microparticle release from mouse aortic endothelial cells results in inflammatory activation that is dependent on EGFR transactivation and lipid raft/caveolae (72). In contrast, activation of proteinase-activated receptor 2 stimulates release of anti-angiogenic soluble VEGF receptor-1 via EGFR in HUVECs (73).
As in VSMCs, hydrogen peroxide activates EGFR and subsequently c-Jun N-terminal kinase in porcine aortic ECs, which appears to require mitochondrial ROS production (74). 20-hydroxyeicosatetraeonic acid has been implicated in the development of vascular complications including cardiac ischemia-reperfusion injury, diabetes mellitus, and hypertension. ECs treated with 20-hydroxyeicosatetraeonic acid experience eNOS uncoupling and ROS production in conjunction with enhanced ACE expression, all being dependent upon EGFR transactivation (75). Moreover, endothelial transactivation of EGFR is also induced with oxLDL (46), unsaturated fatty acids (76), hypoxia (77) and nitric oxide (78). The oxLDL-induced EGFR activation in HUVECs appears to require the AT1R (79). Therefore, EGFR seems to be a point of signal convergence by which various pathophysiological conditions may promote endothelial dysfunction.
Endothelial EGFR Transactivation in Humans and Animal Models
Corroborating in vitro investigation of endothelial EGFR transactivation, numerous in vivo studies exist that illustrate a role for EGFR and related components in specific endothelial phenotypes. In human coronary microarteries, aldosterone potentiates AngII-induced vasoconstriction, which involves counter regulation among eNOS and EGFR (80).
Hypertension-associated Endothelial Dysfunction
In a mouse model of renovascular hypertension, AG1478 treatment restores endothelium-dependent relaxation (81). The signal transduction is also mediated through activity of the gp91- phox subunit of the NADPH oxidase, which is hypothesized to work in a feedback loop with AT1R and EGFR during hypertension (81). L-NG-nitroarginine methyl ester (L-NAME)-induced hypertension in rats promotes increased renal inflammation, ECM accumulation, and decreased endothelial function. The alterations in renal blood flow and endothelial function can be rescued through EGFR antagonism independent of renal inflammation and structural damage (82). Similar protection for endothelial function but not for vascular remodeling is seen in waved-2 mice treated with aldosterone and salt (83).
Diabetes-associated Endothelial Dysfunction
Endothelial dysfunction is a critical component of cardiovascular complications associated with diabetes. In a mouse model of STZ-induced type-1 diabetes, endothelium-dependent relaxation can be restored in the mesenteric resistance arteries treated with AG1478, with potential downstream ablation of ER stress (84). In addition, AG1478 restores eNOS expression and rescues endothelial dysfunction within mesenteric resistance and coronary arteries of db/db mice, which is a widely used model of type-2 diabetes (85).
Neovascularization
Mice injected with erlotinib are protected from oxygen-induced neovascularization (86). To support this notion, mice with endothelial deletion of ADAM17 also show resistance to oxygen–induced retinopathy and neovascularization (87).
Downstream mechanisms of EGFR in mediating endothelium-dependent function and pathophysiology are summarized in Table 3. Cumulatively, these findings highlight a point of emphasis in delineating endothelial EGFR transactivation between physiological responses and pathological insults. Endothelial EGFR appears to be a necessary in normal cardiovascular development and homeostasis (88), but during diseased states where EGFR stimulation is chronic and enhanced, detrimental consequences might occur that contribute to endothelial dysfunction.
Table 3.
Kidney epidermal growth factor receptor pathophysiology
| Model | Intervention | Outcome and intermediate | Reference |
|---|---|---|---|
| Acute kidney injury (mercuric chloride injection) | Waved-2 mice | Resulted in slower recovery from acute renal injury; associated with reduced EGFR activation and DNA synthesis | 150 |
| Acute kidney injury (ischemia/reperfusion) | Proximal tubule EGFR KO mice or erlotinib | Resulted in slower recovery from acute renal injury; associated with reduced ERK and Akt activation | 151 |
| Acute glomerulonephritis (anti-GBM injection) | Podocyte EGFR KO mice or AG1478 | Prevented renal dysfunction, pathology, and inflammation | 153 |
| Hypertensive renal damage (L-NAME infusion) | Gefitinib | Reduced glomerular matrix deposition, ERK activation, and renal dysfunction | 154 |
| Hypertensive renal damage (L-NAME infusion) | Gefitinib upon renal damage | Resulted in nonreversible renal pathology and hypertension, but normalized renal blood flow and endothelial function | 122 |
| Hypertensive renal damage (AngII infusion) | Proximal tubule dnEGFR transgenic | Prevented renal dysfunction and fibrosis; ADAM17 and TGF-α are pivotal components to the EGFR activation | 155 |
| Hypertensive renal fibrosis (AngII infusion) | Proximal tubule EGFR KO mice or erlotinib | Prevented renal fibrosis; the downstream signal of EGFR includes ERK, TGF-β, and SMAD2/3 | 62 |
| Hypertensive renal fibrosis (ISO infusion) | Gefitinib | Reduced hypertrophy and apoptosis; altered βAR-sensitive cytokines including CCL2 and TNF-α | 89 |
| Renal fibrosis (ischemic reperfusion) | Waved-2 mice | Enhanced acute renal damage, but resulted in development of less fibrosis and activated Akt/STAT3 at later stage | 152 |
| Diabetic nephropathy (STZ treatment) | PKI-166 | Reduced kidney enlargement and epithelial cell proliferation, but did not affect hyperfiltration | 157 |
| Diabetic nephropathy (STZ treatment) | eNOS KO mice with erlotinib treatment | Reduced diabetic nephropathy, ER stress, CTGF and collagen; increased AMPK activation and autophagy | 158 |
| Diabetic nephropathy (STZ treatment) | Podocyte EGFR KO mice | Reduced podocyte loss, TGF-β, and mitochondrial ROS; decreased caspase 3 cleavage and restored BCL2 | 159 |
| Chronic renal failure (5/6 nephrectomy) | PKI-166 | Did not prevent renal damage and dysfunction, but attenuated hypertension | 59 |
| Chronic renal failure (3/4 nephrectomy) | Proximal tubule dnEGFR transgenic | Reduced proximal tubule lesions | 160 |
| Chronic renal failure (unilateral ureteral obstruction) | Waved-2 or gefitinib | Reduced renal fibrosis; attenuated expression of TGF-β1 and activation of Smad3, STAT3, and ERK | 161 |
| Polycystic kidney (BPK mice) | EKI-785 | Reduced cystic lesions, improved renal function, and increased life span | 162 |
Abbreviations: ADAM, a disintegrin and metalloprotease; AMPK, AMP-activated protein kinase; AngII, angiotensin II; BCL2, B-cell lymphoma 2; βAR, β-adrenergic receptor; BPK, polycystic kidney mutation of BALB/c origin; CCL2, chemokine C-C motif ligand 2; CTGF, connective tissue growth factor; dnEGFR, dominant-negative EGFR; EGFR, epidermal growth factor receptor; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; GBM, glomerular basement membrane; ISO, isoproterenol; KO, knockout; ROS, reactive oxygen species; SMAD2/3, mothers against decapentaplegic homolog 2/3; STAT3, signal transducer and activator of transcription 3; STZ, streptozotocin; TGF, transforming growth factor; TNF-α/β, tumor necrosis factor alpha/beta.
HEART
Cell Models of Cardiac EGFR Transactivation
Cardiac Myocyte Hypertrophy and Protection
One of the earliest studies regarding the role of EGFR in cardiac myocytes was performed in rat neonatal cardiac myocytes infected with adenovirus encoding the AT1R. The AngII-induced cardiac myocyte hypertrophy, as well as ERK activation, was inhibited with dominant negative Gαq or AG1478 treatment, suggesting the involvement of EGFR transactivation in cardiac hypertrophy in a condition with enhanced AT1R expression. Subsequent studies in this model suggest that AngII-induced EGFR transactivation, ERK activation and myocyte hypertrophy require Gq coupling and metalloprotease–mediated HB-EGF production but not β-arrestin recruitment or AT1R C-terminal Tyr319 phosphorylation (89) (see below). In line with these findings, ADAM12 has been identified as an essential metalloprotease required for AngII-induced HB-EGF shedding, EGFR transactivation and cardiomyocyte hypertrophy- (90). In cardiac myocytes, AG1478 inhibits aldosterone-induced activation of p90RSK and induction of Na+/H+ exchanger-1, which are implicated in cardiac hypertrophy (91).
While these studies support the role of cardiac EGFR for pathological cardiac hypertrophy, recent studies have revealed additional important functions for EGFR in mediating cardiac myocyte survival. Stimulation of β1-Adrenergic Receptors (β1AR) with dobutamine markedly induces EGFR transactivation, internalization and ERK activation in cardiac myocytes. Further, β1AR-mediated transactivation of EGFR requires β-arrestin recruitment through G protein-coupled receptor kinase-5 and -6 and subsequent canonical cascade of EGFR transactivation, including Src, a metalloprotease and HB-EGF shedding (92). Importantly, interruption of this cascade leads to myocyte apoptosis, highlighting the role for EGFR in myocyte protection and survival under conditions of cardiac myocyte stress/overload (92). This G protein-independent mechanism of EGFR transactivation also occurs with certain β-blockers, such as alprenolol and carvedilol, which biased agonism (93). Along with these findings, mechanical stress has been shown to induce β-arrestin-dependent biased agonism of the AT1R leading to EGFR transactivation and protection of apoptosis in cardiac myocytes (94). How one cascade can be considered beneficial to maintaining cell health and the other detrimental is the discrepancy that arises between β-arrestin– and G protein-dependent EGFR transactivation. The answer may lie in the spatial and temporal differences in ERK activation. The G protein-dependent pathway involves immediate and transient ERK1/2 activation and localization to the nucleus, whereas G protein-independent β-arrestin-dependent EGFR transactivation mediates later and sustained ERK activation in the cytosol to form β1AR-EGFR-ERK association working as an anti-apoptotic signaling complex (13).
Cardiac Fibroblast Proliferation
In cardiac fibroblasts, AngII, a PAR-1 agonist and hydrogen peroxide transactivate EGFR (95; 96). It has been demonstrated that the AngII mechanism of EGFR transactivation and subsequent proliferation of cardiac fibroblasts requires recruitment of EGFR to the tyrosine phosphorylated AT1R at Tyr319 (97).
In Vivo Mechanisms of Cardiac EGFR Transactivation
Left Ventricular Hypertrophy and Fibrosis
Just as with in vitro analysis, AngII is a well-known inducer of pathophysiological cardiac remodeling which includes hypertrophy and interstitial fibrosis, and recent evidence has provided a role for EGFR. Earlier studies with antisense infusion demonstrated suppression of left ventricular hypertrophy in SHR and in rats with AngII infusion (49; 50). In addition, transgenic mice overexpressing receptor-associated late transducer, a feedback inhibitor of EGFR signaling, do not develop AngII-induced cardiac hypertrophy (98). As shown in culture, cardiac hypertrophy is attenuated with an ADAM inhibitor in mice with AngII infusion (90). Likewise, wild-type AT1R transgenic mice crossed with mice harboring a dominant negative EGFR mutation in hearts are also protected from AngII-induced cardiac hypertrophy (99). Specifically, transgenic mice with overexpression of an AT1R Y319F mutant within the conserved YIPP motif fail to stimulate EGFR transactivation and are protected from AngII-mediated cardiac hypertrophy, fibrosis and apoptosis within the left ventricular myocardium (99). It should be noted that the function of Tyr319 in activating EGFR is in contradiction to the subsequent work conducted in rat cardiac myocytes (89). In addition, AngII-induced cardiac hypertrophy is markedly prevented in mice infused with erlotinib, while activation of EGFR is mainly observed in coronary arteries (55). Regardless of the mechanism, these data support the role for EGFR in mediating pathophysiological cardiac remodeling via AngII-stimulation of the AT1R.
In contrast, the role of EGFR in aldosterone-dependent cardiac pathophysiology is less clear. In transgenic mice expressing a dominant-negative mutant of EGFR in the heart, aldosterone plus salt-induced cardiac hypertrophy, fibrosis and associated gene expression are unaltered (100). However, this could be explained by the findings that cardiac hypertrophy could be mediated via MR expressed in VSMCs (101) and myeloid cells (102). Further complicating this area, low dose aldosterone/salt treatment appears to be beneficial against cardiac hypertrophy developing in relatively aged male mice lacking both vascular smooth muscle and cardiac myocyte EGFR (see below in detail), whereas the beneficial effects are not seen in female mice (103).
Diabetic Cardiomyopathy
Aside from the enhanced RAAS, diabetes correlates strongly with heart failure (104), and inhibition of EGFR by AG1478 reduces development of cardiac fibrosis in a mouse model of type-1 diabetes (84). In addition to the direct consequence of cardiac EGFR inhibition, this protection may involve improvements in metabolic condition, as AG1478 prevents reduction in body weight and partially reduces plasma glucose concentration (84). To expand this concept, it has been shown that PD153035 improves glucose tolerance, insulin sensitivity and signaling and reduces inflammation in mice fed with high fat diet (105).
Cardiac Homeostasis and Protection
The complexity and controversies of cardiac EGFR observed in in vivo studies are most likely due to its double edged function in cardiac homeostasis and pathophysiology. EGFR is vital for semilunar valvulogenesis, and total body deletion of EGFR results in embryonic lethality in mice (106). In adult C57BL6/J mice, 3 months of treatment with EGFR inhibitors EKB-560 or AG1478 results in increased LV wall thickness and a decline in cardiac function (107). Subsequent studies with waved-2 mice with 2 distinct background strains (C57BL6/J and 129S1/SvImJ) revealed that these mice develop aortic valve stenosis and in C57BL6/J subsequent cardiac hypertrophy, fibrosis, dysfunction and high mortality, whereas 129S1/SvImJ retain normal cardiac structure and function (108). In line with these findings, SM22-mediated smooth muscle and cardiac EGFR deficient mice develop cardiac hypertrophy with high mortality (54).
As mentioned previously, β-arrestin mediated activation of EGFR offers a cardioprotective effect, as cardiac specific transgenic mice expressing β1AR lacking G protein-coupled receptor kinase phosphorylation sites needed in β-arrestin-induced EGFR activation show reduced cardiac function and left ventricular dilation in response to chronic isoproterenol stimulation. Furthermore, mice co-treated with isoproterenol and erlotinib show marked reduction in fractional shortening, increased left ventricular diastolic dimension, and an increase in myocardial apoptosis (92). In contrast, gefitinib blocks cardiac hypertrophy induced by isoproterenol in these mice, but also induces cardiac fibrosis when treated alone (109).
Cardiac Preconditioning, Arrhythmia and Myocardial Infraction
It is well established that brief episodes of ischemia/reperfusion (preconditioning) protect the myocardium from the damage induced by subsequent and more prolonged ischemia. In rat models of cardiac preconditioning, AG1478 treatment attenuates preconditioned protection (110). This concept has also been confirmed in rabbit heart infused with δ-opioid receptor agonist (111) and a rat model of type-1 diabetes that involves downstream activation of survival signals Akt and FOXO transcriptional factors (112). However, treatment with the EGFR inhibitor AG556 reduces ischemia/reperfusion- induced arrhythmias that are associated with a reduction in tyrosine phosphorylation of cardiac Na+ and L-type Ca2+ channels. While EGFR activation may be still beneficial to protect heart from ischemic damage, upon development of myocardial infarction, the role of EGFR appears opposite. Tissue inhibitor of metalloproteinase-3 (TIMP-3) deficient mice show significant increases in cardiac rupture and mortality with left coronary artery ligation. Treatment with an EGFR inhibitor, cetuximab, significantly decreases incidence of cardiac rupture and improved survival (113).
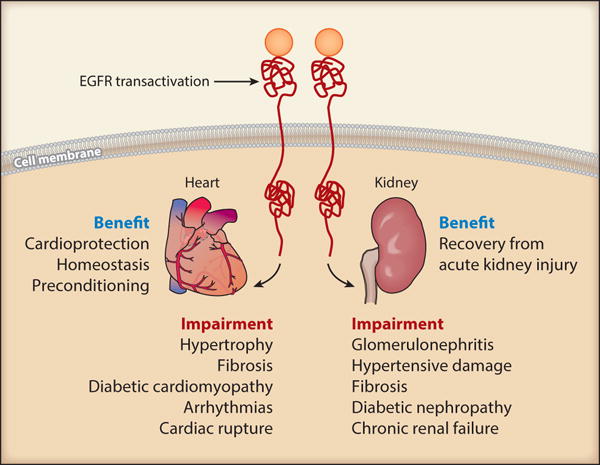
Taken together, while it is evident that EGFR is a necessary component for cardiac development and a needed asset in the cardioprotective response against acute cardiac overload (such as that mimicked by catecholamine stimulation) and myocardial ischemia (angina), chronic activation of EGFR in response to hypertension or diabetes may also promote an environment favoring pathological remodeling and functional alterations (see downstream mediators in Table 4). In summary, cardiac transactivation of EGFR is a diverse topic with implications in both pathology and physiology, and further investigation should be aimed at understanding the balance between the two.
KIDNEY
In normal human kidneys, EGFR is constitutively expressed in glomeruli, tubules, and interstitium with several cell types that include renal epithelial cells, podocytes, renal interstitial fibroblasts, and mesangial cells. EGFR ligands, such as EGF, TGFα, and HB-EGF are also expressed in renal tubules (114).
EGFR Transactivation in Renal Cells
Mesangial Cells
In mouse mesangial cells, AngII stimulates the metalloprotease-dependent processing of HB-EGF and activates EGFR in mediating fibronectin synthesis (115). This process also includes autocrine TGFβ mediated EGFR transactivation (115; 116). Likewise, high glucose-induced TGFβ induction appears to require ADAM17-mediated HB-EGF production and EGFR activation in mesangial cells (117). Src kinase acts upstream of the ADAM17-mediated EGFR transactivation and mediates collagen IV synthesis (118). In addition, aldosterone transactivates EGFR via MR-mediated ROS production. This signaling cascade then promotes proliferation of mesangial cells (119).
Tubular Cells
In the renal proximal tubular cell line LLCPKcl4, transfected with an AT1R expression vector, AngII elicits HB-EGF shedding and EGFR transactivation leading to hypertrophic signal transduction and protein synthesis (120). Further studies on this cell line demonstrate that ROS-dependent Src activation induces phosphorylation of Cav1 and subsequent chronic activation of EGFR in lipid rafts, which seems critical for AngII-induced epithelial-to-mesenchymal cell transition (121). In line with these findings, hydrogen peroxide induces p38 MAPK-dependent EGFR activation in rabbit proximal tubular cells leading to β-catenin tyrosine phosphorylation, a key intermediate step in epithelial-mesenchymal transition (122). In addition, tumor necrosis factor-like weak inducer of apoptosis (TWEAK) transactivates EGFR via ADAM17 and HB-EGF in mediating inflammatory responses in proximal tubular cells (123).
Albuminuria is a well-known marker of renal diseases. Exposure of renal proximal tubular cell to albumin activates EGFR, leading to nuclear factor-κB activation and cell cycle progression (124). In human renal epithelial cells, albumin activates EGFR and ERK via ROS production and this signaling cascade is required for interleukin-8 induction (125). In contrast, in vitro studies also suggest a role for EGFR in tubular cell survival and normal function. Madin-Darby canine kidney epithelial cells expressing a non-cleavable form of membrane anchored HB-EGF showed improved survival from anoikis and, notably, addition of exogenous EGF partially suppresses anoikis (126).
Several cell studies suggest involvement of EGFR signaling in ion transport such as proximal tubule phosphate transportat (127) and magnesium transport in the thick ascending limb of Henle (128). Additionally, EGFR signaling inhibits epithelial sodium channels in the collecting duct to regulate aldosterone-induced nephron sodium transport (129; 130). Bradykinin B2 receptor stimulation is also reported to transactivate EGFR via ADAM17 and decrease podocyte permeability (131).
EGFR Transactivation in Renal Pathophysiology
Acute Kidney Injury
During the recovery from acute renal injury, EGFR activation may play an important role. In a mouse model of acute nephrotoxic injury with injection of mercuric chloride, recovery from injury, as assessed by blood urea nitrogen and creatinine levels, is slower in waved-2 mice, which is associated with reduced renal EGFR activation and DNA synthesis and enhanced tubular injury and apoptosis (132). In line with these findings, proximal tubule EGFR deficient mice or mice treated with elrotinib also show slower recovery in a model of acute kidney injury with bilateral ischemia/reperfusion injury that is associated with reductions in ERK and Akt activation (133). However, while the recovery is slower in waved-2 mice with unilateral ischemia (45 min)-reperfusion, the waved-2 mice develop less renal fibrosis at later stages, suggesting the detrimental role for chronic EGFR activation upon renal injury (134).
In a model of rapidly progressive/crescentic glomerulonephritis with anti-glomerular basement membrane nephrotoxic serum injection, induction of HB-EGF and activation of EGFR are observed in the renal cortex. However, renal pathology, inflammation, increased blood urea nitrogen and albumin urea are markedly prevented in mice deficient in intrinsic glomerular cells/podocyte EGFR or in mice treated with erlotinib, suggesting the therapeutic value of EGFR inhibition for treating inflammatory glomerulonephritis (135).
Hypertensive Renal Damage/Fibrosis
In line with the roles of EGFR in vascular and cardiac fibrosis associated with hypertension, co-treatment of gefitinib prevents EGFR activation, ERK activation in the renal cortex, and glomerular matrix deposition and renal dysfunction but not hypertension in rats treated with L-NAME (136). In transgenic mice expressing dominant-negative EGFR in renal proximal tubular cells, interstitial fibrosis, glomerular and tubular lesions, inflammatory cell infiltration and blood urea nitrogen (BUN) induced by AngII infusion are significantly reduced without attenuating hypertension. The lesion formation is associated with ADAM17 induction. Knockout of TGFα or treatment with an ADAM17 inhibitor also reduces lesions in mouse kidneys suggesting a role for ADAM17-dependent EGFR transactivation by AngII (137). Likewise, targeted deletion of EGFR in proximal tubules or systemic inhibition of EGFR by erlotinib inhibits renal fibrosis induced by AngII infusion in mice. Again, this effect was independent of blood pressure as knockout or pharmacological inhibition of EGFR does not alter blood pressure upon AngII infusion. However, additional experiments suggest roles for Src kinase-dependent EGFR transactivation at lipid rafts and subsequent TGFβ induction in AngII-induced renal fibrosis (56). Interestingly, the effect of EGFR inhibition seems distinct once hypertensive renal pathology is established. Upon confirmation of overt proteinuria in rats with L-NAME treatment, the rats were treated with gefitinib. While geftinib does not affect blood pressure or renal inflammation, it normalizes renal blood flow and afferent arteriole hypertrophy and preserves functional endothelial cells in the kidneys (82). The findings are mechanistically supported by the notion that AngII-induced afferent arteriole contraction and Ca2+ influx are mediated through the EGFR (138).
Diabetic Nephropathy
In a streptozotocin-treated rat model of type-1 diabetes, PKI166 reduces kidney weight, epithelial cell proliferation and increases cell apoptosis, whereas glomerular hyperfiltration remains unchanged (139). In a type-1 diabetic model of streptozotocin-treated endothelial nitric oxide synthase deficient mice, erlotinib treatment reduces development of albuminuria that is associated with EGFR inhibition, with less glomerular collagen deposition and oxidative stress. Moreover, erlotinib attenuates ER stress and stimulates autophagy in these mice (140). In line with these studies, streptozotocin-induced renal pathology and albumin urea are markedly prevented in mice deficient in intrinsic glomerular cells/podocytes EGFR. Further experiments in this model indicated a mechanism involving Smad2/3, TGFβ and mitochondrial ROS downstream of the EGFR (141).
Chronic Renal Failure
In hypertensive 5/6 nephrectomized rats, chronic renal insufficiency is developed by 3 months, with proteinuria, diminished creatinine clearance and increased glomerulosclerosis. PKI-166 does not prevent this progressive renal damage, whereas it attenuates hypertension and maintains cardiac function (53). In strong contrast, nephron reduction and proximal tubule lesions in 3/4 nephrectomized mice are attenuated in transgenic mice expressing dominant-negative EGFR in renal proximal tubular cells (142). Beneficial effects of EGFR inhibition in chronic renal failure are also supported by a study demonstrating reduction in renal fibrosis and associated signal transduction in an obstructive nephropathy model of unilateral ureteral obstruction in waved-2 mice or mice treated with gefitinib (143). In addition, inhibition of EGFR activation seems effective in improving pathological abnormalities in a mouse model of polycystic kidney disease (144).
Overall, inhibition of EGFR may provide considerable efficacy in amelioration of various chronic renal pathologies (see downstream mediators in Table 5), whereas careful attention is needed for the protective role of EGFR in acute renal injury.
CLINICAL ALTERATIONS OF EGFR INHIBITORS
Extrapolating to humans, investigation into EGFR–related mechanisms and cardiovascular disease is limited. Separate from a specific discussion of EGFR, advantages and disadvantages of using receptor tyrosine kinase inhibitors as non-cancer therapeutics and their associations with cardiovascular toxicity have been reviewed elsewhere (145; 146). In particular, EGFR/ErbB1 inhibitors appear to have minimum cardiovascular toxicity compared with other tyrosine kinase inhibitors including those against ErbB2 (145). However, as seen in cell culture study, the use of EGFR targeted antibodies in colorectal cancer patients is associated with decreased serum magnesium concentrations leading to altered renal magnesium retention capacity and hypomagnesaemia (147). Erlotinib has been reported to induce renal toxicity and hypertension if combined with bevacizumab (VEGF-A inhibitor) (148). Thus, further information from patients treated with EGFR inhibitors are warranted regarding their cardiovascular and renal outcomes.
Figure 3.
References
- 1.Zeng F, Harris RC. Epidermal growth factor, from gene organization to bedside. Seminars in cell & developmental biology. 2014;28:2–11. doi: 10.1016/j.semcdb.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature reviews. Molecular cell biology. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65:1566–84. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer cell. 2014;25:282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makki N, Thiel KW, Miller FJ., Jr The epidermal growth factor receptor and its ligands in cardiovascular disease. Int J Mol Sci. 2013;14:20597–613. doi: 10.3390/ijms141020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreier B, Gekle M, Grossmann C. Role of epidermal growth factor receptor in vascular structure and function. Current opinion in nephrology and hypertension. 2014;23:113–21. doi: 10.1097/01.mnh.0000441152.62943.29. [DOI] [PubMed] [Google Scholar]
- 7.George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013;280:5258–68. doi: 10.1111/febs.12509. [DOI] [PubMed] [Google Scholar]
- 8.Leserer M, Gschwind A, Ullrich A. Epidermal growth factor receptor signal transactivation. IUBMB life. 2000;49:405–9. doi: 10.1080/152165400410254. [DOI] [PubMed] [Google Scholar]
- 9.Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nature reviews. Molecular cell biology. 2003;4:651–7. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- 10.Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- 11.Elliott KJ, Bourne AM, Takayanagi T, Takaguri A, Kobayashi T, et al. ADAM17 silencing by adenovirus encoding miRNA-embedded siRNA revealed essential signal transduction by angiotensin II in vascular smooth muscle cells. Journal of molecular and cellular cardiology. 2013;62:1–7. doi: 10.1016/j.yjmcc.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overland AC, Insel PA. Heterotrimeric G Proteins Directly Regulate MMP14/Membrane Type-1 Matrix Metalloprotease: A NOVEL MECHANISM FOR GPCR-EGFR TRANSACTIVATION. J Biol Chem. 2015;290:9941–7. doi: 10.1074/jbc.C115.647073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tilley DG. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res. 2011;109:217–30. doi: 10.1161/CIRCRESAHA.110.231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blobel CP. ADAMs: key components in EGFR signalling and development. Nature reviews. Molecular cell biology. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 15.Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM-mediated shedding in vascular biology. European journal of cell biology. 2012;91:472–85. doi: 10.1016/j.ejcb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 17.Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clinical science. 2007;112:417–28. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- 18.Briet M, Schiffrin EL. Treatment of arterial remodeling in essential hypertension. Curr Hypertens Rep. 2013;15:3–9. doi: 10.1007/s11906-012-0325-0. [DOI] [PubMed] [Google Scholar]
- 19.Mifune M, Ohtsu H, Suzuki H, Frank GD, Inagami T, et al. Signal transduction of betacellulin in growth and migration of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2004;287:C807–13. doi: 10.1152/ajpcell.00436.2003. [DOI] [PubMed] [Google Scholar]
- 20.Montezano AC, Nguyen Dinh Cat A, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16:431. doi: 10.1007/s11906-014-0431-2. [DOI] [PubMed] [Google Scholar]
- 21.Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen- activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273:8890–6. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 22.Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem. 2001;276:7957–62. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- 23.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- 24.Touyz RM, Wu XH, He G, Salomon S, Schiffrin EL. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C- terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 2002;39:479–85. doi: 10.1161/hy02t2.102909. [DOI] [PubMed] [Google Scholar]
- 25.Ohtsu H, Higuchi S, Shirai H, Eguchi K, Suzuki H, et al. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology. 2008;149:3569–75. doi: 10.1210/en.2007-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-Mediated Vascular Growth Requires p42/p44 Mitogen-Activated Protein Kinase and p70 S6 Kinase Cascades via Transactivation of Epidermal Growth Factor Receptor. Endocrinology. 1999;140:4659–68. doi: 10.1210/endo.140.10.7023. [DOI] [PubMed] [Google Scholar]
- 27.Schreier B, Dohler M, Rabe S, Schneider B, Schwerdt G, et al. Consequences of epidermal growth factor receptor (ErbB1) loss for vascular smooth muscle cells from mice with targeted deletion of ErbB1. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1643–52. doi: 10.1161/ATVBAHA.111.223537. [DOI] [PubMed] [Google Scholar]
- 28.Li F, Malik KU. Angiotensin II-induced Akt activation through the epidermal growth factor receptor in vascular smooth muscle cells is mediated by phospholipid metabolites derived by activation of phospholipase D. The Journal of pharmacology and experimental therapeutics. 2005;312:1043–54. doi: 10.1124/jpet.104.076588. [DOI] [PubMed] [Google Scholar]
- 29.Abdallah RT, Keum JS, El-Shewy HM, Lee MH, Wang B, et al. Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors. J Biol Chem. 2010;285:35206–15. doi: 10.1074/jbc.M110.171769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor- receptor pathway. Circ Res. 2004;94:68–76. doi: 10.1161/01.RES.0000109413.57726.91. [DOI] [PubMed] [Google Scholar]
- 31.Hao L, Nishimura T, Wo H, Fernandez-Patron C. Vascular responses to alpha1-adrenergic receptors in small rat mesenteric arteries depend on mitochondrial reactive oxygen species. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:819–25. doi: 10.1161/01.ATV.0000204344.90301.7c. [DOI] [PubMed] [Google Scholar]
- 32.Nagareddy PR, Chow FL, Hao L, Wang X, Nishimura T, et al. Maintenance of adrenergic vascular tone by MMP transactivation of the EGFR requires PI3K and mitochondrial ATP synthesis. Cardiovascular research. 2009;84:368–77. doi: 10.1093/cvr/cvp230. [DOI] [PubMed] [Google Scholar]
- 33.Ushio‐Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–99. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohtsu H, Dempsey PJ, Frank GD, Brailoiu E, Higuchi S, et al. ADAM17 mediates epidermal growth factor receptor transactivation and vascular smooth muscle cell hypertrophy induced by angiotensin II. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:e133–7. doi: 10.1161/01.ATV.0000236203.90331.d0. [DOI] [PubMed] [Google Scholar]
- 35.Takaguri A, Kimura K, Hinoki A, Bourne AM, Autieri MV, Eguchi S. A disintegrin and metalloprotease 17 mediates neointimal hyperplasia in vasculature. Hypertension. 2011;57:841–5. doi: 10.1161/HYPERTENSIONAHA.110.166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, et al. Caveolin 1 is critical for abdominal aortic aneurysm formation induced by angiotensin II and inhibition of lysyl oxidase. Clinical science. 2014;126:785–94. doi: 10.1042/CS20130660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grossmann C, Krug AW, Freudinger R, Mildenberger S, Voelker K, Gekle M. Aldosterone- induced EGFR expression: interaction between the human mineralocorticoid receptor and the human EGFR promoter. American journal of physiology. Endocrinology and metabolism. 2007;292:E1790–800. doi: 10.1152/ajpendo.00708.2006. [DOI] [PubMed] [Google Scholar]
- 38.Mazak I, Fiebeler A, Muller DN, Park JK, Shagdarsuren E, et al. Aldosterone potentiates angiotensin II-induced signaling in vascular smooth muscle cells. Circulation. 2004;109:2792–800. doi: 10.1161/01.CIR.0000131860.80444.AB. [DOI] [PubMed] [Google Scholar]
- 39.Min LJ, Mogi M, Li JM, Iwanami J, Iwai M, Horiuchi M. Aldosterone and angiotensin II synergistically induce mitogenic response in vascular smooth muscle cells. Circ Res. 2005;97:434–42. doi: 10.1161/01.RES.0000180753.63183.95. [DOI] [PubMed] [Google Scholar]
- 40.Montezano AC, Callera GE, Yogi A, He Y, Tostes RC, et al. Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:1511–8. doi: 10.1161/ATVBAHA.108.168021. [DOI] [PubMed] [Google Scholar]
- 41.Meinel S, Gekle M, Grossmann C. Mineralocorticoid receptor signaling: crosstalk with membrane receptors and other modulators. Steroids. 2014;91:3–10. doi: 10.1016/j.steroids.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 42.Limor R, Kaplan M, Sharon O, Knoll E, Naidich M, et al. Aldosterone up-regulates 12- and 15-lipoxygenase expression and LDL oxidation in human vascular smooth muscle cells. Journal of cellular biochemistry. 2009;108:1203–10. doi: 10.1002/jcb.22352. [DOI] [PubMed] [Google Scholar]
- 43.Krug AW, Allenhofer L, Monticone R, Spinetti G, Gekle M, et al. Elevated mineralocorticoid receptor activity in aged rat vascular smooth muscle cells promotes a proinflammatory phenotype via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase and epidermal growth factor receptor-dependent pathways. Hypertension. 2010;55:1476–83. doi: 10.1161/HYPERTENSIONAHA.109.148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frank GD, Mifune M, Inagami T, Ohba M, Sasaki T, et al. Distinct mechanisms of receptor and nonreceptor tyrosine kinase activation by reactive oxygen species in vascular smooth muscle cells: role of metalloprotease and protein kinase C-delta. Molecular and cellular biology. 2003;23:1581–9. doi: 10.1128/MCB.23.5.1581-1589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC, Miller FJ., Jr Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovascular research. 2012;93:406–13. doi: 10.1093/cvr/cvr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suc I, Meilhac O, Lajole-Mazenc I, Vandaele J, Jürgens G, et al. Activation of EGF receptor by oxidized LDL. FASEB J. 1998;12:665–71. doi: 10.1096/fasebj.12.9.665. [DOI] [PubMed] [Google Scholar]
- 47.Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Mechanical stretch stimulates growth of vascular smooth muscle cells via epidermal growth factor receptor. Am J Physiol Cell Physiol. 2000;278:H521–9. doi: 10.1152/ajpheart.2000.278.2.H521. [DOI] [PubMed] [Google Scholar]
- 48.Konishi A, Berk BC. Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J Biol Chem. 2003;278:35049–56. doi: 10.1074/jbc.M304913200. [DOI] [PubMed] [Google Scholar]
- 49.Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips MI. Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation. 2002;106:909–12. doi: 10.1161/01.cir.0000030181.63741.56. [DOI] [PubMed] [Google Scholar]
- 50.Kagiyama S, Qian K, Kagiyama T, Phillips MI. Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy. Hypertension. 2003;41:824–9. doi: 10.1161/01.HYP.0000047104.42047.9B. [DOI] [PubMed] [Google Scholar]
- 51.Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–93. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 52.Nagareddy PR, MacLeod KM, McNeill JH. GPCR agonist-induced transactivation of the EGFR upregulates MLC II expression and promotes hypertension in insulin-resistant rats. Cardiovascular research. 2010;87:177–86. doi: 10.1093/cvr/cvq030. [DOI] [PubMed] [Google Scholar]
- 53.Ulu N, Mulder GM, Vavrinec P, Landheer SW, Duman-Dalkilic B, et al. Epidermal growth factor receptor inhibitor PKI-166 governs cardiovascular protection without beneficial effects on the kidney in hypertensive 5/6 nephrectomized rats. The Journal of pharmacology and experimental therapeutics. 2013;345:393–403. doi: 10.1124/jpet.113.203497. [DOI] [PubMed] [Google Scholar]
- 54.Schreier B, Rabe S, Schneider B, Bretschneider M, Rupp S, et al. Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension. 2013;61:333–40. doi: 10.1161/HYPERTENSIONAHA.112.196543. [DOI] [PubMed] [Google Scholar]
- 55.Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, et al. Role of Epidermal Growth Factor Receptor and Endoplasmic Reticulum Stress in Vascular Remodeling Induced by Angiotensin II. Hypertension. 2015;65:1349–55. doi: 10.1161/HYPERTENSIONAHA.115.05344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen J, Chen JK, Nagai K, Plieth D, Tan M, et al. EGFR signaling promotes TGFbeta- dependent renal fibrosis. J Am Soc Nephrol. 2012;23:215–24. doi: 10.1681/ASN.2011070645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chan SL, Umesalma S, Baumbach GL. Epidermal growth factor receptor is critical for angiotensin II-mediated hypertrophy in cerebral arterioles. Hypertension. 2015;65:806–12. doi: 10.1161/HYPERTENSIONAHA.114.04794. [DOI] [PubMed] [Google Scholar]
- 58.Obama T, Takayanagi T, Kobayashi T, Bourne AM, Elliott KJ, et al. Vascular induction of a disintegrin and metalloprotease 17 by angiotensin II through hypoxia inducible factor 1alpha. American journal of hypertension. 2015;28:10–4. doi: 10.1093/ajh/hpu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lundstam U, Hagg U, Sverrisdottir YB, Svensson LE, Gan LM. Epidermal growth factor levels are related to diastolic blood pressure and carotid artery stiffness. Scand Cardiovasc J. 2007;41:308–12. doi: 10.1080/14017430701439508. [DOI] [PubMed] [Google Scholar]
- 60.Chan AK, Kalmes A, Hawkins S, Daum G, Clowes AW. Blockade of the epidermal growth factor receptor decreases intimal hyperplasia in balloon-injured rat carotid artery. Journal of vascular surgery. 2003;37:644–9. doi: 10.1067/mva.2003.92. [DOI] [PubMed] [Google Scholar]
- 61.Pastore CJ, Isner JM, Bacha PA, Kearney M, Pickering JG. Epidermal growth factor receptor-targeted cytotoxin inhibits neointimal hyperplasia in vivo. Results of local versus systemic administration. Circ Res. 1995;77:519–29. doi: 10.1161/01.res.77.3.519. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, Sunnarborg SW, McNaughton KK, Johns TG, Lee DC, Faber JE. Heparin-binding epidermal growth factor-like growth factor signaling in flow-induced arterial remodeling. Circ Res. 2008;102:1275–85. doi: 10.1161/CIRCRESAHA.108.171728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stanic B, Pandey D, Fulton DJ, Miller FJ., Jr Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2452–60. doi: 10.1161/ATVBAHA.112.256107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dreux AC, Lamb DJ, Modjtahedi H, Ferns GA. The epidermal growth factor receptors and their family of ligands: their putative role in atherogenesis. Atherosclerosis. 2006;186:38–53. doi: 10.1016/j.atherosclerosis.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 65.Oksala N, Levula M, Airla N, Pelto-Huikko M, Ortiz RM, et al. ADAM-9, ADAM-15, and ADAM-17 are upregulated in macrophages in advanced human atherosclerotic plaques in aorta and carotid and femoral arteries–Tampere vascular study. Ann Med. 2009;41:279–90. doi: 10.1080/07853890802649738. [DOI] [PubMed] [Google Scholar]
- 66.Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, et al. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics. 2011;43:993–1003. doi: 10.1152/physiolgenomics.00044.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Satoh H, Nakamura M, Satoh M, Nakajima T, Izumoto H, et al. Expression and localization of tumour necrosis factor-alpha and its converting enzyme in human abdominal aortic aneurysm. Clinical science. 2004;106:301–6. doi: 10.1042/CS20030189. [DOI] [PubMed] [Google Scholar]
- 68.Kaneko H, Anzai T, Horiuchi K, Kohno T, Nagai T, et al. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218:470–8. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 69.Obama T, Tsuji T, Kobayashi T, Fukuda Y, Takayanagi T, et al. Epidermal Growth Factor Receptor Inhibitor Protects Abdominal Aortic Aneurysm in a Mouse Model. Clinical science. 2014 doi: 10.1042/CS20140696. [DOI] [PubMed] [Google Scholar]
- 70.Montiel M, de la Blanca EP, Jimenez E. Angiotensin II induces focal adhesion kinase/paxillin phosphorylation and cell migration in human umbilical vein endothelial cells. Biochemical and biophysical research communications. 2005;327:971–8. doi: 10.1016/j.bbrc.2004.12.110. [DOI] [PubMed] [Google Scholar]
- 71.Maretzky T, Evers A, Zhou W, Swendeman SL, Wong PM, et al. Migration of growth factor- stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. 2011;2:229. doi: 10.1038/ncomms1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burger D, Montezano AC, Nishigaki N, He Y, Carter A, Touyz RM. Endothelial microparticle formation by angiotensin II is mediated via Ang II receptor type I/NADPH oxidase/Rho kinase pathways targeted to lipid rafts. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1898–907. doi: 10.1161/ATVBAHA.110.222703. [DOI] [PubMed] [Google Scholar]
- 73.Al-Ani B, Hewett PW, Cudmore MJ, Fujisawa T, Saifeddine M, et al. Activation of proteinase-activated receptor 2 stimulates soluble vascular endothelial growth factor receptor 1 release via epidermal growth factor receptor transactivation in endothelial cells. Hypertension. 2010;55:689–97. doi: 10.1161/HYPERTENSIONAHA.109.136333. [DOI] [PubMed] [Google Scholar]
- 74.Chen K, Thomas SR, Albano A, Murphy MP, Keaney JF., Jr Mitochondrial function is required for hydrogen peroxide-induced growth factor receptor transactivation and downstream signaling. J Biol Chem. 2004;279:35079–86. doi: 10.1074/jbc.M404859200. [DOI] [PubMed] [Google Scholar]
- 75.Cheng J, Garcia V, Ding Y, Wu CC, Thakar K, et al. Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20- hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1917–24. doi: 10.1161/ATVBAHA.112.248344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vacaresse N, Lajoie-Mazenc I, Auge N, Suc I, Frisach MF, et al. Activation of epithelial growth factor receptor pathway by unsaturated fatty acids. Circ Res. 1999;85:892–9. doi: 10.1161/01.res.85.10.892. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892–7. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oliveira CJ, Schindler F, Ventura AM, Morais MS, Arai RJ, et al. Nitric oxide and cGMP activate the Ras-MAP kinase pathway-stimulating protein tyrosine phosphorylation in rabbit aortic endothelial cells. Free Radic Biol Med. 2003;35:381–96. doi: 10.1016/s0891-5849(03)00311-3. [DOI] [PubMed] [Google Scholar]
- 79.Yamamoto K, Kakino A, Takeshita H, Hayashi N, Li L, et al. Oxidized LDL (oxLDL) activates the angiotensin II type 1 receptor by binding to the lectin-like oxLDL receptor. FASEB J. 2015 doi: 10.1096/fj.15-271627. [DOI] [PubMed] [Google Scholar]
- 80.Batenburg WW, Jansen PM, van den Bogaerdt AJ, AH JD. Angiotensin II-aldosterone interaction in human coronary microarteries involves GPR30, EGFR, and endothelial NO synthase. Cardiovascular research. 2012;94:136–43. doi: 10.1093/cvr/cvs016. [DOI] [PubMed] [Google Scholar]
- 81.Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 82.Helle F, Jouzel C, Chadjichristos C, Placier S, Flamant M, et al. Improvement of renal hemodynamics during hypertension-induced chronic renal disease: role of EGF receptor antagonism. Am J Physiol Renal Physiol. 2009;297:F191–9. doi: 10.1152/ajprenal.00015.2009. [DOI] [PubMed] [Google Scholar]
- 83.Griol-Charhbili V, Fassot C, Messaoudi S, Perret C, Agrapart V, Jaisser F. Epidermal growth factor receptor mediates the vascular dysfunction but not the remodeling induced by aldosterone/salt. Hypertension. 2011;57:238–44. doi: 10.1161/HYPERTENSIONAHA.110.153619. [DOI] [PubMed] [Google Scholar]
- 84.Galan M, Kassan M, Choi SK, Partyka M, Trebak M, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012;60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Belmadani S, Palen DI, Gonzalez-Villalobos RA, Boulares HA, Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–37. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hewing NJ, Weskamp G, Vermaat J, Farage E, Glomski K, et al. Intravitreal injection of TIMP3 or the EGFR inhibitor erlotinib offers protection from oxygen-induced retinopathy in mice. Investigative ophthalmology & visual science. 2013;54:864–70. doi: 10.1167/iovs.12-10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weskamp G, Mendelson K, Swendeman S, Le Gall S, Ma Y, et al. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ Res. 2010;106:932–40. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wilson CL, Gough PJ, Chang CA, Chan CK, Frey JM, et al. Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mechanisms of development. 2013;130:272–89. doi: 10.1016/j.mod.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith NJ, Chan HW, Qian H, Bourne AM, Hannan KM, et al. Determination of the exact molecular requirements for type 1 angiotensin receptor epidermal growth factor receptor transactivation and cardiomyocyte hypertrophy. Hypertension. 2011;57:973–80. doi: 10.1161/HYPERTENSIONAHA.110.166710. [DOI] [PubMed] [Google Scholar]
- 90.Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nature medicine. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- 91.De Giusti VC, Nolly MB, Yeves AM, Caldiz CI, Villa-Abrille MC, et al. Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. Hypertension. 2011;58:912–9. doi: 10.1161/HYPERTENSIONAHA.111.176024. [DOI] [PubMed] [Google Scholar]
- 92.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, et al. β-Arrestin– mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. The Journal of Clinical Investigation. 2007;117:2445–58. doi: 10.1172/JCI31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008;105:14555–60. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3:ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang D, Yu X, Cohen RA, Brecher P. Distinct effects of N-acetylcysteine and nitric oxide on angiotensin II-induced epidermal growth factor receptor phosphorylation and intracellular Ca(2+) levels. J Biol Chem. 2000;275:12223–30. doi: 10.1074/jbc.275.16.12223. [DOI] [PubMed] [Google Scholar]
- 96.Sabri A, Short J, Guo J, Steinberg SF. Protease-activated receptor-1-mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: distinct PAR-1 signaling pathways in cardiac fibroblasts and cardiomyocytes. Circ Res. 2002;91:532–9. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 97.Seta K, Sadoshima J. Phosphorylation of tyrosine 319 of the angiotensin II type 1 receptor mediates angiotensin II-induced trans-activation of the epidermal growth factor receptor. J Biol Chem. 2003;278:9019–26. doi: 10.1074/jbc.M208017200. [DOI] [PubMed] [Google Scholar]
- 98.Cai J, Yi FF, Yang L, Shen DF, Yang Q, et al. Targeted expression of receptor-associated late transducer inhibits maladaptive hypertrophy via blocking epidermal growth factor receptor signaling. Hypertension. 2009;53:539–48. doi: 10.1161/HYPERTENSIONAHA.108.120816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhai P, Galeotti J, Liu J, Holle E, Yu X, et al. An angiotensin II type 1 receptor mutant lacking epidermal growth factor receptor transactivation does not induce angiotensin II-mediated cardiac hypertrophy. Circ Res. 2006;99:528–36. doi: 10.1161/01.RES.0000240147.49390.61. [DOI] [PubMed] [Google Scholar]
- 100.Messaoudi S, Zhang AD, Griol-Charhbili V, Escoubet B, Sadoshima J, et al. The epidermal growth factor receptor is involved in angiotensin II but not aldosterone/salt-induced cardiac remodelling. PloS one. 2012;7:e30156. doi: 10.1371/journal.pone.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nature medicine. 2012;18:1429–33. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li C, Zhang YY, Frieler RA, Zheng XJ, Zhang WC, et al. Myeloid mineralocorticoid receptor deficiency inhibits aortic constriction-induced cardiac hypertrophy in mice. PloS one. 2014;9:e110950. doi: 10.1371/journal.pone.0110950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schreier B, Rabe S, Winter S, Ruhs S, Mildenberger S, et al. Moderate inappropriately high aldosterone/NaCl constellation in mice: cardiovascular effects and the role of cardiovascular epidermal growth factor receptor. Sci Rep. 2014;4:7430. doi: 10.1038/srep07430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Eckel RH, Kahn R, Robertson RM, Rizza RA. Preventing cardiovascular disease and diabetes: a call to action from the American Diabetes Association and the American Heart Association. Circulation. 2006;113:2943–6. doi: 10.1161/CIRCULATIONAHA.106.176583. [DOI] [PubMed] [Google Scholar]
- 105.Prada PO, Ropelle ER, Mourao RH, de Souza CT, Pauli JR, et al. EGFR tyrosine kinase inhibitor (PD153035) improves glucose tolerance and insulin action in high-fat diet-fed mice. Diabetes. 2009;58:2910–9. doi: 10.2337/db08-0506. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 106.Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, et al. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nature genetics. 2000;24:296–9. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- 107.Barrick CJ, Yu M, Chao HH, Threadgill DW. Chronic pharmacologic inhibition of EGFR leads to cardiac dysfunction in C57BL/6J mice. Toxicology and applied pharmacology. 2008;228:315–25. doi: 10.1016/j.taap.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, et al. Reduced EGFR causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in C57BL/6J but not 129S1/SvImJ mice. American journal of physiology. Heart and circulatory physiology. 2009;297:H65–75. doi: 10.1152/ajpheart.00866.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grisanti LA, Repas AA, Talarico JA, Gold JI, Carter RL, et al. Temporal and gefitinib-sensitive regulation of cardiac cytokine expression via chronic beta-adrenergic receptor stimulation. American journal of physiology. Heart and circulatory physiology. 2015;308:H316–30. doi: 10.1152/ajpheart.00635.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Benter IF, Juggi JS, Khan I, Yousif MH, Canatan H, Akhtar S. Signal transduction mechanisms involved in cardiac preconditioning: role of Ras-GTPase, Ca2+/calmodulin- dependent protein kinase II and epidermal growth factor receptor. Mol Cell Biochem. 2005;268:175–83. doi: 10.1007/s11010-005-3895-1. [DOI] [PubMed] [Google Scholar]
- 111.Forster K, Kuno A, Solenkova N, Felix SB, Krieg T. The delta-opioid receptor agonist DADLE at reperfusion protects the heart through activation of pro-survival kinases via EGF receptor transactivation. American journal of physiology. Heart and circulatory physiology. 2007;293:H1604–8. doi: 10.1152/ajpheart.00418.2007. [DOI] [PubMed] [Google Scholar]
- 112.Akhtar S, Yousif MH, Chandrasekhar B, Benter IF. Activation of EGFR/ERBB2 via pathways involving ERK1/2, P38 MAPK, AKT and FOXO enhances recovery of diabetic hearts from ischemia-reperfusion injury. PloS one. 2012;7:e39066. doi: 10.1371/journal.pone.0039066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hammoud L, Lu X, Lei M, Feng Q. Deficiency in TIMP-3 increases cardiac rupture and mortality post-myocardial infarction via EGFR signaling: beneficial effects of cetuximab. Basic research in cardiology. 2011;106:459–71. doi: 10.1007/s00395-010-0147-7. [DOI] [PubMed] [Google Scholar]
- 114.Liu N, He S, Ma L, Ponnusamy M, Tang J, et al. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PloS one. 2013;8:e54001. doi: 10.1371/journal.pone.0054001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Uchiyama-Tanaka Y, Matsubara H, Nozawa Y, Murasawa S, Mori Y, et al. Angiotensin II signaling and HB-EGF shedding via metalloproteinase in glomerular mesangial cells. Kidney Int. 2001;60:2153–63. doi: 10.1046/j.1523-1755.2001.00067.x. [DOI] [PubMed] [Google Scholar]
- 116.Uchiyama-Tanaka Y, Matsubara H, Mori Y, Kosaki A, Kishimoto N, et al. Involvement of HB- EGF and EGF receptor transactivation in TGF-beta-mediated fibronectin expression in mesangial cells. Kidney Int. 2002;62:799–808. doi: 10.1046/j.1523-1755.2002.00537.x. [DOI] [PubMed] [Google Scholar]
- 117.Uttarwar L, Peng F, Wu D, Kumar S, Gao B, et al. HB-EGF release mediates glucose-induced activation of the epidermal growth factor receptor in mesangial cells. Am J Physiol Renal Physiol. 2011;300:F921–31. doi: 10.1152/ajprenal.00436.2010. [DOI] [PubMed] [Google Scholar]
- 118.Taniguchi K, Xia L, Goldberg HJ, Lee KW, Shah A, et al. Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes. 2013;62:3874–86. doi: 10.2337/db12-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang S, Zhang A, Ding G, Chen R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol. 2009;296:F1323–33. doi: 10.1152/ajprenal.90428.2008. [DOI] [PubMed] [Google Scholar]
- 120.Chen J, Chen JK, Neilson EG, Harris RC. Role of EGF receptor activation in angiotensin II- induced renal epithelial cell hypertrophy. J Am Soc Nephrol. 2006;17:1615–23. doi: 10.1681/ASN.2005111163. [DOI] [PubMed] [Google Scholar]
- 121.Chen J, Chen JK, Harris RC. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Molecular and cellular biology. 2012;32:981–91. doi: 10.1128/MCB.06410-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhuang S, Yan Y, Han J, Schnellmann RG. p38 kinase-mediated transactivation of the epidermal growth factor receptor is required for dedifferentiation of renal epithelial cells after oxidant injury. J Biol Chem. 2005;280:21036–42. doi: 10.1074/jbc.M413300200. [DOI] [PubMed] [Google Scholar]
- 123.Rayego-Mateos S, Morgado-Pascual JL, Sanz AB, Ramos AM, Eguchi S, et al. TWEAK transactivation of the epidermal growth factor receptor mediates renal inflammation. J Pathol. 2013;231:480–94. doi: 10.1002/path.4250. [DOI] [PubMed] [Google Scholar]
- 124.Lee YJ, Han HJ. Albumin-stimulated DNA synthesis is mediated by Ca2+/PKC as well as EGF receptor-dependent p44/42 MAPK and NF-kappaB signal pathways in renal proximal tubule cells. Am J Physiol Renal Physiol. 2008;294:F534–41. doi: 10.1152/ajprenal.00408.2007. [DOI] [PubMed] [Google Scholar]
- 125.Reich H, Tritchler D, Herzenberg AM, Kassiri Z, Zhou X, et al. Albumin activates ERK via EGF receptor in human renal epithelial cells. J Am Soc Nephrol. 2005;16:1266–78. doi: 10.1681/ASN.2004030222. [DOI] [PubMed] [Google Scholar]
- 126.Singh AB, Sugimoto K, Harris RC. Juxtacrine activation of epidermal growth factor (EGF) receptor by membrane-anchored heparin-binding EGF-like growth factor protects epithelial cells from anoikis while maintaining an epithelial phenotype. J Biol Chem. 2007;282:32890–901. doi: 10.1074/jbc.M702677200. [DOI] [PubMed] [Google Scholar]
- 127.Arar M, Zajicek HK, Elshihabi I, Levi M. Epidermal growth factor inhibits Na-Pi cotransport in weaned and suckling rats. Am J Physiol. 1999;276:F72–8. doi: 10.1152/ajprenal.1999.276.1.F72. [DOI] [PubMed] [Google Scholar]
- 128.Groenestege WM, Thebault S, van der Wijst J, van den Berg D, Janssen R, et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest. 2007;117:2260–7. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grossmann C, Freudinger R, Mildenberger S, Krug AW, Gekle M. Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. Am J Physiol Renal Physiol. 2004;286:F1226–31. doi: 10.1152/ajprenal.00378.2003. [DOI] [PubMed] [Google Scholar]
- 130.McEneaney V, Harvey BJ, Thomas W. Aldosterone rapidly activates protein kinase D via a mineralocorticoid receptor/EGFR trans-activation pathway in the M1 kidney CCD cell line. The Journal of steroid biochemistry and molecular biology. 2007;107:180–90. doi: 10.1016/j.jsbmb.2007.03.043. [DOI] [PubMed] [Google Scholar]
- 131.Dey M, Baldys A, Sumter DB, Gooz P, Luttrell LM, et al. Bradykinin decreases podocyte permeability through ADAM17-dependent epidermal growth factor receptor activation and zonula occludens-1 rearrangement. The Journal of pharmacology and experimental therapeutics. 2010;334:775–83. doi: 10.1124/jpet.110.168054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang Z. Importance of Functional EGF Receptors in Recovery from Acute Nephrotoxic Injury. Journal of the American Society of Nephrology. 2003;14:3147–54. doi: 10.1097/01.asn.0000098681.56240.1a. [DOI] [PubMed] [Google Scholar]
- 133.Chen J, Chen JK, Harris RC. Deletion of the epidermal growth factor receptor in renal proximal tubule epithelial cells delays recovery from acute kidney injury. Kidney Int. 2012;82:45–52. doi: 10.1038/ki.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tang J, Liu N, Tolbert E, Ponnusamy M, Ma L, et al. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am J Pathol. 2013;183:160–72. doi: 10.1016/j.ajpath.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bollee G, Flamant M, Schordan S, Fligny C, Rumpel E, et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nature medicine. 2011;17:1242–50. doi: 10.1038/nm.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Francois H, Placier S, Flamant M, Tharaux PL, Chansel D, et al. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J. 2004;18:926–8. doi: 10.1096/fj.03-0702fje. [DOI] [PubMed] [Google Scholar]
- 137.Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, et al. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nature medicine. 2005;11:867–74. doi: 10.1038/nm1275. [DOI] [PubMed] [Google Scholar]
- 138.Che Q, Carmines PK. Angiotensin II triggers EGFR tyrosine kinase-dependent Ca2+ influx in afferent arterioles. Hypertension. 2002;40:700–6. doi: 10.1161/01.hyp.0000035524.10948.93. [DOI] [PubMed] [Google Scholar]
- 139.Wassef L, Kelly DJ, Gilbert RE. Epidermal growth factor receptor inhibition attenuates early kidney enlargement in experimental diabetes. Kidney Int. 2004;66:1805–14. doi: 10.1111/j.1523-1755.2004.00955.x. [DOI] [PubMed] [Google Scholar]
- 140.Zhang MZ, Wang Y, Paueksakon P, Harris RC. Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63:2063–72. doi: 10.2337/db13-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen J, Chen JK, Harris RC. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J Am Soc Nephrol. 2015;26:1115–25. doi: 10.1681/ASN.2014020192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Terzi F, Burtin M, Hekmati M, Federici P, Grimber G, et al. Targeted expression of a dominant-negative EGF-R in the kidney reduces tubulo-interstitial lesions after renal injury. J Clin Invest. 2000;106:225–34. doi: 10.1172/JCI8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu N, Guo JK, Pang M, Tolbert E, Ponnusamy M, et al. Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J Am Soc Nephrol. 2012;23:854–67. doi: 10.1681/ASN.2011050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- 145.Chen MH, Kerkela R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev Drug Discov. 2010;9:956–70. doi: 10.1038/nrd3297. [DOI] [PubMed] [Google Scholar]
- 147.Tejpar S, Piessevaux H, Claes K, Piront P, Hoenderop JG, et al. Magnesium wasting associated with epidermal-growth-factor receptor-targeting antibodies in colorectal cancer: a prospective study. The Lancet. Oncology. 2007;8:387–94. doi: 10.1016/S1470-2045(07)70108-0. [DOI] [PubMed] [Google Scholar]
- 148.Izzedine H, Rixe O, Billemont B, Baumelou A, Deray G. Angiogenesis inhibitor therapies: focus on kidney toxicity and hypertension. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2007;50:203–18. doi: 10.1053/j.ajkd.2007.04.025. [DOI] [PubMed] [Google Scholar]