

Abstract
Protein folding is well known to be supervised by a dedicated class of proteins called chaperones. However, the core mode of action of these molecular machines has remained elusive due to several reasons including the promiscuous nature of the interactions between chaperones and their many clients, as well as the dynamics and heterogeneity of chaperone conformations and the folding process itself. While troublesome for traditional bulk techniques, these properties make an excellent case for the use of single‐molecule approaches. In this review, we will discuss how force spectroscopy, fluorescence microscopy, FCS, and FRET methods are starting to zoom in on this intriguing and diverse molecular toolbox that is of direct importance for protein quality control in cells, as well as numerous degenerative conditions that depend on it.
Keywords: chaperones, molecular machines, single‐molecule, trigger factor, Hsp90, Hsp70, GroEL
Introduction
To perform their role within cells, proteins typically interact with a limited set of the proteome. Histidine kinases recognize specific response regulators in order to transmit detected signals, while kinesins move along microtubules to deliver neurotransmitters in axons. Molecular chaperones are a notable exception to this general rule. This class of proteins is involved in assisting a wide range of proteins throughout their life cycle. As soon as a newly synthesized polypeptide emerges from the ribosomal tunnel, chaperones bind and protect it against aggregation with other polypeptides and proteins, promote proper folding into a functional structure, and pass it on to other chaperone systems.1, 2 However, the function of chaperones is not limited to de‐novo folding. They act to disrupt already formed aggregates,3 help the formation of multiprotein complexes,4 regulate the activity of large numbers of receptors and kinases,5 and are involved in a range of other tasks. As such, chaperones are implicated in many normal cellular processes such as the cell cycle and apoptosis, but also in numerous medical conditions ranging from cancer to neurodegeneration diseases.6, 7
Most chaperones are constitutively present but overexpressed at high temperatures, as well as under oxidative stress,8 deviating pH, and various other conditions.9 Many chaperones owe their name to this effect, and since their discovery in 1974 are hence referred to as heat shock proteins followed by their molecular weight (e.g., Hsp70).10 Some chaperones undergo important structural changes triggered by the ATP hydrolysis cycle. For instance, Hsp60, known in bacteria as GroEL, is a barrel‐like structure that can accommodate (mis)folded proteins and is closed‐off by the GroES cap. Also, Hsp70 is known to bind exposed polypeptides in a groove that can be covered by a helical lid. Chaperones have therefore been referred to as “folding machines,” though this term does not do justice to their far wider range of cellular functions.
Despite the huge amount of knowledge acquired in the last decades, it is striking that many of the most basic questions remain unresolved to this day. For instance, it is still debated whether chaperones can directly guide and promote folding beyond suppressing aggregation. Merely detecting whether chaperones interact with partially folded chains along their folding pathway is already a challenge. When they do appear to promote folding, the physical principle is obscure, and may range from affecting chain entropy to recognizing key transition states of the folding protein.11 The list of open questions is endless: interaction sites on chaperone and client protein are often unknown, as is the interplay between ATP hydrolysis, chaperone and client conformational changes. The reason for these gaps in our knowledge is clear: conformational changes and folding transitions are hard to measure in ensemble measurements. Indeed, in bulk refolding assays it is even a challenge to distinguish (reversible) aggregation from intrinsically delayed folding. Other complicating factors are the transient nature of chaperone‐client complexes, the conformational dynamics of the chaperone, and the involvement of numerous cofactors.
These technical challenges can in principle be addressed by zooming into single client‐chaperone complexes. The past decade has witnessed a rapid development of novel single‐molecule approaches that are now beginning to address these crucial questions. Diverse techniques have been employed, ranging from single‐molecule FRET to optical tweezers and atomic force microscopy. Here we discuss a number of example studies that reflect these efforts—without aiming to systematically cover this field—and mention complementary bulk approaches where appropriate. We have organized the studied chaperone systems by their complexity, starting with the ATP independent chaperones trigger factor and SecB, and then moving to the ATP dependent chaperone classes Hsp70, Hsp90, and GroEL. This exciting first look at the action of chaperones at the single‐molecule level is revealing a range of unexpected mechanisms, and first answers to big open questions.
Trigger Factor, a Cradle for Nascent Chains
The chaperone trigger factor (TF) is the first protein that most newly synthesized proteins interact with in bacteria.12 This dragon‐shaped13 protein [Fig. 1(A)] associates with the ribosome with its tail bound close to the ribosome exit site, and the body and arms forming a cradle that receives the nascent chain when it emerges from the exit tunnel. TF can leave the ribosome while bound to the nascent chain17 and suppress their aggregation.18 With the latter function, TF exhibits functional overlap with the chaperones DnaK, GroEL,19, 20 and SecB,12, 21 which do not directly bind the ribosome. A number of key questions have remained difficult to address with approaches used so far. Specifically, it is difficult to obtain structural information on the client‐chaperone complex owing to the conformational dynamics of the unfolded polypeptide clients. We also lack information on how TF affects these conformational dynamics, and the process of folding into active proteins with tertiary structure. These questions, which go to the heart of chaperone functions, are now beginning to be addressed by single‐molecule methods, as well as by computational approaches and NMR spectroscopy. Here we discuss a few of these recent studies, and contrast them to findings on another independent chaperone, SecB.
Figure 1.
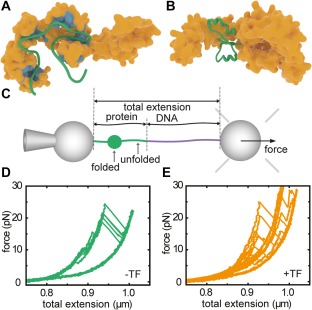
Interactions between trigger factor and client proteins. (A) Interaction sites on TF for MBP as derived from NMR experiments.14 (B) Interaction of TF with a partial fold of MBP, as determined by MD simulations,15 and observed by optical tweezers experiments (panels c–e). (C) Single‐molecule optical tweezers experimental setup with MBP tethered between two polystyrene beads. One bead is held on a pipette, while the other is held by an optical trap that is also used to determine the applied force. Pulling experiments on MBP in isolation (D) and MBP with TF present (E) show an increased presence of partially folded states for the latter, during pulling and also during refolding at low force in between pulling cycles. Panel A is redrawn from Saio et al.,14 panel B from Singhal et al.,15 panels C–E from Mashaghi et al.16
Structural data of TF‐substrate complexes has been lacking until recently, due to the transient nature of the underlying interactions and conformations. Pushing the envelope of the size of protein systems addressable by NMR spectroscopy, Saio et al.14 studied the interaction between TF and alkaline phosphatase PhoA. PhoA is a periplasmic protein that remains in an unfolded state under reducing conditions. The data indicated four substrate binding‐sites on TF for unfolded PhoA: three on the body and arms and one on the head, all highly enriched in nonpolar residues. They found the same binding sites, and an additional one on one of the TF arms, for an unfolded fragment of maltose binding protein [MBP, see Fig. 1(A)] and the trans membrane region of OmpA, suggestion some commonality in where substrates bind to TF. The authors also probed the interaction sites on the substrate PhoA, and found that they are—in addition to nonpolar residues—rich in aromatic residues. In contrast, hydrophobic stretches lacking aromatic residues seemed to have low affinity for TF. The binding sites were promiscuous: each site could bind some or even all of the TF sites with relatively low affinities. An encounter between unfolded PhoA and TF should thus result in a dynamic search for the combination of TF‐PhoA binding sites that have the lowest energy.
The dynamic nature of the TF‐bound protein chain presses the question whether it can form tertiary structure. This issue requires the ability to trigger folding, and the ability to follow it in time, which can be achieved with optical tweezers. Using this single molecule technique, Mashaghi et al. tethered MBP to beads using DNA linkers [Fig. 1(C)], and unfolded and refolded them in repeated cycles of pulling and relaxation,16 showing a reproducible folding behavior of the protein [Fig. 1(D)]. These force‐extension curves changed dramatically upon addition of TF [Fig. 1(E)]. First, unfolded proteins remained unfolded for longer, consistent with TF‐chain binding, but then did form tertiary structure. More surprisingly, these partially folded states were stable for seconds and against applied force, and folding now proceeded via these intermediate states that were promoted by TF. Thus, this approach provided direct evidence for how folding guidance by chaperones is sometimes imagined: to continue interacting with a protein chain during the process of folding into tertiary structure. Local conformational plasticity of TF is relevant to this behavior; the flexibility of TF's two arms facilitates the binding of folded substrates of a variety of sizes.22 Moreover, TF was found to not only promote refolding of MBP monomers within a 4xMBP repeat‐construct, but also to suppress misfolding interactions between them. These data suggested a generic mechanism to separate the good from the bad: multiple TF molecules bound to the different domains within multi‐domain proteins that suppress erroneous interactions between domains while allowing native interactions within domains.
To gain further structural insight into substrate‐TF complexes in these different stages of folding, MD simulations have been employed.15 Simulations on unfolded MBP conformations revealed some of the same sites as detected by NMR. Intermediate folded states initially formed a ‘touching complex’ with the flexible tips of two TF domains. Interestingly, subsequent transfer to the TF cradle and embrace by its flexible arms provided a structural explanation of the experimentally observed stabilization of folded structures [Fig. 1(B)]. Substrate‐TF interactions became weaker for more fully folded states, which makes sense functionally. A next step would be to assess the chaperoning action of TF on the ribosome. Biochemical assays on stalled ribosomes suggest that TF can then also delay folding of large multidomain proteins23 and even partially unfold some substrates.24 Course grained simulations suggested that this folding delay is caused by kinetic trapping of unfolded ensembles, while smaller proteins could fold in between TF and the ribosome without delay. In this manner, the chaperone effectively lengthens the tunnel of the ribosome with its space‐limited cradle.25 Single‐molecule experiments on stalled ribosomes have shown that formation of tertiary structure in nascent chains is suppressed due to confinement by proximity of the ribosome.26
The chaperone SecB presents interesting similarities and differences with TF. SecB also interacts and stabilizes unfolded chains,21 which here facilitates their transport across the membrane by the SecA translocation machinery. A recent NMR study27 revealed how an unfolded PhoA chain wraps around the chaperone SecB. Long, hydrophobic grooves on the chaperone tetramer facilitate binding of the substrate. The parts of PhoA in contact with SecB are fully unfolded with no secondary structure present, and the interaction surface—as deduced from the modeled structure of the complex—turns out to be much larger than that of PhoA with TF (250 vs. 25 interacting residues). This larger interaction surface might explain the stronger antifolding properties of SecB compared to TF. The latter was also consistent with single‐molecule force spectroscopy, which indicated that SecB keeps MBP substrates in an unfolded state by preventing the formation of stable tertiary interactions.28
A picture emerges of TF as a more versatile chaperone than commonly assumed. It forms the first line of defense against aggregation of the nascent chain, and protects freshly synthesized hydrophobic stretches from aggregation. TF also binds and transiently stabilizes partially folded structures, which protects them from long‐range interactions at the cost of reduced folding rates. By holding unfolded as well as partially folded states in a transient and ATP‐independent manner, TF can deepen corresponding energy valleys and guide folding trajectories along them. These initial insights into the structure and dynamics of TF‐substrate complexes raise a host of novel questions. For instance, it remains unclear how the conformational dynamics of the substrate chain is affected when TF is bound, whether TF remains fully bound during folding transitions, or rather leaves transiently, or how TF can partially compensate for deletions of ATP‐dependent chaperones DnaK and GroEL.19, 20 Another open question is how the role of TF differs at the ribosome.24 A recent study suggests additional substrate binding sites on the tail of TF (ribosome binding domain) that only becomes available upon ribosome binding.29 Single‐molecule studies of ribosome‐client‐chaperone complexes are within reach,26 and could shed light on these important questions on cotranslational chaperone action. A suggested hydrophilic binding mode of TF15, 18 may also stimulate further structural and single‐molecule investigation. Finally, it is of interest to determine how TF differs mechanistically from eukaryotic chaperone systems that fulfill similar functions (see30 for an overview). These insights may also find practical use, for instance in helping to reduce the misfolding rate of bacterially produced eukaryotic proteins.23, 31
Hsp70, a Clamp for Unfolded and Folded Protein Structures
The 70 kDa heat shock proteins (Hsp70s) are one of the most ubiquitous families of chaperones, and are highly conserved across species. They are involved in a remarkably diverse range of cellular processes, well beyond assisting in de novo protein folding. Other roles are for instance the disaggregation of already formed aggregates,3 assistance in protein trafficking across membranes, and regulating the activity of kinases and receptors.5 Hsp70s are thought to interact with unfolded peptide chain segments extending from substrate proteins, which may be in (partially) unfolded of misfolded conformations. In addition, auxiliary cochaperones interact with Hsp70s and regulate their activity.32
Hsp70s consist of two distinct domains, a C‐terminal substrate binding domain (SBD, 27 kDa) and an N‐terminal nucleotide‐binding domain (NBD, 43 kDa), connected through a highly conserved linker. An important feature of the SBD is its two subdomains, a twisted β‐sandwich (SBDβ) and an α‐helical (SBDα) subdomain ending in an unstructured stretch of about 30 residues, widely referred to as the chaperone lid.33, 34 High‐resolution crystal structure studies revealed two conformations of the chaperone that have been very instructive in understanding peptide binding, as discussed further below [Fig. 2(A)]. Hsp70 acts as a clamp: in the closed conformation, observed in the nucleotide‐free and ADP‐bound states, the lid (SBDα) is positioned closely against the peptide binding cleft on SBDβ and both subdomains are spatially separated from the NBD.33 In the open conformation, the lid is detached from SBDβ, and both subdomains dock to different parts of the NBD.36
Figure 2.
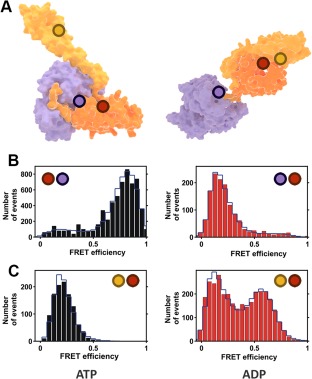
Single‐molecule FRET experiments with Hsp70 (A) Crystal structures of Hsp70 open (left) and closed (right) conformations. Purple corresponds to the NBD subdomain and orange and yellow to the SBDβ and SBDα subdomains, respectively. The circles denote the approximate location of the donor and acceptor labels described in Ref. 35. (B) FRET histograms for the inter‐domain dynamics under ATP (left panel, docked domains) and ADP (right panel, undocked domains) conditions. (C) FRET histograms for the lid dynamics under ATP (left panel, open lid) and ADP (right panel, heterogeneous state) conditions. (B) and (C) are adapted with permission from Ref. 35.
The ATP cycle is important for its peptide‐binding function: in the open ATP‐bound state, association and dissociation rates are high, resulting in low substrate affinity. In the closed ADP‐bound state, both rates are several orders of magnitude lower, leading to a higher affinity for polypeptides.37 The chaperone‐polypeptide interaction and the nucleotide state of the chaperone are strongly coupled and affect each other reciprocally. For instance, peptide binding catalyzes ATP hydrolysis, which is otherwise a rather slow process. ATP conversion to ADP, in return, stabilizes peptide binding.38 Certain cochaperones, such as DnaJ in E. coli or Mdj1 in mitochondria, play important roles in these interactions.39
Crystal structures provide a detailed yet static picture, without information on dynamics. For instance, they do not reveal whether the two Hsp70 conformations correspond strictly to a particular nucleotide state, or whether other intermediate conformations exist. This problem was addressed by Mapa et al., choosing the chaperone Ssc1, a mitochondrial member of the Hsp70 family, using ensemble and single‐pair Förster Resonance Energy Transfer (FRET) as experimental techniques.35 The authors engineered two FRET‐constructs using strategically selected cysteine residues for labeling [Fig. 2(A)]. For the first construct, two dyes were introduced in the NBD and the SBD in order to study the interaction between the two domains. The second contained cysteine residues in the lid and the base of the SBD, to study the dynamics of these subdomains. For the single‐pair FRET experiments, a very low concentration of Ssc1 (20–40 pM) was used, ensuring that only individual proteins were probed with a confocal microscope and pulsed interleaved excitation. Both donor and acceptor emissions of at least 500 different molecules were independently recorded and combined in a FRET‐efficiency histogram (Fig. 2). The results demonstrate that the conformation of Ssc1 in the ATP‐bound state is well defined, with the lid detached from SBDβ and the NBD and SBD docked [Fig. 2(B,C), left panels], in agreement with structural data. In contrast, the ADP state of Ssc1 is much more heterogeneous, both in the SBD–NBD interaction and in the lid conformation. Similar behavior was observed by the same authors for the bacterial Hsp70 (DnaK), with the NBD–SBD domains largely separated in the ADP state [Fig. 2(B), right panel] as the only difference with its mitochondrial counterpart, showing similar heterogeneous lid dynamics [Fig. 2(C), right panel).35
As mentioned, substrate‐Hsp70 interaction and nucleotide cycle are tightly coupled and strongly dependent on cochaperones. The details of this link were also explored.35 The addition of a peptide substrate greatly accelerated ATP hydrolysis and resulting domain undocking and lid‐closure; with evidence of these two processes happening virtually simultaneously. In presence of the Hsp40 Mdj1, a cochaperone of Ssc1, both events were further accelerated. The lid was found to adopt a stable closed conformation over the binding cleft in presence of both the substrate and Mdj1. Interestingly, when the substrate is absent, Mdj1 is able to trigger domain undocking and lid closure, but after a short period of time the conformation changes back to that of the heterogeneous ADP‐bound state. Another cofactor, nucleotide exchange factor (NEF), also plays a role in accelerating the ATP cycle by promoting the exchange of ADP by ATP in the NBD of Hsp70s.
These chaperone dynamics press an urgent question: how is the substrate affected? Single‐molecule FRET was employed by Kellner et al. to study the conformation of different rhodanese‐fluorophore constructs when they interact with the bacterial Hsp70 (DnaK) and its cochaperone DnaJ.40 Five different FRET pairs were engineered to monitor changes in different parts of the rhodanese polypeptide. A small probing volume was illuminated with pulsed excitation, and the emission of single molecules detected to obtain FRET histograms. Upon chemical denaturation, the FRET efficiency of all variants remained fairly high, suggesting a rather compact state. Importantly, refolding to the native state occurred spontaneously on a timescale of minutes without chaperones. Presence of DnaJ resulted in a broadening of the FRET distributions, indicating the blocking of refolding and the formation of heterogeneous nonfolded conformations. Addition of DnaK to preformed DnaJ‐rhodanese complexes led to a shift toward lower FRET values. This observation, supported by molecular dynamics simulations, suggested that several DnaK molecules bind to the peptide chain, resulting in its expansion by means of volume exclusion.40 Interestingly, DnaK was not enough to drive substrate expansion, as the presence of DnaJ was essential for this process.
One of the most enigmatic aspects of Hsp70s is the role of the lid. While it is generally assumed to serve to stabilize peptide binding only,32 recent single‐molecule studies have demonstrated a broader functional role. Mashaghi et al. recently employed optical tweezers41 to mechanically control the folding state of MBP substrates and studied the response upon addition of DnaK. Surprisingly, these data showed that not only exposed peptide segments were stabilized, but also near‐native folded protein structures: in presence of the bacterial Hsp70, the folded structures displayed high unfolding forces, or at times they could not be unfolded within the force limits of this method (up to 65 pN). Fully folded native structures were not stabilized—a minor unfolding transition that removes a number of external MBP alpha‐helices was required to trigger stabilization by Hsp70. The authors further showed that both the lid and ADP are key to the stabilizing function. This mode of binding and stabilizing folded structures extends the longstanding canonical model of Hsp70, in which only extended peptides are bound and released. Notably, it has essentially the opposite effect to the known binding mode, as it stabilizes folded rather than unfolded states. Co‐chaperones and nucleotide concentration may play an important role in regulating the different modes of Hsp70s binding, as stabilization occurs only in the ADP state and is thus transient.
This extended role of the lid is consistent with observations of its conformational dynamics. Marcinowski et al. used single‐pair FRET, in a similar fashion as Mapa et al.,35 to resolve the conformational changes of the mammalian Hsp70 BiP (heavy chain‐binding protein).42 The chaperone conformational dynamics during the nucleotide cycle was analogous to that of Ssc1 and DnaK, including lid closure upon peptide binding. When the peptide substrate was replaced by a larger unstructured client protein, however, the lid remained predominantly open, while cross‐linking experiments revealed physical interactions between lid and bound substrate. Interestingly, it was also shown that the addition of ERdJ3, a cochaperone of BiP, primes the conformation of the latter for protein substrate binding, while hindering peptide binding. Again, these results manifest the intricate interaction between chaperone, nucleotide, substrate and cochaperones. Similar evidence for the lid versatility of Hsp70s was recently shown by Banerjee et al., which used smFRET to study the dynamics of the lid from DnaK.43 Here, the lid remained mainly open in the presence of proteins in a molten globule state, in contrast to its closure upon peptide binding.
A number of intriguing questions arise from these findings, such as how Hsp70s can discriminate between partial folds that are native‐like or misfolded, and more generally whether this direct binding of folded structures allows Hsp70 to actively fold proteins. The findings also suggest that in episodes of stress, when ATP levels are low and ADP levels are high, Hsp70‐mediated stabilization may keep key proteins intact, as supported by increased unfolding temperatures of the protein RepE in the presence of Hsp70 and ADP.41 It will also be interesting to determine how this novel binding mode affects the cooperation between Hsp70 and its cochaperones, as well as other chaperones such as Hsp90.
Hsp90, a Versatile Regulatory Chaperone
The 90‐kDa heat shock proteins (Hsp90s) constitute an essential chaperone family in bacteria and eukaryotic organisms. Like Hsp70, they participate in a broad spectrum of cellular processes, including heat stress protection, signal transduction and protein trafficking.44 While the bacterial homologue HtpG is not essential in normal conditions, Hsp90s are critical in eukaryotic cells and make up to 1–2% of total soluble cell protein.45 Hsp90 also plays an important role in certain disorders, including cancer, and has in the recent years emerged as a potential target for tumor treatment strategies.7
Hsp90 forms a high affinity dimer [Fig. 3(A)], with each monomer consisting of three domains: a highly conserved N‐terminal nucleotide binding domain (NTD), a middle domain (MD), and a C‐terminal dimerization domain (CTD). An interesting feature of Hsp90 is its unique ATPase activity. A series of early studies reported contradictory observations, suggesting both the existence and absence of the ATPase activity of Hsp90.47, 48 This controversy was resolved by a crystallographic study that revealed a nucleotide binding site on the N‐terminal domain.49 Subsequent work revealed that Hsp90 dimer adopts at least two conformations in a clip‐like manner [Fig. 3(A)]: an ATP‐free open state in which only the C‐terminal domains interact, and an ATP‐bound closed state, in which the other domains also interact [Fig. 3(A)].50, 51 In other studies, three to four states have been suggested.52, 53 Biochemical assays have suggested that, in the particular case of bacterial HtpG, these structural changes are tightly coupled to the nucleotide cycle, contrary to eukaryotic Hsp90s, for which conformational dynamics are more subtle and yet not clearly related to the remarkably slow ATPase activity.54 However, such heterogeneous ensembles of molecular states makes are difficult to characterize properly using bulk assays.
Figure 3.
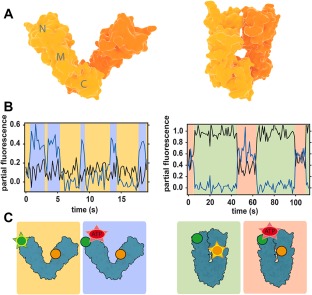
Conformational changes of Hsp90 studied with FRET. (A) Crystal structures of open (left) and closed (right) conformations of bacterial Hsp90 dimer (monomers are indicated by different color shades). (B) Partial fluorescence traces of two acceptors in 3‐colour FRET experiments: black line corresponds to the NTD acceptor, blue line to nucleotide acceptor. The traces are calculated by dividing acceptor intensity by the total fluorescence signal.46 Data shows that nucleotides can bind Hsp90 dimer in both open and closed conformations. (C) Scheme of the conformations and labeling of Hsp90. Green circle is donor, yellow is acceptor monitoring NTD dynamics and red is the nucleotide acceptor. Emission is represented by a star. Background colors link each conformation to the corresponding portion of the fluorescence traces in (B). Figures (B) and (C) are redrawn from Ref. 46.
This problem has been addressed in several single‐molecule studies, the first of which used FRET to investigate the dynamics of the N‐terminus dimerization.55 Here, the authors created two different single‐cysteine mutants of the yeast Hsp90, labeling each of them with an acceptor and a donor fluorophore, respectively. The formation of heterodimers produced an increase of the acceptor signal, further amplified when the chaperone adopted a closed state. Saturating ATP conditions led to a continuous switching between open and closed conformations on the seconds timescale, much faster than the expected 100 s ATPase cycle. Analysis of dwell times showed that the dynamics was best described by four states, two open and two closed, revealing eight different kinetic rates between them, as well as a simplified energy landscape. In the presence of ATP, two of the kinetic barriers were lowered, but all states could be accessed spontaneously even in the absence of nucleotide. Together, all these results imply that the large conformational changes of the NTDs and the ATP‐cycle are only weakly coupled for yeast Hsp90, and mostly driven by thermal fluctuations.
The same authors extended these observations in a second study using 3‐color FRET.46 Combining the labeled Hsp90 heterodimer with a second acceptor dye attached to the nucleotide (either ATP or ADP) allowed simultaneous detection of conformational changes and nucleotide binding events. Both ATP and ADP were found to bind open and closed states of Hsp90 with slightly different rates, strengthening the importance of thermal fluctuations [Fig. 3(B,C)]. In addition, it was found that ATP binds on and off multiple times before it is hydrolyzed, contrasting with the long established idea that the chaperone remains in an ATP‐bound “waiting state” until hydrolysis occurs.
In a parallel study, the differences between yeast and bacterial Hsp90 were investigated. For HtpG, it was found that the NTD conformational dynamics and the ATP cycle are strongly coupled by a mechanical ratchet mechanism.56 These observations suggest that Hsp90s evolved from the rigid bacterial, nucleotide‐regulated chaperone to its more flexible eukaryotic counterpart. This feature may have allowed the chaperone to adapt to a larger range of substrates without additional energy cost. It may also enable a more versatile and sophisticated regulation by cochaperones, in addition to the ATPase activity. This might explain the extensive number of cochaperones for eukaryotic Hsp90, while none has been found for the bacterial HtpG.
To investigate regulation of yeast Hsp90 by the cochaperone p23, the FRET strategy was extended to four colors. The observations suggested that the ATP turnover regulates p23 binding, without a direct impact on the large NTD conformational changes, and is the interaction with the cochaperone what provided the Hsp90 machinery its directionality.57 Further evidence of cochaperone regulation was found using single‐molecule photoinduced electron transfer (PET) to study the intra‐subunit domain interactions.58 These local conformational changes in the NTD and MD, though more coupled to the ATPase activity than the inter‐subunit NTD dimerization, are strongly catalyzed by Aha‐1, another cochaperone of yeast Hsp90.
Together, these results illustrate the novel insights that single‐molecule experiments can provide. Key mechanisms have been revealed on the conformational dynamics of Hsp90 and its relation to the ATPase cycle and cochaperones. An additional and critical element involved in the chaperone regulation is substrate binding, but its detailed impact on Hsp90 dynamics remains elusive. Conversely, how Hsp90 affects substrate conformations also remains largely unknown. A recent study showed that it is possible to monitor both monomer and dimer Hsp90 folding states using optical tweezers,59 while the same tool has been used to investigate how chaperones affect protein folding.41 The extraordinary versatility of Hsp90 makes this an outstanding challenge, as diverse substrates may affect and be affected in different fashion.
GroEL, a Confining Barrel
The GroEL‐GroES system is arguably the most studied molecular chaperone. The GroEL barrel‐like structure is composed of 14 identical subunits of 57 kDa each that are stacked as two heptameric rings [see Fig. 4(A)].61, 62 Each monomer has three domains: an apical domain that binds with polypeptides and GroES, an intermediate domain, and the nucleotide binding equatorial domain.63 GroEL functions with its co‐chaperonin GroES that acts as a lid for encapsulating nonnative proteins [see Fig. 4(A), left‐top panel] and is composed of 7 subunits of 10 kDa each. ATP binding in the equatorial domain of GroEL results in large structural movements in the apical domain and exposure of hydrophobic residues that facilitate GroES binding, in turn doubling the size of the GroEL cavity, such that proteins of up to 60 kDa size can be encapsulated.64
Figure 4.
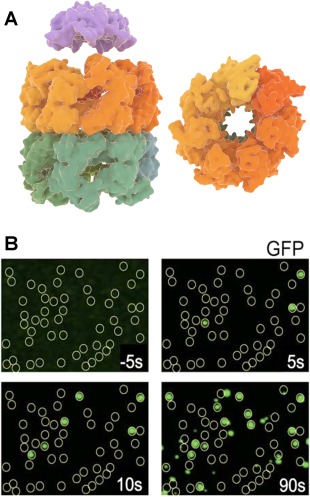
GroEL–GroES structure and folding of GFP by the complex. (A) GroEL side view (left–bottom) and top view (right) with its two heptameric rings and cochaperonin GroES (left–top) (B) Fluorescence images acquired by total internal reflection fluorescence microscopy (TIRFM), showing GroEL positions as yellow circles and folded GFP molecules as green dots co‐localized with GroEL.60 Folding kinetics of individual GFP molecules was measured by acquiring the fluorescence images at different times. Panel B is adapted from Ref. 60.
Early stopped flow fluorescence anisotropy and enzymatic activity studies have been instrumental in revealing many aspects of the ATP hydrolysis cycle,65 – 67 while cryo‐EM and X‐ray crystallography pushed understanding of the corresponding GroEL structural changes.68, 69, 70 Non‐native polypeptides are thought to first bind the GroEL apical domain, after which the binding of GroES drives them into the central GroEL cavity where folding takes place. Binding of a second substrate and GroES on the other ring of the double‐barrel GroEL structure triggers release of the folded substrate protein. Despite the detailed biochemical and structural knowledge that has been amassed, the core folding mechanism remains contested.71 GroEL‐GroES could act as a passive folding cage by physically protecting substrates from aggregation, actively catalyze the folding of individual substrates, or exert pulling forces on misfolded conformers in order to unfold them and allow autonomous refolding to the native state. Detailed questions also remain unanswered on various other aspects, including the cooperation between the two rings, the precise role of GroES, and the substrate‐accepting state of GroEL.
Compared to the other chaperones, the GroEL‐GroES system has been extensively probed with single‐molecule fluorescence approaches. One recent example is by Lin et al.,72 where using intra‐molecular FRET the authors observed the binding of nonnative Rubisco to a nucleotide free ring of GroEL with ATP and GroES bound to the other ring (also termed as ATP bullet). The results suggested that the ATP bullet is the polypeptide accepting state, which is also consistent with the asymmetric functioning of GroEL. In this asymmetric model, release of ADP from the trans‐ring is catalyzed by peptide binding, which in turn triggers ATP hydrolysis in the cis ring.73 This notion was supported by recent single‐molecule studies on symmetric or football‐shaped GroEL‐GroES complexes.74, 75 Saturated substrate concentrations and slow ATP hydrolysis were found to promote symmetric complexes over asymmetric ones. Most studies on the symmetric complexes have been performed using the GroEL variant D398A that hydrolyzes ATP more slowly. Takei et al.76 studied the football complex with a fluorescently labeled D398A variant and using GFP as the substrate protein. Single‐molecule total internal reflection fluorescence (TIRF) microscopy was used to localize the positions of GFP molecules at the GroEL‐GroES positions60 [Fig. 4(B)]. The authors interestingly observed that two GFP molecules can fold simultaneously within the two GroEL cavities capped by GroES, with refolding kinetics similar to those observed in the asymmetric complex.
The physiological significance of symmetric complexes has been a matter of debate, with a number of bulk studies considering only the asymmetric complexes as part of the functional cycle in vivo.66, 77 On the other hand, recent single‐molecule studies have provided observations of symmetric complexes working as parallel folding machines, which may be a more productive protein folding state than the asymmetric complexes.76, 78, 79 Moreover, during stress conditions when concentrations of nonnative proteins are high and negative cooperativity between the rings decreases,80 the formation of symmetric complexes should be favored.
Another conundrum about GroEL‐GroES chaperonin is the process of protein folding itself. One debate is whether GroEL‐GroES acts only as a passive or Anfinsen cage that simply prevents protein aggregation,81 with the encapsulated protein folding essentially autonomously,82 or whether the complex actively assist and accelerates protein folding, for instance by smoothening the folding landscape.83, 84, 85 Another model, known as iterative annealing, proposes that GroEL‐GroES functions by unfolding misfolded proteins, which subsequently fold autonomously inside or outside the GroEL cavity.86 Ensemble measurements have limitations when aiming to eliminate the effect of protein aggregation on the overall folding rate.81 A recent study85 used FRET on a slow‐folding Maltose Binding Protein variant (DM‐MBP) to measure spontaneous and GroEL‐GroES assisted folding rates.83 The results suggested accelerated refolding rates by eightfold. Using fluorescence correlation spectroscopy the authors estimated that at 100 pM DM‐MBP, the observation volume contains only monomeric substrates. Consistently, a constant number of DM‐MBP was detected in the observation volume, which indicated limited reversible aggregation that can confound the quantification of folding rates. We note that others have put forward arguments against active acceleration models.87 Experiments on the single ring GroEL (SR‐1) variant that goes through just one round of ATP cycle without dissociating GroES65 displayed similar folding environment for the substrate.
Theoretical models suggest that acceleration could be afforded by the confinement itself, as this can lower the entropic barrier that limits access to folded states, or an optimal range of hydrophobic interactions of the substrate GroEL cavity, which may restrict the formation of misfolded states.88, 89, 90 The iterative annealing model has been considered for stringent substrates such as Rubisco, which are prone to form misfolded, kinetically trapped structures. GroEL‐mediated unfolding could then provide another chance to refold from a high energy state.86 The study by Lin et al.91 found two different phases in the interaction between Rubisco monomers and ADP GroEL bullets. The first phase—before addition of ATP—displayed slowly decreasing FRET signals indicating passive unfolding by the trans ring, while ATP addition led to a rapid decrease in the FRET signal, consistent with unfolding of the monomer by GroEL. Using single‐molecule FRET and rapid microfluidic mixing experiments, Hofmann et al.92 studied the folding kinetics of the protein Rhodanese. Interestingly, the N and L regions of Rhodanese displayed similar refolding rates with or without chaperonin, while the C domain refolded two orders more slowly with the chaperonin. A possible reason for folding deceleration was postulated to be the lower diffusion constant of the polypeptide caused by interactions with the GroEL cavity surface.
Despite the large body of work on GroEL, many questions are still elusive. For instance, it is unclear whether GroEL–GroES functions typically as asymmetric complexes, with symmetric complexes favored under stress conditions. It also remains unknown how general the acceleration and stimulated annealing mechanisms are. The ability of GroEL to accelerate folding of proteins with diverse structure and folding kinetics would raise intriguing questions on how this is achieved. The specific ability of single‐molecule methods to reveal individual conformational transitions will be central to resolving these important questions.
Outlook
Polypeptide chains that emerge from the ribosomal tunnel are bound for a multi‐faceted journey guided by chaperones. The single‐molecule approaches discussed here have begun to provide a glimpse of the intricate dynamics that these companions engage in. At the same time, these first results also underscore how much we do not yet know. Elementary questions are for instance whether and how chaperones such as Hsp70 directly promote folding, and how they switch to contrasting roles such as disaggregation and membrane translocation. The observed diverse modes of action also press questions on cooperation between chaperones and cochaperones. Existing models follow a rather hierachical view, with some chaperones acting upstream near the ribosome, and others downstream on mature or aggregated and damaged proteins. Observed action on near‐mature proteins of supposedly upstream actors such as trigger factor challenges this logic. How, when and why different chaperones interact with a client is a crucial issue to begin addressing protein homeostasis at the cellular level. Another intriguing question is what happens early on, at the ribosome itself. Nascent chains emerge vectorially, and hence can begin to fold before synthesis is complete. But the ribosome is also a busy platform that recruits a host of chaperones and other factors. The purpose of these actions and complex dynamics is filled with interesting open questions that are amenable to single‐molecule approaches, as has been demonstrated.17, 26 Small heat‐shock proteins are a distinct class of chaperones not reviewed here but with important roles in preventing protein damage and aggregation, which can also be studied at the single‐molecule level.93 Another intriguing aspect of chaperones is their direct regulatory role. Specifically, Hsp70 and Hsp90 are involved in modulating the activity of a host of receptors and kinases, with key implications for medical conditions. Yet, how these roles are fulfilled is still obscure. Resolving these issues remains a key outstanding challenge, and single‐molecule methods will be central in adressing them. At the same time, current methods are far from providing the full picture. Other rapidly advancing methods such as NMR, hydrogen exchange mass spectrometry, as well as combined fluorescence‐manipulation techniques, will be crucial to arrive at the next level of models of these intriguing systems.
Acknowledgments
Work in the Tans group is part of the Netherlands Organization for Scientific Research (NWO). The authors declare that there is not conflict of interest.
References
- 1. Young JC, Agashe VR, Siegers K, Hartl FU (2004) Pathways of chaperone‐mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 5:781–791. [DOI] [PubMed] [Google Scholar]
- 2. Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125:443–451. [DOI] [PubMed] [Google Scholar]
- 3. Liberek K, Lewandowska A, Zietkiewicz S (2008) Chaperones in control of protein disaggregation. EMBO J 27:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meunier L (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell 13:4456–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pratt WB, Toft DO (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70‐based chaperone machinery. Exp Biol Med 228:111–133. [DOI] [PubMed] [Google Scholar]
- 6. Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22. [DOI] [PubMed] [Google Scholar]
- 7. Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772. [DOI] [PubMed] [Google Scholar]
- 8. Arrigo A‐P (2001) Hsp27: novel regulator of intracellular redox state. IUBMB Life 52:303–307. [DOI] [PubMed] [Google Scholar]
- 9. Kern Re Malki A, Abdallah J, Tagourti J, Richarme G (2007) Escherichia coli HdeB is an acid stress chaperone. J Bacteriol 189:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tissieres A, Mitchell HK, Tracy UM (1974) Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol 84:389–398. [DOI] [PubMed] [Google Scholar]
- 11. Clark PL (2004) Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci 29:527–534. [DOI] [PubMed] [Google Scholar]
- 12. Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, Weissman JS, Bukau B (2011) Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147:1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N (2004) Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature 431:590–596. [DOI] [PubMed] [Google Scholar]
- 14. Saio T, Guan X, Rossi P, Economou A, Kalodimos CG (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344:1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singhal K, Vreede J, Mashaghi A, Tans SJ, Bolhuis PG (2015) The trigger factor chaperone encapsulates and stabilizes partial folds of substrate proteins. PLoS Comput Biol 11:e1004444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mashaghi A, Kramer G, Bechtluft P, Zachmann‐Brand B, Driessen AJM, Bukau B, Tans SJ (2013) Reshaping of the conformational search of a protein by the chaperone trigger factor. Nature 500:98–101. [DOI] [PubMed] [Google Scholar]
- 17. Kaiser CM, Chang H‐C, Agashe VR, Lakshmipathy SK, Etchells SA, Hayer‐Hartl M, Hartl FU, Barral JM (2006) Real‐time observation of trigger factor function on translating ribosomes. Nature 444:455–460. [DOI] [PubMed] [Google Scholar]
- 18. Martinez‐Hackert E, Hendrickson WA (2009) Promiscuous substrate recognition in folding and assembly activities of the trigger factor chaperone. Cell 138:923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deuerling E, Schulze‐Specking A, Tomoyasu T, Mogk A, Bukau B (1999) Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature 400:693–696. [DOI] [PubMed] [Google Scholar]
- 20. Vorderwülbecke S, Kramer G, Merz F, Kurz TA, Rauch T, Zachmann‐Brand B, Bukau B, Deuerling E (2004) Low temperature or GroEL/ES overproduction permits growth of Escherichia coli cells lacking trigger factor and DnaK. FEBS Lett 559:181–187. [DOI] [PubMed] [Google Scholar]
- 21. Ullers RS, Ang D, Schwager F, Georgopoulos C, Genevaux P (2007) Trigger factor can antagonize both SecB and DnaK/DnaJ chaperone functions in Escherichia coli . Proc Natl Acad Sci USA 104:3101–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singhal K, Vreede J, Mashaghi A, Tans SJ, Bolhuis PG (2013) Hydrophobic collapse of trigger factor monomer in solution. PLoS One 8:e59683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Agashe VR, Guha S, Chang H‐C, Genevaux P, Hayer‐Hartl M, Stemp M, Georgopoulos C, Hartl FU, Barral JM (2004) Function of trigger factor and DnaK in multidomain protein folding: increase in yield at the expense of folding speed. Cell 117:199–209. [DOI] [PubMed] [Google Scholar]
- 24. Hoffmann A, Becker AH, Zachmann‐Brand B, Deuerling E, Bukau B, Kramer G (2012) Concerted action of the ribosome and the associated chaperone trigger factor confines nascent polypeptide folding. Mol Cell 48:63–74. [DOI] [PubMed] [Google Scholar]
- 25. O'Brien EP, Christodoulou J, Vendruscolo M, Dobson CM (2012) Trigger factor slows co‐translational folding through kinetic trapping while sterically protecting the nascent chain from aberrant cytosolic interactions. J Am Chem Soc 134:10920–10932. [DOI] [PubMed] [Google Scholar]
- 26. Kaiser CM, Goldman DH, Chodera JD, Tinoco I, Jr , Bustamante C (2011) The ribosome modulates nascent protein folding. Science 334:1723–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang C, Rossi P, Saio T, Kalodimos CG (2016) Structural basis for the antifolding activity of a molecular chaperone. Nature 537:202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bechtluft P, van Leeuwen RGH, Tyreman M, Tomkiewicz D, Nouwen N, Tepper HL, Driessen AJM, Tans SJ (2007) Direct observation of chaperone‐induced changes in a protein folding pathway. Science 318:1458–1461. [DOI] [PubMed] [Google Scholar]
- 29. Deeng J, Chan KY, van der Sluis EO, Berninghausen O, Han W, Gumbart J, Schulten K, Beatrix B, Beckmann R (2016) Dynamic behavior of trigger factor on the ribosome. J Mol Biol 428:3588–3602. [DOI] [PubMed] [Google Scholar]
- 30. Preissler S, Deuerling E (2012) Ribosome‐associated chaperones as key players in proteostasis. Trends Biochem Sci 37:274–283. [DOI] [PubMed] [Google Scholar]
- 31. Baneyx F, Mujacic M (2004) Recombinant protein folding and misfolding in Escherichia coli . Nat Biotechnol 22:1399–1408. [DOI] [PubMed] [Google Scholar]
- 32. Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272:1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morshauser RC, Hu W, Wang H, Pang Y, Flynn GC, Zuiderweg ER (1999) High‐resolution solution structure of the 18 kDa substrate‐binding domain of the mammalian chaperone protein Hsc70. J Mol Biol 289:1387–1403. [DOI] [PubMed] [Google Scholar]
- 35. Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CaM Neupert W, Lamb DC, Mokranjac D (2010) The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell 38:89–100. [DOI] [PubMed] [Google Scholar]
- 36. Sousa R (2012) A dancer caught midstep: the structure of ATP‐bound Hsp70. Mol Cell 48:821–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schmid D, Baici A, Gehring H, Christen P (1994) Kinetics of molecular chaperone action. Science 263:971–973. [DOI] [PubMed] [Google Scholar]
- 38. McCarty JS, Buchberger A, Reinstein J, Bukau B (1995) The role of ATP in the functional cycle of the DnaK chaperone system. J Mol Biol 249:126–137. [DOI] [PubMed] [Google Scholar]
- 39. Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B (1999) Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci USA 96:5452–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kellner R, Hofmann H, Barducci A, Wunderlich B, Nettels D, Schuler B (2014) Single‐molecule spectroscopy reveals chaperone‐mediated expansion of substrate protein. Proc Natl Acad Sci USA 111:13355–13360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mashaghi A, Bezrukavnikov S, Minde DP, Wentink AS, Kityk R, Zachmann‐Brand B, Mayer MP, Kramer G, Bukau B, Tans SJ (2016) Alternative modes of client binding enable functional plasticity of Hsp70. Nature 539:448–451. [DOI] [PubMed] [Google Scholar]
- 42. Marcinowski M, Höller M, Feige MJ, Baerend D, Lamb DC, Buchner J (2011) Substrate discrimination of the chaperone BiP by autonomous and cochaperone‐regulated conformational transitions. Nat Struct Mol Biol 18:150–158. [DOI] [PubMed] [Google Scholar]
- 43. Banerjee R, Jayaraj GG, Peter JJ, Kumar V, Mapa K (2016) Monitoring conformational heterogeneity of the lid of DnaK substrate binding domain during its chaperone cycle. FEBS J 283:2853–2868. [DOI] [PubMed] [Google Scholar]
- 44. Young JC (2001) Hsp90: a specialized but essential protein‐folding tool. J Cell Biol 154:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lai B‐T, Chin WNW, Stanek AE, Keh W, Lanks KW (1984) Quantitation and intracellular localization of the 85K heat shock protein by using monoclonal and polyclonal antibodies. Mol Cell Biol 4:2802–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ratzke C, Berkemeier F, Hugel T (2012) Heat shock protein 90's mechanochemical cycle is dominated by thermal fluctuations. Proc Natl Acad Sci USA 109:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nadeau K, Sullivan Ma Bradley M, Engman DM, Walsh CT (1992) 83‐kilodalton heat shock proteins of trypanosomes are potent peptide‐stimulated ATPases. Protein Sci 1:970–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheibel T, Neuhofen S, Weikl T, Mayr C, Reinstein J, Vogel PD, Buchner J (1997) ATP‐binding properties of human Hsp90. J Biol Chem 272:18608–18613. [DOI] [PubMed] [Google Scholar]
- 49. Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH (1997) Identification and structural characterization of the ATP/ADP‐binding site in the Hsp90 molecular chaperone. Cell 90:65–75. [DOI] [PubMed] [Google Scholar]
- 50. Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH (2006) Crystal structure of an Hsp90‐nucleotide‐p23/Sba1 closed chaperone complex. Nature 440:1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shiau AK, Harris SF, Southworth DR, Agard DA (2006) Structural analysis of E. coli hsp90 reveals dramatic nucleotide‐dependent conformational rearrangements. Cell 127:329–340. [DOI] [PubMed] [Google Scholar]
- 52. Weikl T, Muschler P, Richter K, Veit T, Reinstein J, Buchner J (2000) C‐terminal regions of Hsp90 are important for trapping the nucleotide during the ATPase cycle. J Mol Biol 303:583–592. [DOI] [PubMed] [Google Scholar]
- 53. Bron P, Giudice E, Rolland J‐P, Buey RM, Barbier P, Díaz JF, Peyrot V, Thomas D, Garnier C (2008) Apo‐Hsp90 coexists in two open conformational states in solution. Biol Cell 100:413–425. [DOI] [PubMed] [Google Scholar]
- 54. Pearl LH, Prodromou C (2006) Structure and mechanism of the Hsp90 molecular chaperone machinery. Ann Rev Biochem 75:271–294. [DOI] [PubMed] [Google Scholar]
- 55. Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T (2009) The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat Struct Mol Biol 16:281–286. [DOI] [PubMed] [Google Scholar]
- 56. Ratzke C, Nguyen MNT, Mayer MP, Hugel T (2012) From a ratchet mechanism to random fluctuations evolution of Hsp90's mechanochemical cycle. J Mol Biol 423:462–471. [DOI] [PubMed] [Google Scholar]
- 57. Ratzke C, Hellenkamp B, Hugel T (2014) Four‐colour FRET reveals directionality in the Hsp90 multicomponent machinery. Nat Commun 5:4192–4192. [DOI] [PubMed] [Google Scholar]
- 58. Schulze A, Beliu G, Helmerich DA, Schubert J, Pearl LH, Prodromou C, Neuweiler H (2016) Cooperation of local motions in the Hsp90 molecular chaperone ATPase mechanism. Nat Chem Biol 12:628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jahn M, Buchner J, Hugel T, Rief M (2016) Folding and assembly of the large molecular machine Hsp90 studied in single‐molecule experiments. Proc Natl Acad Sci USA 113:1232–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ueno T, Taguchi H, Tadakuma H, Yoshida M, Funatsu T (2004) GroEL mediates protein folding with a two successive timer mechanism. Mol Cell 14:423–434. [DOI] [PubMed] [Google Scholar]
- 61. Braig K, Otwinowski Z, Hegde R, Boisvert DC, Joachimiak A, Horwich AL, Sigler PB (1994) The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature 371:578–586. [DOI] [PubMed] [Google Scholar]
- 62. Boisvert DC, Wang J, Otwinowski Z, Horwich AL, Sigler PB (1996) The 2.4 A crystal structure of the bacterial chaperonin GroEL complexed with ATP gamma S. Nat Struct Biol 3:170–177. [DOI] [PubMed] [Google Scholar]
- 63. Saibil HR, Fenton WA, Clare DK, Horwich AL (2013) Structure and allostery of the chaperonin GroEL. J Mol Biol 425:1476–1487. [DOI] [PubMed] [Google Scholar]
- 64. Clare DK, Vasishtan D, Stagg S, Quispe J, Farr GW, Topf M, Horwich AL, Saibil HR (2012) ATP‐triggered conformational changes delineate substrate‐binding and ‐folding mechanics of the GroEL chaperonin. Cell 149:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL (1996) Characterization of the active intermediate of a GroEL‐GroES‐mediated protein folding reaction. Cell 84:481–490. [DOI] [PubMed] [Google Scholar]
- 66. Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL (1997) Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature 388:792–798. [DOI] [PubMed] [Google Scholar]
- 67. Rye HS, Roseman AM, Chen S, Furtak K, Fenton WA, Saibil HR, Horwich AL (1999) GroEL‐GroES cycling: ATP and nonnative polypeptide direct alternation of folding‐active rings. Cell 97:325–338. [DOI] [PubMed] [Google Scholar]
- 68. Xu ZH, Horwich AL, Sigler PB (1997) The crystal structure of the asymmetric GroEL‐GroES‐(ADP)(7) chaperonin complex. Nature 388:741–750. [DOI] [PubMed] [Google Scholar]
- 69. Ranson NA, Clare DK, Farr GW, Houldershaw D, Horwich AL, Saibil HR (2006) Allosteric signaling of ATP hydrolysis in GroEL‐GroES complexes. Nat Struct Mol Biol 13:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clare DK, Bakkes PJ, van Heerikhuizen H, van der Vies SM, Saibil HR (2009) Chaperonin complex with a newly folded protein encapsulated in the folding chamber. Nature 457:107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hayer‐Hartl M, Bracher A, Hartl FU (2016) The GroEL‐GroES chaperonin machine: a nano‐cage for protein folding. Trends Biochem Sci 41:62–76. [DOI] [PubMed] [Google Scholar]
- 72. Lin Z, Puchalla J, Shoup D, Rye HS (2013) Repetitive protein unfolding by the trans ring of the GroEL‐GroES chaperonin complex stimulates folding. J Biol Chem 288:30944–30955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Madan D, Lin Z, Rye HS (2008) Triggering protein folding within the GroEL‐GroES complex. J Biol Chem 283:32003–32013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sameshima T, Iizuka R, Ueno T, Funatsu T (2010) Denatured proteins facilitate the formation of the football‐shaped GroEL‐(GroES)2 complex. Biochem J 427:247–254. [DOI] [PubMed] [Google Scholar]
- 75. Ye X, Lorimer GH (2013) Substrate protein switches GroE chaperonins from asymmetric to symmetric cycling by catalyzing nucleotide exchange. Proc Natl Acad Sci USA 110:E4289–E4297. USA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Takei Y, Iizuka R, Ueno T, Funatsu T (2012) Single‐molecule observation of protein folding in symmetric GroEL‐(GroES)2 complexes. J Biol Chem 287:41118–41125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hayer‐Hartl MK, Ewalt KL, Hartl FU (1999) On the role of symmetrical and asymmetrical chaperonin complexes in assisted protein folding. Biol Chem 380:531–540. [DOI] [PubMed] [Google Scholar]
- 78. Yang D, Ye X, Lorimer GH (2013) Symmetric GroEL:GroES2 complexes are the protein‐folding functional form of the chaperonin nanomachine. Proc Natl Acad Sci USA 110:E4298–E4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fei X, Ye X, LaRonde NA, Lorimer GH (2014) Formation and structures of GroEL:GroES2 chaperonin footballs, the protein‐folding functional form. Proc Natl Acad Sci USA 111:12775–12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Llorca O, Galan A, Carrascosa JL, Muga A, Valpuesta JM (1998) GroEL under heat‐shock. Switching from a folding to a storing function. J Biol Chem 273:32587–32594. [DOI] [PubMed] [Google Scholar]
- 81. Apetri AC, Horwich AL (2008) Chaperonin chamber accelerates protein folding through passive action of preventing aggregation. Proc Natl Acad Sci USA 105:17351–17355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Horwich AL, Apetri AC, Fenton WA (2009) The GroEL/GroES cis cavity as a passive anti‐aggregation device. FEBS Lett 583:2654–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chakraborty K, Chatila M, Sinha J, Shi Q, Poschner BC, Sikor M, Jiang G, Lamb DC, Hartl FU, Hayer‐Hartl M (2010) Chaperonin‐catalyzed rescue of kinetically trapped states in protein folding. Cell 142:112–122. [DOI] [PubMed] [Google Scholar]
- 84. Georgescauld F, Popova K, Gupta AJ, Bracher A, Engen JR, Hayer‐Hartl M, Hartl FU (2014) GroEL/ES chaperonin modulates the mechanism and accelerates the rate of TIM‐barrel domain folding. Cell 157:922–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gupta AJ, Haldar S, Milicic G, Hartl FU, Hayer‐Hartl M (2014) Active cage mechanism of chaperonin‐assisted protein folding demonstrated at single‐molecule level. J Mol Biol 426:2739–2754. [DOI] [PubMed] [Google Scholar]
- 86. Thirumalai D, Lorimer GH (2001) Chaperonin‐mediated protein folding. Ann Rev Biophys Biomol Struct 30:245–269. [DOI] [PubMed] [Google Scholar]
- 87. Tyagi NK, Fenton WA, Deniz AA, Horwich AL (2011) Double mutant MBP refolds at same rate in free solution as inside the GroEL/GroES chaperonin chamber when aggregation in free solution is prevented. FEBS Lett 585:1969–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Betancourt MR, Thirumalai D (1999) Exploring the kinetic requirements for enhancement of protein folding rates in the GroEL cavity. J Mol Biol 287:627–644. [DOI] [PubMed] [Google Scholar]
- 89. Thirumalai D, Klimov DK, Lorimer GH (2003) Caging helps proteins fold. Proc Natl Acad Sci USA 100:11195–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jewett AI, Baumketner A, Shea JE (2004) Accelerated folding in the weak hydrophobic environment of a chaperonin cavity: creation of an alternate fast folding pathway. Proc Natl Acad Sci USA 101:13192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lin Z, Madan D, Rye HS (2008) GroEL stimulates protein folding through forced unfolding. Nat Struct Mol Biol 15:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hofmann H, Hillger F, Pfeil SH, Hoffmann A, Streich D, Haenni D, Nettels D, Lipman EA, Schuler B (2010) Single‐molecule spectroscopy of protein folding in a chaperonin cage. Proc Natl Acad Sci USA 107:11793–11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ungelenk S, Moayed F, Ho C‐T, Grousl T, Scharf A, Mashaghi A, Tans S, Mayer MP, Mogk A, Bukau B (2016) Small heat shock proteins sequester misfolding proteins in near‐native conformation for cellular protection and efficient refolding. Nat Commun 7:13673–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]