

Abstract
Helicases are a broad family of enzymes that separate nucleic acid double strand structures (DNA/DNA, DNA/RNA, or RNA/RNA) and thus are essential to DNA replication and the maintenance of nucleic acid integrity. We review the picture that has emerged from single molecule studies of the mechanisms of DNA and RNA helicases and their interactions with other proteins. Many features have been uncovered by these studies that were obscured by bulk studies, such as DNA strands switching, mechanical (rather than biochemical) coupling between helicases and polymerases, helicase‐induced re‐hybridization and stalled fork rescue.
Keywords: magnetic traps, helicases, polymerase, primase, fork regression, DNA unwinding, Holliday junction migration, ds‐DNA fork, active unwinding, helicase/polymerase coupling, replisome, primosome, strand annealing
Abbreviations
- ATP
adenosine triphosphate
- bp
base pair
- DNA
deoxyribonucleic acid
- dsDNA
double‐stranded DNA
- HJ
Holliday junction
- kB
Boltzmann constant
- kcat
turnover number under optimum conditions (saturated enzyme)
- kM
Michaelis constant
- LNA
locked nucleic acid
- NA
nucleic acid
- NTP
nucleoside triphosphate
- µm
micrometer
- nm
nanometer
- nts
nucleotides
- Pm
maximum processivity under saturated condition
- pN
pico‐Newtons
- RNA
ribonucleic acid
- SF
superfamily
- SM
single molecule
- ssDNA
single stranded DNA
- ss‐dsNA
nucleic acid forming a junction between single‐stranded and double stranded molecule
- SSB
single strand binding
- vm
maximum speed under saturated condition.
Introduction
Helicases are enzymes whose role is to unwind the double helix of nucleic acid to provide other enzymes with an ssDNA or ssRNA substrate. DNA helicases are involved in DNA replication, recombination and repair,1, 2, 3, 4, 5 whereas RNA helicases are involved in transcription termination, translation initiation and RNA splicing among other roles.6 Since helicases translocate along their substrate using the energy of NTP hydrolysis, they are molecular motors: they use chemical energy to perform mechanical work. Their activity is so crucial in many key elements of living systems, that they are ubiquitous in the tree of life3 and their genetic sequences are hypothesized to represent about 1% of each individual genome.7
Complete descriptions of DNA and RNA helicase families can be found respectively in the books of M. Spies8 and E. Jankowsky9 and the citations therein. Helicases can be classified according to diverse parameters. One classification is based on the polarity of translocation along the DNA strand, which has classically been deduced from the overhang required to load the enzyme at the ss‐dsNA (nucleic acid) junction where unwinding is initiated. Helicases that translocate along NAs from the 3′ to the 5′ end are designated as 3′‐5′ helicases and often need a 3′‐overhang to load onto the junction. Similarly, enzymes that translocate and unwind in the 5′ to 3′ direction are designated as 5′‐3′ helicases and often require a 5′‐overhang loading site.
Several groups also suggested wider and more unifying classifications of DNA and RNA helicases encompassing unwinding polarity with sequence and structural features.10, 11, 12, 13 The most recent classification thus sorts helicases into superfamilies 1 to 6 (SF1 to 6), with toroidal ring forming enzymes sorted into SF3 to 6, while non‐ring forming are found in SF1 and SF2.
SF1 and SF2 helicases share a conserved helicase core formed of two similar RecA‐like domains resembling the fold of RecA recombination protein. These folds notably comprise at least 12 signature helicases motifs shared by both SF1 and SF2 but not present in each family and group. Such characteristic motifs are either expected or experimentally proven to share similar functions despite sequence variability among different helicase families. Highest conservation between SF1 and SF2 is observed in the residues coordinating binding and hydrolysis of triphosphates (motifs I, II and VI). The Q‐motif, which coordinates the ATP adenine base is less conserved between both SFs, and not present in all families. Other motifs coordinating NTP and nucleic acid binding are highly conserved within each superfamily but not across both, indicating mechanistic variations in the communication between the two functional sites. Several motifs are also known to contact nucleic acids on the face opposite the ATP binding site, and are well conserved between both SFs.
SF1 enzymes, with members such as UvrD, Rep or Upf1, exhibit a large contact area with ssNA. For this reason, they are classified as ssNA translocases that melt downstream dsNA as they proceed along one of the strands, see below.
Conversely, SF2 enzymes, with members such as RecG, RecQ, NS3, or DEAD‐box helicases, interact nonspecifically with the NA phosphodiester backbone and are assumed to be dsNA translocases, see below.
SF3 comprises helicases from small DNA and RNA viruses, while SF4 and SF5 contain 5′‐3′ hexameric helicases related either to the E. coli replicative helicase DnaB (SF4) or the bacterial termination factor, Rho (SF5).
Beyond such classifications, a full understanding of the activity of these motors in a living cell requires an understanding of their assembly, structure and conformation(s) of their nucleic acid target, their sequence specificity if any, and their binding modality. Equally important is the mechanism used during their translocation, their directionality, the number of bases they can unwind before dissociating, their velocity, their step size and stoichiometry of NTP hydrolysis (how many base‐pairs are unwound per NTP consumed) and, of course, their coupling with other proteins. Many of these elements are related to the mechanical aspects of their activity and thus single molecule micromanipulations are very useful tools to investigate these issues together with classical bulk assays.
From a mechanistic point of view the most important issue concerning the function of helicases is the coupling between ATP hydrolysis and DNA unwinding.
The simplest model assumes that the helicase is “passive”: ATP hydrolysis is required only for translocation of the helicase which advances upon unbinding of the DNA junction [see Fig. 1(A)]. DNA fraying maybe thermally induced, or the junction may be destabilized by its interactions with the helicase or other enzymes (e.g., polymerases active with the helicase at the replication fork).
Figure 1.
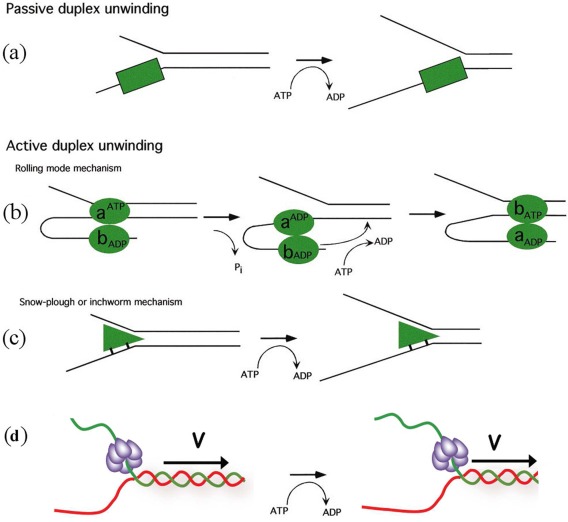
Possible translocation mechanisms of helicases. (A) The helicase can be passive, trapping fork fluctuations through an ATP‐driven progression. Alternatively, helicases can actively unwind DNA (B) as they progress along by a “rolling model” where each monomer takes turn at unwinding the molecule or (C) by driving a “plow” through the DNA fork or (D) pulling the DNA through the helicase as in some hexameric helicases. So far all these mechanisms are found in helicase with the exception of the rolling model which had not been demonstrated experimentally for any helicase.
In other models the helicase is “active”: it uses part of the energy from ATP hydrolysis to melt the dsDNA and translocate [see Fig. 1(B,C)]. One such model assumes that ATP hydrolysis switches the affinity of the binding sites of a dimeric enzyme from a single strand to double strand DNA. In this “rolling model” of DNA unwinding [see Fig. 1(B)], in the absence of ATP, both subunits have a high affinity for ssDNA. Binding of ATP to the subunit upstream of the junction causes it to detach from ssDNA and rebind to the adjacent (downstream) dsDNA, which melt apart when ATP is hydrolyzed. The release of products revert the system to its initial state with the subunits having switched order. In this model, the enzymatic step size is at least as large as the number of DNA bases contacted by each monomer. However it seems that this mechanism is not used by any of the helicases studied so far.14, 15, 16, 17, 18
A second active model, the “inchworm model,” assumes that ATP hydrolysis is used to translocate the enzyme along single or double stranded DNA, with the opening of the junction a consequence of that translocation. For example, if the helicase translocates along ssDNA (SF1 enzymes), it will unwind the junction like a snowplow [see Fig. 1(B)]. If the enzyme translocates on dsDNA (SF2 enzymes), it may use a wedge domain positioned at the junction to open the DNA like a wire‐stripper. This model is supported by observations on various helicases: RecBCD.18, 19, 20, 21 Another possibility to form an active helicase is to force a kink in the NA duplex that act as a wedge destabilizing locally increasing the unpeeling fluctuations.22
In spite of these precise descriptions of the enzymatic mechanism, whether an enzyme is active or passive, has rested on problematic estimations of the free energy of the interaction of the enzyme with the DNA fork. To cut this Gordian knot, we have defined a passive enzyme as one whose unwinding rate is very sensitive to the DNA sequence and increases significantly when a force is acting to unzip the two DNA strands (see below). By contrast the unwinding rate of an active enzyme is insensitive to the DNA sequence and unchanged (or even possibly decreased) when a force is acting to separate the two strands.
When unwound by the helicase the two single‐stranded nucleic acids are in close proximity and may thus re‐anneal in the wake of the enzyme. In vivo to prevent this re‐annealing cofactors act on the ssDNA generated by the helicase activity:1, 2, 23 proteins that bind ssDNA to stabilize it (e.g., Single Strand Binding (SSB) proteins) or enzymes that process the ssDNA (e.g., Single Strand Nucleases or the cell replication machinery). In fact most helicases function poorly (inefficiently and with reduced processivity) when isolated from the macromolecular machinery and coupling factors with which they are intended to operate.23 The inefficiency of isolated helicases is not entirely surprising, as unwinding dsDNA could be potentially dangerous for the cell if it was not tightly regulated. The cell further regulates the activity of its helicases in two ways: by using specific enzymes to load the helicase on its template (this is the case for replicative helicases for example) or by using a cofactor to switch the helicase between different modes. One of the challenges of studying helicases as molecular machines is thus to understand not only the mechanisms of these helicases but also their coupling with other proteins and their alteration in different in vitro and in vivo contexts.
The re‐annealing of the unwound strands behind the helicase makes it challenging to understand their mechanism in vitro. Indeed bulk in vitro assays are limited by the fact that once dsDNA has been opened by the helicase under study the re‐hybridization of the two strands proceeds spontaneously and “erases” the action of the helicase unless very specific actions are taken to prevent this. The re‐hybridization problem is a major issue which has been solved only partially. A classical approach is to use Single Strand Binding (SSB) proteins that are supposed to stabilize ssDNA but as we shall see single molecule approaches show this strategy to be problematic: the characterization of a helicase depends on the experimental conditions.23 Due to this re‐hybridization issue, most bulk assays of helicases report a lower processivity than expected from their role in vivo. The only exceptions are helicases that work in conjunction with a polymerase that synthesizes a strand complementary to the unwound template2 or destroy one of the strands via the action of a coupled endonuclease as in RecBCD24 which we discuss first. In these two cases, the rehybridization issue has been abolished leading to strong processivity.
Mechanism of RecBCD
RecBCD consists of two helicases RecB and RecD. The RecB subunit is a helicase with 3′‐5′ directionality. Its first two domains define it as a member of the SF1 family. Its third domain is a nuclease domain, connected to the remainder of the protein by a long linker region of about 70 amino acids.15, 25
The RecD subunit is also a SF1 helicase but with a rather uncommon 5′‐3′ unwinding activity. The RecC protein contacts both strands of DNA and splits them (like the shovel of a snowplow) before feeding the 3′ strand to the RecB protein and the 5′ tail to the RecD subunit. The two helicase motors apparently work independently of each other and because of their opposite polarity they both pull‐in the anti‐parallel DNA strands. A detailed picture of the mechanism of coupling between these units has emerged from single‐molecule experiments and structural data.
Bulk measurements of the activity of RecBCD (estimated by the amount of hydrolyzed DNA) yield a very large unwinding rate26 (vm∼930 bps/s), a huge processivity27 (about Pm=27000 ±3000 bps) and a rate of ATP hydrolysis28 (with kcat ∼ 740 s−1) all characterized by the same Michaelis‐Menten coefficient26, 28, 29, 30 (kM∼100 µM). These results imply that the unwinding rate and processivity are controlled by ATP hydrolysis. Assuming a tight coupling between ATP consumption and translocation, one deduces that about 1 ATP is required to unwind a single (∼v m/k cat) base‐pair. Furthermore, single‐turnover DNA unwinding experiments31 yield an estimate of the helicase step size of about 3.9 ± 1.3 bps. However in these bulk experiments an estimated 30% of the enzymatic population was inactive (either because of misfolding or for not being on their substrate). Therefore, it was not clear how the previously quoted numbers were affected by the presence of this population of inactive enzymes.
To address that issue Bianco et al.32 have directly visualized the motion of RecBCD on single DNA molecules (see Fig. 2). In their experiment, a DNA molecule was tethered at one end to a small polystyrene bead held in an optical tweezers. The DNA molecule was stained with a fluorescent dye (YOYO1), stretched by the flow of the enzyme containing buffer and visualized on a sensitive camera.
Figure 2.
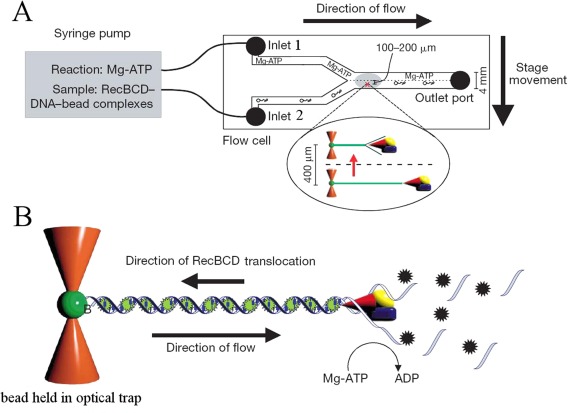
(A) The flow cell introduced by the Kowalczykowski group to study the interaction between DNA and various motors. It consists of a microfluidic cell with two (or more) inlets. Through one inlet a solution of ATP, dye, Mg++, etc. can be introduced and through the other, DNA bound to transparent beads and possibly proteins (such as RecBCD). In this laminar flow regime, the two solutions do not mix. One bead is captured by optical tweezers. The cell is then translated and the trapped DNA molecule brought in contact with ATP (to launch the unwinding reaction) and/or a dye (to stain the molecule). (B) as the molecule is unwound by the progression of RecBCD, it is degraded by the RecB exonuclease activity. Its length can be deduced by the fluorescence of the stained and stretched remaining dsDNA. Hence the progression of the helicase complex on the molecule can be monitored in real time. (Figure taken from Figure 1 of [32] with permission)
As it is unwound and degraded by the RecB nuclease domain, its extension shortens (see Fig. 2). By monitoring the change in extension as a function of time, one can directly measure the rate and processivity of RecBCD. A surprising result of these single‐molecule investigations was the extreme variability in the unwinding rates measured for various enzymes on the same substrate. Although the rate of DNA unwinding by any individual RecBCD was uniform (within experimental error) on any given DNA molecule, the rate for different enzyme molecules deviated by up to fivefold. Subtle changes in the conformation of the complex as it is loaded on DNA may result in this large difference in unwinding rates. In spite of these large enzyme to enzyme fluctuations, the average rate (v m ∼ 970 bps/s at 37°C), average processivity (P m = 38,000 ± 5700 bps) and Michaelis‐Menten coefficient (k M ∼ 150 µM) were all compatible with bulk measurements. This is a comforting result since for this enzyme, no coupling factors are required to observe its unwinding activity, i.e., to prevent reannealing of the two DNA strands. Hence direct observations of DNA unwinding and degradation by a single enzyme and bulk measurements of DNA unwinding are expected to yield similar results (as we shall see that need not always be the case).
Another result from these studies concerns the activity of RecBCD after it encounters a χ sequence (5′‐GCTGGTGG‐3′). Recognition of χ was known to reduce both nuclease activity and translocation speed of RecBCD and to activate RecA‐loading. What single‐molecule experiments have shown is that upon encountering a χ sequence, the enzyme paused for a few seconds and then continued to translocate and unwind DNA as a full trimeric complex,33 but on average at about half the previous rate (for some enzyme molecules the rate after χ recognition could be one tenth of the prior rate). While before χ, both single strands were degraded, after χ the 3′ strands is not degraded: its end is sequestered by the enzyme and as RecBCD translocates a ssDNA loop is extruded. In the presence of DNA specific dyes in solution this increasing loop is visible as an increasingly bright spot translocating along a fluorescently labeled dsDNA.34
A full mechanistic picture of the working of RecBCD emerges when these single‐molecule results are combined with the crystallographic data of RecBCD.15 The location of the nuclease active site (the third domain of RecB) allows processive hydrolysis of the 3′ tail, as it emerges from the RecC subunit. However, proximity of the 5′ tail would enable it to compete with the 3′ tail for binding at the nuclease. Thus, before χ, both strands can be degraded by the nuclease activity of RecB. Upon encountering χ, the RecC subunit binds tightly to the 3′ tail, preventing its further digestion. This induces a conformational change in RecD that slows it down. The 5′ tail is now able to access the nuclease site more frequently and is degraded more fully. The enzyme continues to advance along the DNA extruding a loop out from the RecB subunit that can be loaded with RecA proteins.
Single Molecule Mechanical Assays of Helicases
The RecBCD study is a rather singular example as it is not plagued by the re‐hybridization of the separated strands in the wake of the enzyme. As we shall see below, single molecule micro‐manipulation approaches offer ways to study a variety of helicases, by avoiding re‐hybridization using a stretching force that hinders the reannealing of the strands. This force, however, is not completely neutral and may influence the enzymatic behavior by, for example, favoring unpeeling of DNA. Extrapolation of the results to zero force is a way to address that issue.
The general approach for single molecule mechanical assays of helicases is to stretch a DNA molecule and observe the change in its extension resulting from the transformation of double‐stranded DNA (dsDNA) into single‐stranded DNA (ssDNA). This can be implemented mainly by two different configurations: the unpeeling configuration where a tension is applied between the two ends of the DNA molecule and the unzipping configuration where the force is applied between the two complementary strands at the same DNA extremity.
Force, the mechanical parameter introduced in these studies of helicases by micromanipulation techniques, has two roles:
It prevents re‐hybridization of the strands in the wake of the helicase. By separating the two complementary strands, the force hinders their re‐hybridization. As this effect increases with the force, the probability of re‐hybridization is strongly dependent on the force, displaying an all or none behavior. Above a critical force F r (∼25 pN for the unpeeling assay, ∼3 to 5 pN for the unzipping assay), re‐hybridization is virtually impossible while it occurs readily below F r (as reannealing in the wake of the enzyme becomes more frequent). This sets a lower limit to the force that can be used in these assays.
-
The force determines the molecule's extension and its fluctuations via the bead's Brownian motion, thus it sets the signal to noise ratio of the assay. The larger the difference between the ssDNA and dsDNA extensions the better is the signal (Fig. 3), in the same spirit reducing the bead size also reduces Brownian fluctuations. As the extension of both DNA forms is zero if no force is applied, the signal is null at zero force (and in the unpeeling situation at a force F ∼ 5 pN where the extensions of both are equal, see below). Considering the elastic response of ssDNA and dsDNA the sensitivity of the measurements increases with the force, so it is tempting to apply strong forces to improve the signal to noise ratio of the experiment but this comes at the cost of applying a mechanical tension that might be out of the physiological range.
Figure 3.
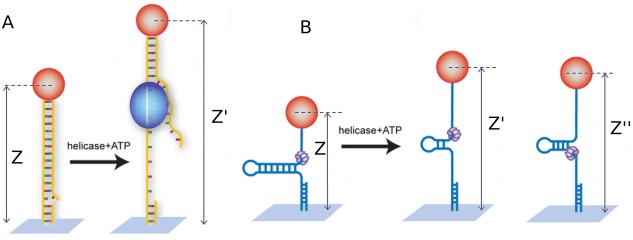 (A) Schematics of the unpeeling configuration used to study helicases: a helicase (blue blob) loads on a nick or gap in the dsDNA molecule under tension. Unwinding of the molecule results in an increase (Δz = z′ – z) of the overall extension. The two ssDNA (one under tension and one free) are unable to match in the wake of the enzyme due to a mismatch in their extension. (B) Schematics of the unzipping assay: a helicase (the violet hexamer) loads at the fork of a DNA hairpin under tension. Unwinding of the hairpin results in an increase (Δz = z′ – z) of the molecule's extension. The tension on the released ssDNA strands prevents their reannealing in the wake of the enzyme. As the helicase reaches the hairpin apex, its continuing translocation on a ssDNA template allows for reannealing of hairpin in its wake, monitored by the decreasing change in extension (Δz = z″ – z)
(A) Schematics of the unpeeling configuration used to study helicases: a helicase (blue blob) loads on a nick or gap in the dsDNA molecule under tension. Unwinding of the molecule results in an increase (Δz = z′ – z) of the overall extension. The two ssDNA (one under tension and one free) are unable to match in the wake of the enzyme due to a mismatch in their extension. (B) Schematics of the unzipping assay: a helicase (the violet hexamer) loads at the fork of a DNA hairpin under tension. Unwinding of the hairpin results in an increase (Δz = z′ – z) of the molecule's extension. The tension on the released ssDNA strands prevents their reannealing in the wake of the enzyme. As the helicase reaches the hairpin apex, its continuing translocation on a ssDNA template allows for reannealing of hairpin in its wake, monitored by the decreasing change in extension (Δz = z″ – z)
Indeed a large applied tension may destabilize the dsDNA. The mechanical unfolding of a hairpin construct in the absence of a helicase can be characterized, as seen in Figure 4: above a critical force of F u = 15 ± 1 pN the hairpin spontaneously unfolds, while it remains otherwise stably folded at forces F c < 12 ± 1 pN. Thus at forces F < F c, any unfolding observed in the presence of a helicase is a result of its activity. Indeed in its absence, the extension of the DNA molecule remains constant and equals to the folded hairpin. In the unpeeling configuration the destabilizing force is F u ∼ 60 pN. However, in both configuration, there exists a substantial range of forces (F r < F < F u) where the opening of the dsDNA can only be caused by the helicase and where the tension on the molecule prevents re‐hybridization of the strands in the wake of the enzyme.
Figure 4.
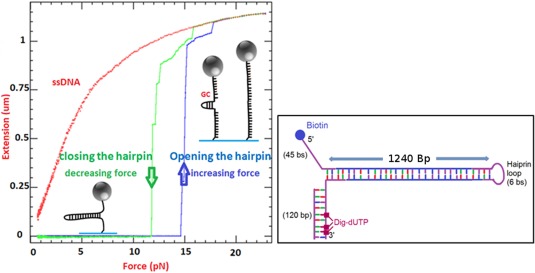
Extension of a DNA hairpin molecule versus the applied force in the unzipping configuration. The molecular design is given in the box on the right: a 1.2 kb hairpin is made of a dsDNA stem closed at one end by a loop and with a fork at the second end. The arm ending in 5′ has a biotin while the other arm holds multiple digoxigenin. This molecule is attached to a 1‐micron size magnetic bead coated with streptavidin and to its second extremity to the flow cell via a digoxigenin/anti‐dig bound. At low force this molecule remains closed and its extension is null. As the magnets are brought closer to the bead, the force can exceed 15 pN leading to the molecule unfolding. The DNA sequence of this molecule presents a GC rich zone close to the apex: this region is more difficult to open and remains folded until the force reaches 17 pN. At that force the molecule is fully open and its extension is nearly 1.2 μm. Further increase of the force leads to a small stretching of the ssDNA molecule. Upon decreasing the force, the molecule refolds with a hysteresis of typically 3 pN. The refolding process nucleates at the molecule apex. If one introduces a 18 nts oligonucleotide that hybridizes to the apex, the refolding of the hairpin is hindered and one observes the red curve corresponding to ssDNA elasticity. When the force is decreased to very low values, the oligonucleotide can be expelled and the hairpin refolds
In the unpeeling configuration, the molecule is a stretched dsDNA [Fig. 3(A)]. A nick or a gap in one of the strands of the DNA serves as a loading site for the helicase which proceeds by unpeeling one strand from the other. To prevent the reannealing in the wake of the helicase a large enough tension on the molecule has to be applied (F > 25 pN = 25 × 10−12 N). In these conditions the mismatch in the inter‐nucleotide distance in the peeled (free) strand and in the strand under tension is large enough: the product of the force (25 pN) by the distance (about 1 nm) between the just separated complementary strands blocked by the helicase leads to an estimate of the energy cost that thermal fluctuations should overcome to re‐hybridize the strands in the wake of the helicase. At F > 25 pN this energy exceeds several times the typical scale of thermal energies, k B T (≈ 4.1 × 10−21 J, at room temperatures) resulting in a rare encircling of the helicase. As the extension of ssDNA is longer than that of dsDNA (for force > 5 pN) the result of helicase activity is an increase in the distance between the bead and the surface (the extension of the molecule) proportional to the amount of peeled ssDNA. Typically, the change of extension is a fraction of the extent of a base‐pair (0.34 nm) depending on the force. Thus, in this configuration one has to apply quite a large force to get a good signal albeit with a poor sensitivity. The main advantage of this assay is its simplicity.
In the unzipping assay a tension is applied between the two ends of a hairpin structure. This configuration mimics a DNA replication fork structure [Fig. 3(B)]. The tension applied on the molecule acts on the two arms of the fork. The molecule's opening by the helicase increases the length of these arms while the tension prevents their re‐hybridization in the wake of the enzyme. When the enzyme reaches the apex of the hairpin the molecule is completely unzipped. Since nothing prevents helicase translocation on the extended ssDNA strand, further translocation of the enzyme along that ssDNA allows the hairpin to refold in its wake, regenerating a fork that may push on the enzyme. In this configuration, the separation between the two arms of the fork is about twice the extension of ssDNA: 0.93 nm/bp at 10 pN. Consequently, the signal (i.e., the change in the bead/surface distance) is very sensitive to DNA unwinding. With a typical precision in the measurement of extensions of a few nanometers, this unzipping assay approaches single base resolution. In addition the critical force to prevent re‐hybridization, F r is low, typically 5 pN which allows an investigation of these enzymes in a more reasonable range of forces.
The magnetic trap
To stretch the DNA molecule in the unpeeling or unzipping configurations, the magnetic trap is a very convenient method for high throughput single molecule assays.35 Briefly, it consists of stretching a single DNA molecule bound at one end to a surface and at the other to a magnetic micro‐bead (1 to 4.5 µm in diameter, a bigger size allowing to reach higher forces but with more noise) (see Fig. 5).
Figure 5.

Right, schematic representation of a magnetic trap. Small super‐paramagnetic beads are bound to the surface of a flow cell by one (or a few) DNA molecules. Magnets positioned above the sample exert a force on the beads and thus on the tethering molecules. DNA hairpins attached at their free ends by one strand to a magnetic bead and at the opposite one to a surface can be unzipped at high enough force (typically F > 15pN). About 50 beads are simultaneously tracked on an inverted microscope. A typical image is shown on the left. Analysis of the successive images of the beads on a camera allows deducing their 3D position and from it the distance of the bead to the surface (i.e., the extension of the tethering molecule) and the exerted force (from the bead's fluctuations, see text)
Small rare earth magnets, whose position is controlled, are used to pull on the micro‐bead and thus stretch the molecule. This system allows applying and measuring forces ranging from a few femtoNewtons (f N = 10−15N) to more than 25 pN (25 pN for 1 µm beads, 200 pN for 2.8 µm beads and 1nN for 4.5 µm beads),35 with a relative absolute accuracy of ∼10%. In contrast with other techniques, the force measurement is absolute and does not require a calibration of a force sensor (once the microscope magnification has been calibrated). It is based on the analysis of the Brownian fluctuations of a tethered bead, whose 3D position can be measured by an analysis of its image on a CCD camera at frequencies up to few hundreds Hertz and in some cases to kHz.36, 37, 38 The center of the bead allows for the determination of its x, y‐coordinates, while the size of its diffraction rings allows for a determination of its distance along the optical axis to the focal point, i.e., its z‐coordinate. The DNA‐bead system is completely equivalent to a damped pendulum of length l =<z> pulled by a magnetic force F (along the vertical axis). Its longitudinal fluctuations (δz 2=< z 2> – < z >2) and transverse fluctuations δx 2 are characterized by effective rigidities k ǁ= ∂z F and k ⊥= F/l. By the equipartition theorem, they satisfy:39, 40
Thus from the bead's Brownian fluctuations (δx 2; δy 2), one can deduce the force pulling on the molecule (the smaller the fluctuations, the larger F) and from δz 2 one deduces its first derivative ∂z F. This measurement method is valid with magnetic (but not optical) traps since the variation of the trapping gradients occurs on a scale (∼ few hundreds of µm, linked to the separation of the two pulling magnets) much larger than the scale on which the elasticity of the molecule changes (∼ 0.1 µm)). In other words, the stiffness of a magnetic trap is very small compared to F/l (the reverse is true for optical tweezers). A further advantage of the magnetic trap technique is that measurements of DNA at constant force are trivial (simply fix the position of the magnets).41 In contrast, for optical tweezers to work at a constant force an appropriate feedback is required to ensure that the displacement of the sensor is kept constant. The accuracy of the absolute force measured by Brownian motion is typically 10%. This number depends on the accuracy of the measurement of δx2: better accuracy is possible but at the expense of longer experiments (more data points). Conversely, for a fixed magnet position the force is constant and its magnitude can be adjusted with extreme accuracy: it is possible to increase the force F by 0.1%, though its precise value is more difficult to determine.
The force deduced from Brownian motion assumes that the measuring device (the camera) has a bandwidth larger than the characteristic frequency of the Brownian fluctuations which may not be the case when the force is large and/or the DNA tether is small. In such instances, special care must be taken in analyzing these fluctuations. Analyzing them in Fourier's space is usually the best strategy.42, 43, 44
A great advantage of magnetic traps is their intrinsic parallelism as the magnets apply a constant force on a large surface and as a consequence, on a large number of beads, i.e., DNA molecules. The increasing size and speed of the new CCD and CMOS cameras offer the possibility to visualize a large number of beads, while fast acquisition cards and parallel computing allow one to track their position (i.e., the molecules' extension) simultaneously.45, 46 It is thus possible to track several hundreds to thousands of beads with a few nanometers accuracy. The inherent parallelism of magnetic traps is extremely valuable when monitoring enzymatic activity. Helicases can be delicate molecular motors which do not work 100% of the time in these in vitro conditions. With hundreds of parallel assays (DNA bound beads), it is far easier to characterize them. Magnetic tweezers are now used by many groups who have extended and refine the technic, the reader may find complementary useful information in Refs. 38 and 47.
Although the magnetic tweezers offer many interesting features they also have limitations: some of them are related to single molecule micromanipulation in general, others are specific to magnetic tweezers. Devices achieving single molecule micromanipulation rely on the measure of the molecule extension, thus they require a finite force to lead to good measurements. This force applied to the NA molecule does not correspond to the in vivo situation and may also help (or hinder in rare case) the enzyme activity. Although the force constrain helps understanding molecular motors function, extrapolating results to the zero force regime is always a challenge. A second issue is the requirement for single molecule events: all the helicase activity bursts present in this article correspond to well identify events surrounded by signals at rest. That is events represent at most 10% of the signal. This condition implies that a single enzyme is working, this is fine but it constrains enzyme concentration to be low and again in some case not close to in vivo conditions. A third issue appears when investigating several enzymes collaborating in a biological process (see below), one would like to have a signal characterizing each enzyme but we only have a single signal: the molecule extension. This implies supplemental control experiments to analyze this signal and to find which enzyme this signal is related. An alternative to this issue is to couple micromanipulation and fluorescence. The beauty of the magnetic tweezers is to be a very simple force clamp system applied to many molecules in parallel, but in some occasions one may want to move things around at a specific position, then optical tweezers are better adapted. Finally, good signal resolution is often difficult to achieve: while you typically get a resolution of three bases in the molecule extension measurement from one video frame to the next, one would like to reach the single base resolution. This is not impossible to achieve36, 38, 47 but this is not done with simple and easy to use systems, technical improvements might help in this direction.
DNA Unwinding in the Unpeeling Configuration
UvrD, a member of the SF1 superfamily, plays a crucial role in nucleotide excision repair and methyl directed mismatch repair.3, 4, 5 It has been shown to initiate unwinding from a 3′ end ssDNA tail, a gap or a nick and to translocate along ssDNA in a 3′‐5′ direction,48, 49 a loading configuration that can be used in the unpeeling assay previously described. Most bulk assays of its activity require the presence of single‐stranded stabilizing factors, proteins such as SSB or a high concentration of UvrD (stoichiometric ratio of enzyme/nucleotide49) which by binding to ssDNA in the wake of the enzyme prevent reannealing of the separated strands behind the advancing helicase complex. Clearly, in these conditions, it is difficult to assess the intrinsic unwinding rate, processivity or efficiency of an active enzymatic complex.
As already mentioned, in the unpeeling configuration [see Fig. 3(A)], at a high enough tension (F > F r = 25 pN) the reannealing of the strands in the wake of the enzyme is greatly slowed down.50 This allows one to study the activity of a single enzymatic complex at very low concentration (well below the enzyme's dissociation constant Kd ∼ 10 nM) and without processivity factors. As explained before [see Fig. 3(A,B)] and displayed in Figure 6, DNA unwinding (U) results in a continuous (ATP dependent) increase with time of the molecule's extension. The slope of this signal (the derivative of the extension with respect to time) yields the unwinding rate v U, its spatial extent—the number of base‐pairs unwound NU and its duration—the on‐time τ U of the enzymatic complex. As the tension on the dsDNA is not strong enough to destabilize it, upon dissociation, the DNA fork serves as a nucleation point for the quick re‐hybridization (H) of the two unwound strands, resulting in a rapid (ATP independent) shortening of the molecular extension. Bursts of increase in the molecule's extension (upon enzymatic Unwinding) followed upon enzyme dissociation by rapid shortening (re‐Hybridization) are thus often observed. We call these activity bursts, UH bursts. Figure 6 displays such a burst observed in the unzipping configuration. The large temporal separation between activity bursts (relative to their duration) makes it very unlikely that they are due to the simultaneous action of many enzymatic complexes.
Figure 6.
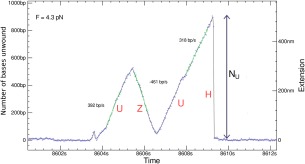
A typical signal observed with UvrD in the unzipping configuration: Unwinding (U) of the dsDNA results in a continuous increase of the extension by N U bps during a time τ U from which the mean processivity < N U>, mean rate of unwinding v U =<N U/τ U> can be determined. Unbinding of the enzyme results in re‐hybridization (H) of the two strands. Strand switching by UvrD results in an ATP dependent reZipping (Z) of the two strands in the wake of the helicase
These single‐molecule experiments yield the probability distribution of unwinding rates, which is Gaussian with a mean v U≈ 248 bps/s and standard deviation σ U ≈ 100 bps/s at 25°C. By varying the ATP concentration, the various parameters characterizing enzymatic activity (rate, processivity, on time, step‐size, etc.) may vary. While the measurement of these parameters in single‐molecule experiments can be time consuming, the parallelism of magnetic traps greatly facilitates that task.
For UvrD helicases the average rate follows a Michaelis‐Menten (MM) kinetics:
With = 275 bp/s and K M= 53 μM. The value of is about three times larger than quenched flow estimates,51 a significant but not unusual difference when comparing single‐molecule and bulk assays. Considering the caveats accompanying bulk measurements (for example the proportion of inactive enzymes or reannealing of the ssDNAs in the back of the complex) and the fact that the SM data were obtained at a rather large stretching force (F = 35pN) this discrepancy is reasonable. More to the point, the value of KM (which should be less sensitive to systematic experimental errors) is consistent with the value deduced from bulk ATPase assays.
UvrD Strands Switching and Complex Unwinding Dynamics
Surprisingly and unexpectedly, UvrD helicase exhibits another type of unwinding bursts. In many events, unwinding (U) is followed by a slow, ATP dependent re‐zipping (Z) of the two separated strands (see Fig. 6). The re‐zipping rate v Z obeys the same Michaelis‐Menten kinetics as the unwinding rate vU, suggesting that these (UZ) events are due to the enzyme switching strands, moving 3′‐5′ on the complementary ssDNA and limiting thereby the reannealing rate of the two separated strands in its wake. The similarity of the unwinding and re‐zipping rates suggests that the rate of UvrD on DNA is only slightly affected by the enzyme having to open the strands or by the fork closing in its wake and points to an active mechanism of unwinding,52 as for RecQ (see below).
The strand switching mechanism could not be deduced from bulk assays. It was first considered as a possible peculiarity of UvrD, but in fact many of the helicases studied so far in single molecule conditions display similar behavior, the exceptions being hexameric enzymes that encircle a single strand of DNA and thus cannot switch strands.53 Single molecule analysis has also revealed that helicases may lose their grip on the ssDNA on which they move leading to a slippage or rapid backward motion.54
The strand switching behavior of helicases is compatible with an inchworm mechanism: the structure of the helicase involves two RecA domains articulated with a hinge domain. The enzyme binds to ssDNA in two regions whose distance varies upon ATP binding. The inchworm mechanism implies that the binding sites of the two helicase domains on ssDNA alternate. In the normal situation, the enzyme steps on the same strand. However, if one of the domains binds to the opposite strand, the enzyme will switch strands and invert its propagation direction. Though these events are rare on a per cycle basis, they become common when the number of enzymatic cycles grows (i.e., as the enzyme processivity increases). Occasionally, some complex types of events are observed, such as Unwinding, followed by slow re‐Zipping(Z), then Unwinding and finally a rehybridization may also occur, see Figure 7. Single molecule approaches have thus revealed that helicases are molecular motors working in a very stochastic manner.
Figure 7.
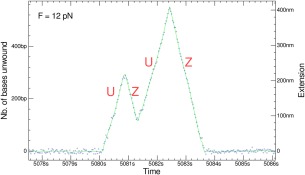
(A) More complex signals observed with UvrD resulting from a combination of the elementary processes described in the text: Unwinding (U), re‐zipping (Z). Signal obtained in the unzipping configuration
DNA Unwinding in the Unzipping Configuration
As said in paragraph 2, in the unzipping assay the helicase activity results in a larger signal than in the unpeeling assay which allows one to study helicases at much lower tensions on the molecule, in a force range that is likely more compatible with in vivo assays. Under these conditions DNA tethers also last much longer, which helps to collect more data.
UvrD has also been studied in the unzipping configuration as shown in Figures 6, 7, 8. The unwinding signal to noise is larger than in the unpeeling configuration since the sensitivity of the former is larger by a factor 2 to 5 as compared to the latter. In Figure 8, the trace in which the DNA extension increases as a function of time corresponds to hairpin unwinding by UvrD (see sketch 1 in Fig. 8), while the trace in which the DNA extension decreases as a function of time (see sketch 3 in Fig. 8) is due to the enzyme translocating on ssDNA after reaching the hairpin apex [see sketch 2 in Figs. 8 and 3(B)]. Note that in the extension decreasing trace the helicase blocks the fork that is reforming in its wake which could push the enzyme forward. The two traces thus correspond to different thermodynamic situations: in the increasing trace the helicase is working to unwind the dsDNA while in the decreasing trace the helicase delivers no work but is pushed in the back by the fork. For UvrD we observe that the rates in both situations are similar which means that the enzyme is not slowed down as it unwinds the molecule. Moreover, both rates are independent of the applied force which can help or hinder dsDNA opening. Such a behavior is the hallmark of an active helicase.52
Figure 8.
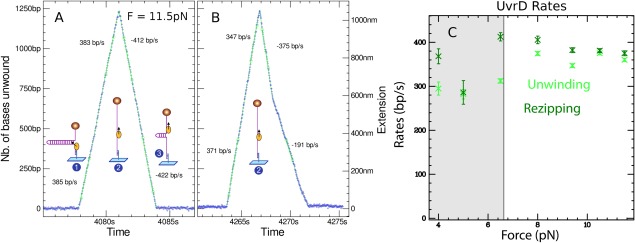
(A) Signal produced by a UvrD helicase unwinding a 1.2 kbps DNA hairpin at T = 29°C and F = 11.5 pN. Before t = 4078 s the hairpin is closed. Then (1) a UvrD helicase loads on the hairpin and starts unwinding it in a processive and fast rate (about 380 bp/s). The UvrD helicase reaches the hairpin apex (2) and pursues its translocation as it essentially travels on a ssDNA molecule. Once the helicase has passed the apex, the hairpin starts refolding until the fork bumps on the helicase (3). The extension in this phase reproduces the position of the helicase as it translocates along ssDNA at a rate of about 400 bp/s. Note that the motion is extremely regular (each point corresponds to a video frame acquired at 30 Hz). (B) A trace obtained in conditions similar to (A) but where the helicase changes speed as it re‐zips the molecule. The events with lower speed are not very frequent but do occur repeatedly, suggesting that a helicase might display different rates perhaps due to a change in its conformation. (C) Unzipping and re‐zipping rates show no significant dependence on the applied force (the gray area corresponds to forces where the statistics on traces is weaker)
These traces also illustrates the very processive behavior of UvrD. In this experiment the enzyme has traveled along 2.4 knts without falling off and with a remarkable regularity. The strand‐switching ability of UvrD has also been observed in this configuration as the enzyme can switch directions before reaching the hairpin apex. Complex bursts were also observed. The unzipping assay with UvrD also demonstrates an interesting feature of this helicase: its ability to unbind biotin from streptavidin.55 As the enzyme translocates past the hairpin apex, it can reach the end of the molecule where biotins present on the DNA are bound to streptavidin present on the magnetic bead. We observed that many beads that were anchored to a surface by a DNA hairpin would detach in the presence of UvrD. When tracking several tens of beads, a small subset remains bound and provides useful and reproducible unwinding signals (in these instances the biotin link is presumably inaccessible to UvrD).
The possibility of observing on the same trace helicase‐mediated DNA unwinding in the rising part of the extension signal and helicase‐translocation on ssDNA on the decreasing part opens interesting possibilities. The replicative helicase of the T4 replisome, gp41, behaves in a very different way56 [Fig. 9(A)] upon unwinding and upon translocation on ssDNA. The rezipping (translocation) rate is constant and independent of the tension on the DNA molecule, while the unwinding rate increases exponentially with the force, equaling the ssDNA translocation rate at a force (F ∼14 pN) large enough to open the hairpin. This behavior is compatible with the behavior of a passive helicase which is not capable of melting the DNA fork but relies on spontaneous (thermal) fluctuations of the fork to proceed forward.57
Figure 9.
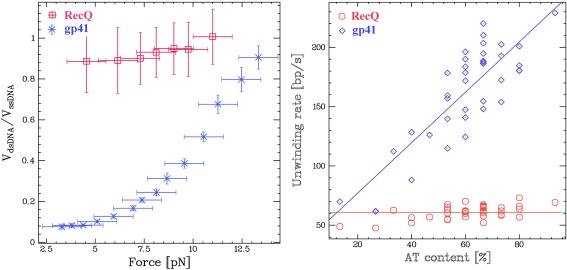
(A) Comparison of the rates of unwinding of RecQ and gp41 as a function of the tension on the hairpin molecule in the unzipping assay at saturating ATP concentrations. (B) Comparison of the rates of unwinding of RecQ and gp41 as a function of the AT content of the hairpin molecule (at F = 9 pN). The independence of the rates of unwinding by RecQ on force and AT content are compatible with an active unwinding mechanism. On the other hand, the strong dependence of the rates of unwinding by gp41 on force and AT content are indicative of a passive unwinding mechanism, whereby the helicase progression is limited by the probability of spontaneous fork opening.52
The magnetic tweezer helicase assay has also been used to study RecQ, a 3′‐5′ helicase belonging (as NS3, PriA and RecG) to the SF2 superfamily of helicases. Adding RecQ in a solution with hairpin molecules under moderate tension (F r≤ F <F c), results in processive unwinding of the hairpin at a constant rate v RecQ ≈ 80 bps/s (Fig. 9). As for UvrD, the unwinding rate of RecQ varies little with force [see Fig. 9(A)] or enzymatic concentration.52 Conversely, and in contrast with UvrD, RecQ is never observed in a situation where the fork pushes on the back of the enzyme once it has passed the hairpin apex. As the enzyme passes the apex, the force applied by the closing fork induces the enzyme to switch strands reverting into an unwinding mode.58
The rate of DNA unwinding by helicases has also been studied as a function of the local AT (or GC) content of the hairpin.59, 60, 61, 62 In that case one measures the local rate of unwinding (averaged over many unwinding traces, such as the increasing trace in Fig. 8) as a function of the molecule's extension (i.e., the position of the enzyme along the molecule). One can use that assay to distinguish [see Fig. 8(B)], between an active helicase (such as RecQ) whose rate is insensitive to the AT‐content and a passive helicase (such as gp41) whose rate increases with the local AT content of the molecule. Similar results have been obtained on the E. coli replicative helicase DnaB in the two unpeeling and unwinding configuration.63, 64 E1 helicase has also been characterized by single molecule FRET assay.65
Helicase Translocation on ssDNA
One can use the magnetic trap force modulations on a hairpin substrate to measure the rate of translocation of a helicase on an ssDNA substrate (without pushing by the closing fork as before). By mechanically and transiently increasing the force (to a value F > Fu), the hairpin can be completely opened during an unwinding event allowing the enzyme to translocate on ssDNA at mean rate vss (see Fig. 10). As the force is reduced (down to its initial value F < Fc), the hairpin rewinds. However it is blocked by the helicase and its length is shorter by an amount corresponding to the distance traveled by the enzyme. Notice that in the case of RecQ the translocation rate on ssDNA is the same as the dsDNA unwinding rate (Fig. 10), confirming the interpretation of this enzyme as an active helicase which can melt the DNA fork.56, 66, 67
Figure 10.
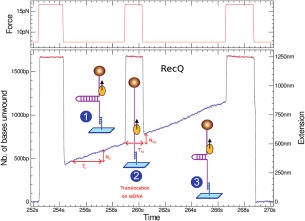
To investigate the translocation of a helicase on ssDNA the following assay has proved useful. Once the enzyme has started to unwind the hairpin fork, the force is increased such as to mechanically unzip the hairpin. The enzyme thus proceeds by translocating on ssDNA for a time T SS determined by the time span over which the hairpin is maintained open. Upon reducing the tension of the DNA molecule, the hairpin rewinds until its fork encounters the helicase. At that point the increase in the DNA's extension, N bpS, allows one to deduce the rate of translocation of the enzyme on ssDNA: v SS= N bp/T SS
Coupling between Helicase and Primase in the Primosome
DNA replication is a fundamental process in the cell, the fact that the double helix implies two strands that are anti‐parallel and that the polymerase enzyme can only copy a template along one direction leads to a complex process. In the replication fork that is opened by the replicative helicase, one strand (the leading strand) is in the correct orientation to be synthesized continuously, but the other (the lagging) strand is in the opposite orientation. To replicate it the cell uses a discontinuous synthesis process whereby it is synthesized in small stretches known as Okazaki segments. These segments are synthetized by a polymerase working opposite to the direction of the replication fork. This situation cannot proceed for long and the lagging polymerase detaches after finishing one segment to start a next one. However, the polymerase is unable to initiate the replication of its template, it needs a primer that it elongates. This primer is synthesized by a special enzyme, a primase that in the T4 virus is part of a complex with the helicase (the primosome) that encircles the lagging strand. In the T7 virus, the primase is actually a subdomain of the helicase. Whenever the helicase opens a specific sequence, the primase detects this event and starts synthesizing an RNA primer complementary to the specific sequence. This primer will eventually be used by the polymerase to start an Okazaki segment. The primase is thus a type of RNA polymerase which surprisingly synthesizes its primer opposite to direction of the helicase motion. How these two opposite activities are coupled has long been an open question. Several models had been proposed (see Fig. 11) that suggested that either the activity of the primase was blocking the translocation of the helicase [pausing model, Fig. 11(B)], or that the two enzymes dissociated transiently [disassembly model, Fig. 11(C)] or that an ssDNA loop was extruded between the two enzymes during priming [looping model, Fig. 11(D)].
Figure 11.
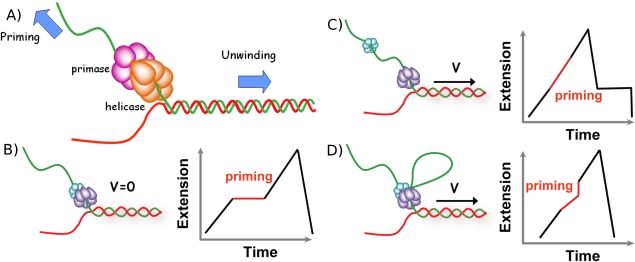
Models of primosome behavior during primer synthesis. (A) In the T4 virus, the helicase and the primase work as a complex encircling the lagging strand. However, their respective displacement directions are just opposite raising the question of how they collaborate. (B, C, and D) Schematic representation of three possible models for helicase and primase interaction during primer synthesis (left) and the real‐time DNA extension traces expected for each model (right). (B) In the pausing model the helicase temporarily stop translocating during priming. (C) In the disassembly model the primase dissociates from the helicase to synthesize a primer while the helicase continues unwinding DNA. (D) In the DNA looping model the primosome remains intact and DNA unwound during priming forms a loop
The unzipping assay has provided a means to address that issue. As we have seen, the T4 replicative helicase gp41 unwinds a DNA hairpin in a few seconds. Since the helicase encircles the DNA it displays a burst with a rising edge corresponding to the helicase unwinding the hairpin and a falling edge corresponding to the helicase translocating on the ssDNA while a DNA fork reforms in its wake. The primase is performing a task that is less easy to observe: if we supply NTPs, the primase will lay down a 5mer RNA oligonucleotide each time it encounters a DNA sequence starting by GTT or GCT. We have constructed two hairpins with a small number of priming sites located in only one of the strands.68 If we add the primase to a buffer containing helicase and ATP only, the primase is unable to initiate a primer since that requires CTP (Cytidine triphosphate). In this situation, we observe bursts of helicase activity similar to those observed with helicase alone. This test shows that a primase does not alter the helicase motion when it is inactive.
If we add the NTPs and use a hairpin with priming sites in the lagging strand only that is with priming sites that are encountered by the primosome when opening the DNA, we observe two noticeable features in the activity bursts. As shown in Figure 12(B), the rising edge of the burst is similar to a helicase burst but on the falling edge we observe a blockage where the hairpin remains open at a given position for sometimes. This phenomenon is not seen if we use a hairpin having no priming sites on the lagging strand. The blockage positions occur randomly in time but at positions corresponding to the expected priming sites. The interpretation of this observation is as follows: as the helicase opens the hairpin, whenever a priming site is detected, the primase synthesizes a primer that remains hybridized to its template and is blocking the rehybridization of the hairpin in the wake of the helicase (once it has passed the apex). The hybridized primer is in fact linked to a part of the primase that stabilizes it on its DNA template. This observation validates the disassembly model for the primosome whereby the primase dissociates from the helicase as it synthesizes its primer. This priming activity is clearly a stochastic process where the possible priming sites randomly trigger the priming activity: in many instances a priming site is not associated with a priming event. Thus, the blockage positions although very well localized appear in random order and the duration of the blockages follows a Poisson distribution with a mean duration of a few seconds.
Figure 12.
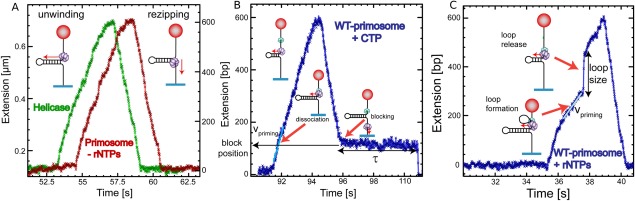
Primer synthesis by primosome depends on rNTP concentration. (A) Experimental traces corresponding to the gp41 helicase activity (green) and the wt primosome activity (red) in the absence of rNTPs. (B) Experimental trace displaying characteristics (unwinding velocity during priming, position and lifetime of the block) for the primosome disassembly model. DNA looping mechanism for primer synthesis. (C) Experimental trace displaying characteristics (unwinding velocity during priming and the loop size) for the DNA looping model.68
We also observe a second type of events where the rising edge of the burst displays a special feature shown in Figure 14(C). Starting at a priming site, the measured increase in extension slowed down by a factor two, before presenting a jump in extension and recovering its original rate of lengthening. This event may be explained by the extrusion of a loop between the helicase and the primase. During this loop extrusion, the lengthening of the molecule is only due to the other opened strand, as the sequestered loop does not participate in the extension signal. As the primase dissociates from the helicase, the extension increases suddenly. This event is compatible with the looping model. The size of the loop is typically 200 or 300 nts.
Figure 14.
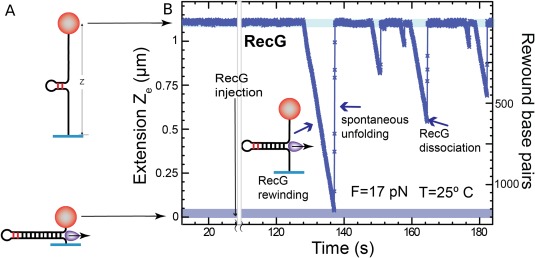
(A) The substrate for the single molecule study of the rewinding activity of a helicase is an opened hairpin with a small stem loop at its end (that serves as a loading site for the enzyme). (B) Upon enzymes rewinding the extension of the hairpin (maintained under a high tension) is observed to decrease. Sudden increases in extension are due to dissociation of the enzyme from its substrate and subsequent force‐induced unfolding of the hairpin.80
This single molecule assay of the primosome has provided valuable information which nicely complement bulk assays, in particular we can definitely rule out the pausing model which was never seen. The stochastic nature of the primosome was not fully expected. While it seems reasonable that not all priming sites lead to a priming event, it is surprising that two priming processes occur in a random manner. We found that in our conditions, upon priming a primase will more often dissociate from the helicase that extrude a loop, but the ratio between the two behaviors may depend on replisome cofactors.
Coupling between Helicases and Polymerases during Replication
During the replication of a DNA molecule, the rate of DNA unwinding and DNA polymerization must be tightly coupled within the replication complex. The situation has been first investigated by De Lagoutte and Von Hippel, more recently the T4 replisome description is discussed in Refs. 69 and 70, while the T7 replisome has been also extensively studied.71, 72, 73, 74 In the case of the T4 replisome1, 2 using as a template, a dsDNA molecule ending with an open fork and a primer on the leading strand (mimicking the natural replication fork) they have shown the following surprising result:
The helicase gp41 alone is very inefficient in opening DNA, its rate is a few tens of bp/s and its processivity is reduced to ∼100 bases
The T4 polymerase alone is not active since it lacks strand‐displacement activity
But the combination of the helicase and the polymerase (holoenzyme) together results in a rapid elongation of the primer at 400 bp/s over several kbs.
While the inactivity of the T4 holoenzyme alone is understood, the inefficiency of the helicase alone is more surprising: one would imagine that a replicative helicase which eventually opens the entire genome, is a heavy‐duty enzyme that does not require the collaboration of the polymerase to unwind DNA. An understanding of the coupling between helicase and polymerase was made possible using single molecule assays.
Single molecule manipulations, particularly in the unzipping configuration, offers a convenient and versatile assay to study that coupling. First, each component of the complex (e.g., helicase, polymerase, primase, etc.) can be studied in isolation from the others. As shown previously the replicative helicase from phage T4, gp41, is a passive helicase with a slow unwinding rate at low tensions, a rate that can be 10 times slower than its unhindered translocation rate on ssDNA of 600nt/sec [see Fig. 9(B)]. Thus, the single molecule approach confirmed the bulk results that the helicase alone is inefficient at low forces, but it also shows that the full speed of the helicase can be recovered provided it is assisted by a strong force.75 Conversely, the T4 polymerase, gp43, cannot work in a strand displacement mode. However, studying the T4 polymerase in the unzipping configuration with a primer hybridized on one strand revealed a surprising result: If a pulling force F > 10 pN is applied, the polymerase can elongate the primer with a substantial rate of 250bp/s, which suggests that if the fork is slightly destabilized (by the tension) the polymerase can synthesize a new complementary strand by displacing the old one in front of it. At lower tension on the hairpin the polymerase stalls and can even go into exonuclease mode, removing the strand it has just synthesized [see Fig. 13(C)]. When the two enzymes (helicase and polymerase) are assembled on a DNA hairpin,76 at low forces their joint action allows the helicase to unwind at a rate similar to the maximal rate of the gp43 polymerase [Fig. 13(A)]. At higher tensions, the two enzymes decouple and each proceeds at the rate measured in isolation [Fig. 13(B)].
Figure 13.
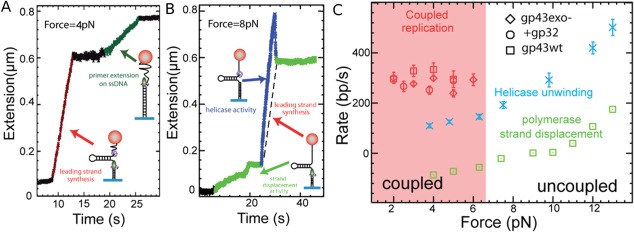
Single molecule studies of the coupling between the T4 helicase (gp41) and the T4 polymerase (gp43). (A) At low forces (F < 7pN), the polymerase synthesizes the complementary strand as the helicase unwinds the molecule, resulting in a fast increase (red) of the molecule extension. As the helicase and polymerase meet head‐on at the hairpin apex, they stall (black). The helicase then falls off and the polymerase resumes its activity on the remaining ssDNA (the upper complementary strand of the hairpin). (B) At higher forces (F > 7pN), the helicase unwinds DNA (blue) independently of the proximity of the polymerase which synthesizes the complementary strand on the unwound ssDNA. As the helicase falls off (at t = 30 s), the hairpin re‐hybridizes up to the point where the polymerase is (the dashed line represents the polymerase rate during the uncoupled helicase unwinding). The polymerase resumes polymerization in the very inefficient strand displacement mode (green). (C) Measured rates of unwinding and strand‐synthesis by gp41 and gp43 alone or when they are coupled (with or without the SSB protein, gp32). Notice that at low forces (F < 5pN) and in the absence of helicase the polymerase switches into exonuclease mode (its rate is negative).76
A simple mechanical model accounts for these observations. It assumes that at low forces the polymerase destabilizes the fork enough to allow the passive gp41 helicase to proceed with DNA unwinding. Its rate is then determined by the maximal rate of polymerization (it cannot unwind faster as it will decouple from the polymerase and slow down). At higher tensions, the fork is sufficiently destabilized that the helicase does not need the polymerase to unwind the molecule. It unwinds DNA at a rate which is the one measured at that tension in the absence of polymerase [Fig. 13(B)]. In the wake of the helicase, on the generated ssDNA the polymerase synthesizes the complementary strand [see Fig. 13(B)]. As the helicase unbinds from its substrate, the hairpin reforms up to the polymerase whose action is slowed down or stalled by the fork ahead.
This purely mechanical coupling between helicase and polymerase, in contrast with a biochemical (for example allosteric) coupling, suggests that helicases and polymerases from different species could work together. This prediction has been validated by showing that the phage T4 helicase and the phage T7 polymerase could indeed complement each other to form an efficient replicative complex. These results suggest the presence in the T4 polymerase of a specific region that destabilizes the fork, increasing unpeeling fluctuations that speed up the gp41 helicase.
Finally, this mechanical coupling provides a rational for the difference existing between passive and active helicases: the proximity of the T4 polymerase effectively switches the gp41 passive helicase into an active mode, a coupling that prevents opening of the fork without DNA synthesis activity. Very similar coupling has been observed in the T7 replisome.73, 77, 78
Helicase Rewinding of DNA Hairpin
Some helicases, such as RecG and UvsW, possess the surprising ability to rewind an unzipped DNA hairpin against a considerable force (up to about 30 pN).79, 80 This activity is required in the rescue of a stalled replication fork. If a DNA damage exists on the leading strand of a replication fork, it will stop the leading strand polymerase posing a serious threat to the cell. Even a phage such as T4 has a special helicase devoted to address that issue. If such damage blocks the leading polymerase, one way to bypass the problem is to replace the defective template by the copy of its complementary strand done by the lagging strand polymerase, this implies regressing the stalled fork and generating a so‐called “chicken‐foot” DNA structure. Regenerating the DNA fork allows the replication complex to bypass the damaged area and to proceed with replication. This fascinating mechanism led to a strong debate as it seemed that the coordination of the different events was so crucial that its occurrence would be highly improbable. Indeed, it was difficult to understand how an enzyme would reverse the replication fork when it is stalled, how the reverse process will happen once the elongation of the critical bases had occurred and how many different enzymes were required. Using a single molecule approach, we found that this complex process is in fact the result of the collaboration of just two enzymes engaged in a stochastic ballet. For the T4 virus, these enzymes are the UvsW helicase and the gp43 polymerase (in E. coli, RecG replaces UvsW)
To study these enzymes an unzipping assay was used. The DNA hairpin was designed to have a GC‐rich segment close to its apex. The tension in the hairpin (F >14 pN) was adjusted such as to partially open it (up to the more stable GC‐rich segment close to the apex, see the rise from 15 to 17 pN in the hairpin opening trace of Fig. 4) leaving a small stem loop at its end that serves as a loading site for the helicase [Fig. 14(A)]. The rewinding (re‐zipping) activity of the enzyme against the tension pulling on the DNA can be monitored via the decrease in extension of the molecule. Detachment of the enzyme from its substrate is observed as a rapid increase in extension resulting from the tension‐induced opening of the hairpin [Fig. 14(B)].
Except for the inversion in the extension signal, the processivity and the rate of these enzymes can be deduced in a similar manner as for the unwinding activity of more common helicases.
Converting a Three‐Way Junction to a Holliday Junction
Helicases not only translocate on ssDNA but are also involved in the conversion of a three‐way junction into a “chicken‐foot” Holliday junction (HJ) and in the migration of this junction. This process is a key aspect of the fork regression error correction mechanism. Using a variant of the unzipping configuration it has been possible to study this process in real time.
The preparation of a DNA template mimicking a stalled replication is sketched in Figure 15(A). First a DNA hairpin with a built‐in primer (1) is used to anchor a bead to a surface. The DNA hairpin contains three consecutive Locked Nucleic Acids (LNA) bases a distance 40 bps from its apex. Adding the T4 polymerase and increasing the force (2) allows for primer extension until the polymerase bumps into the LNA bases that stop the elongation. An oligonucleotide with a sequence complementary to the lagging strand (3) is then injected. As the tension on the molecule is reduced the hairpin refolds partially mimicking a stalled replication fork (4) with the LNA playing the role of the DNA damage.
Figure 15.
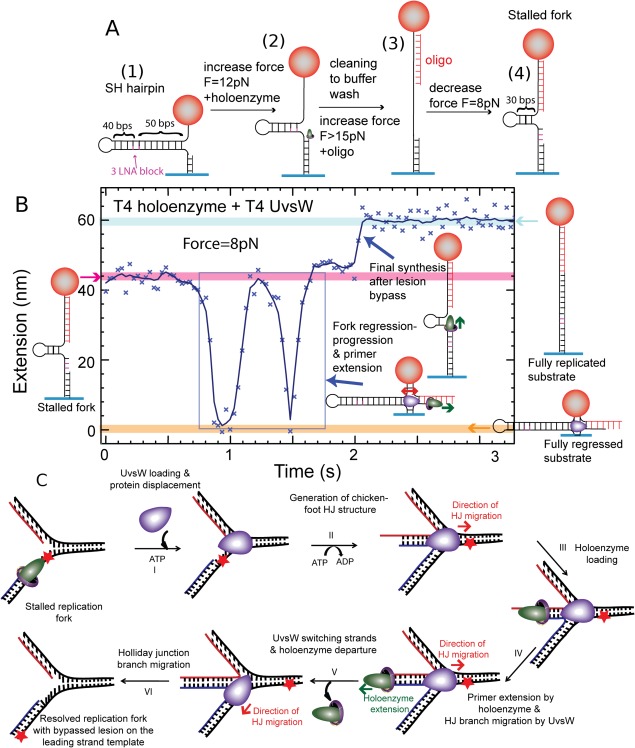
Reconstruction of the template switching pathway. (A) Construction of a stalled fork substrate with a LNA block at the leading strand, mimicking a damage. (B) Experimental trace at F = 8 pN displaying the extension z(t) with ATP, dNTPs, UvsW and T4 holoenzyme starting with the stalled fork and ending with the fully replicated substrate. (C) Schematic of the repair of a stalled replication fork by its regression.79
To test the efficiency of a helicase in the rescue of this stalled replication fork by converting this three‐way junction into a Holliday junction, the UvsW helicase, the gp43 polymerase and ATP were flowed in. As in the previous experiment, UvsW starts refolding the hairpin. But here the two arms are in fact dsDNA and the helicase first converts the three‐way junction into a Holiday junction that it displaces. This is evidenced by the rapid decrease of the molecule's extension as the bead approaches the glass surface. During this process, the strand which elongation was blocked by the presence of improper LNA bases, is re‐hybridized to a strand that has no damage. This offers the possibility for the elongation to restart by polymerase elongation of a proper template. UvsW displays an interesting behavior: it reverts the direction of the Holiday junction migration in a random manner. Thus, the bead goes up again after its first downwards motion. On Figure 15(B), we observe two consecutive motions up and down illustrating this behavior. There is no special coordination between UvsW and the T4 polymerase, so that the polymerase will not load automatically as the fork is reverted but it can do so. In Figure 15(B), on the second fork reversal, the T4 polymerase was able to load and elongate its template so that the lesion was actually passed. As UvsW switches back the chicken foot to a three‐way junction, the fork replication can restart.
This error correction mechanism is a perfect illustration of a stochastic collaboration between two enzymes: by moving the Holiday junction forward and backward, UvsW provides an opportunity for the gp43 polymerase to elongate the strand that was blocked. When the proper complementary strand has been synthesized the fork is ready to reload the replication helicase gp41 and to re‐initiate replication on a proper template. We observed that the gp41 helicase has priority over UvsW so that in normal fork progression, UvsW does not revert the fork. When a damage blocks the replication fork, the uncoupling between the polymerase and the replicative helicase leads to the unbinding of the helicase, which allows for the action of UvsW.79, 81
Helicase Unwinding of RNA/DNA Duplexes
The world of RNA helicases is more diverse than the DNA helicases homologs. The RNA helicase activities target specific RNA structures which are not simply double‐stranded RNA, a structure which is rather uncommon in the cell. The RNA helicase substrates might be specific secondary structures (local hairpins or bulges) but also ribonucleoprotein complexes. In eukaryotes, RNA helicases are implicated in every molecular process involving RNAs and notably transcription and all the post‐transcriptional events including pre‐mRNA processing, mRNA transport, translation and degradation.82, 83 Some RNA helicases are really surprising as they may have sometimes extremely low processivities;84 for instance, DEAD box helicases are involved in disrupting just a few nucleotides and do not translocate on their template while others simply serve as RNA clamp.85
We can also test RNA helicases with our magnetic tweezers using the same protocol as that for DNA helicases. However, preparing an RNA template either in the peeling or unzipping configuration is more difficult than the equivalent DNA template. The panel of enzymes handling RNA in the laboratory is not as rich as for DNA, no restriction enzymes are available, ligases are less efficient, and incorporating modified bases is not obvious, though one can find interesting recipes.86 RNA also has the ability to form more complex secondary structures than those expected from Watson‐Crick pairings, leading to a difficult molecular design. Finally, RNAs are prone to degradation by very ubiquitous ribonucleases. Preparing a complete RNAse‐free single molecule flow cell is a challenge. Consequently, the lifetime of an RNA construct in a magnetic tweezers set‐up is usually substantially shorter than a DNA one, at least in our hands. A useful alternative when the helicase works on a RNA/DNA hybrid is to use a DNA template molecule as a holding device and to hybridize RNA single stranded molecules to the DNA.
We have recently studied the human Upf1 RNA helicase87 that is essential to survey the integrity of eukaryotic mRNAs by the process of nonsense‐mediated mRNA decay (NMD).88 This helicase has some interesting features: it translocates slowly but presents a surprisingly high processivity (Fig. 16). It works both on RNA and DNA templates without significant differences. Upf1 translocates at a few bases per second with an irregular rate. The Upf1 enzyme is regulated by several factors including the internal CH domain [Fig. 16(A)] which inhibits the helicase activity when not in the correct configuration and the NMD cofactor Upf2 which is needed for helicase activity. The existence of these two mechanisms is presumably responsible for the irregular activity of Upf1. The single molecule approach allowed us to characterize the regulatory effect of Upf2. First, in the absence of the regulatory CH domain, Upf1‐HD binds to its template very firmly and keeps on moving for very long distance: it is able to completely unwind a 156 bp RNA hairpin [Fig. 16(B)] as well as a 1.2kbps DNA hairpin. When we tested the enzyme with its regulatory CH domain, Upf1‐CH‐HD was seen mostly binding ssDNA and blocking the DNA fork (a small fraction of the enzymes shown some helicase activity presumably because the CH domain may have different configurations and thus did not block all the enzyme). Interestingly, adding Upf2 clearly changed the pattern as most complexes became active with a normal helicase behavior. The activity is compatible with Upf2 flipping the CH domain in the active configuration. This was confirmed by using the Upf1‐CH‐HD‐F192E mutant where the CH domain is flipped in the active configuration permanently by the mutation.89 The ability of testing helicase co‐factors is fundamental to better characterize RNA helicases which often act as part of protein complexes in which RNA helicases are regulated by one or several cofactors. Using our single molecule assay, we estimated that the Upf1 processivity is extremely large reaching 16 kbps. This very high processivity suggests that Upf1 might remodel numerous protein‐RNA interactions to trigger the degradation of aberrant mRNAs by NMD.87
Figure 16.
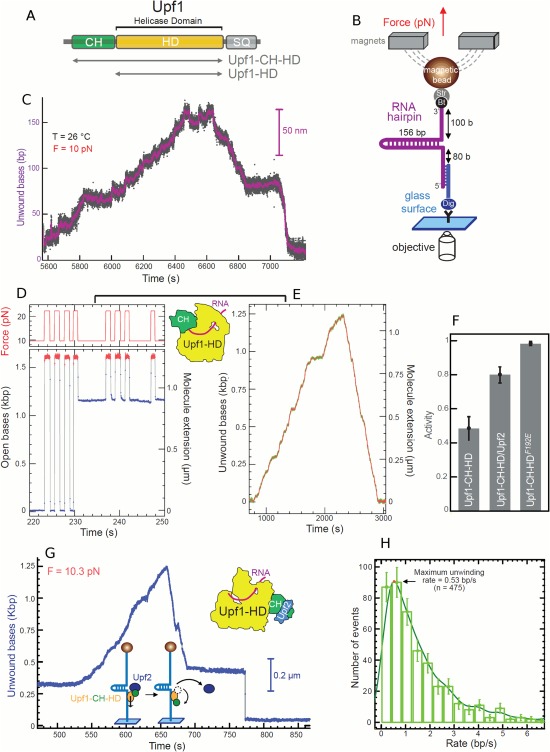
Upf1 helicase activity on RNA. (A) Organization of Upf1 and truncated versions (gray arrows) used in our study, Upf1‐HD and Upf1‐CH‐HD. (B) Schematic representation of the RNA used for the MT set up. (C) Experimental trace showing the activity of Upf1‐HD in saturating concentration of ATP. The number of unwound bases is deduced from the molecular extension Z(t) obtained at F = 10pN. From 5600 s to 6440 s, the helicase unwound the 156bp RNA hairpin. From 6640 s to 7200 s, the RNA hairpin refolded, while Upf1‐HD translocated on ssRNA reaching the 3′extremity. (D, E, and F) The binding of Upf2 to CH domain activates Upf1‐CH‐HD unwinding and translocation. (D, E) Experimental traces corresponding to the two types of enzymatic activity detected for Upf1 on a DNA hairpin. (D) Upf1 binds on ssDNA (starting at 231 s), blocking the re‐zipping of the hairpin. (E) The enzyme is active. (F) Histogram of relative activity of Upf1‐CH‐HD, Upf1‐CH‐HD/Upf2 complex and Upf1F192E mutant. (G) Trace of human Upf1‐CH‐HD/Upf2 complex unwinding steadily and completely the DNA molecule, passing the apex, pursuing its translocation and refolding the DNA hairpin. At t = 675 s, the complex makes two strand switching events before finally stopping its activity leaving the hairpin blocked (at t = 687 s). This trace is atypical since the Upf1‐CH‐HD/Upf2 complex translocates for some time before stalling; for most bursts, the complex stops very quickly after passing the apex. (H) Distribution of instantaneous unwinding rate of the Upf1‐CH‐HD/Upf2 complex.87
Conclusions and Perspectives
Single molecule manipulations are now used either to characterize helicase stepping90 or the role of their subdomains and their link91 or to understand the coupling between enzymes required in many biological situations. Here we have shown examples where two enzymes interact: this is the case of a primase and a helicase, a helicase and a polymerase in leading strand synthesis and in the fork regression mechanism. The obvious advantage of these single molecule studies is the ability to follow in real time the different interactions, revealing their underlying dynamics. More complex systems have also been investigated which pave the way for further deeper studies. The Van Oijen group has studied the entire replisome of T792 and E. coli. These experiments involve many enzymatic partners and the probability of their successful assembly is low. Hence these authors have used extensive parallelism (tracking several 10,000 beads) to observe a small set of clear events where all the partners in the complex were active. This has allowed them to study the collaborative behavior of the different elements of the replisome.
In the same spirit, the collision of the replication fork with the Tus‐Ter replication termination has been investigated using single molecule micromanipulation.93 DNA repair mechanism is also a subject where micromanipulation is bringing valuable information such as the way used by polymerase to overcome DNA damage94 or how stalled RNA‐pol are restarted.95 The observation of helicase activity on a specific DNA quadruplex96 opens new avenues of investigation.97 However, as soon as several enzymes are involved in a biological process, the probability of assembly of a functional complex in single molecule assays decreases dramatically. The success of this approach thus relies on the development of high throughput single‐molecule manipulation assays98 which the inherent parallelism of magnetic traps is ideally poised to provide.
Acknowledgments
We thank N. Desprat, J. Ouellet, M.M. Spiering, S.J. Benkovic and C. Andre, for useful comments on the manuscript.
References
- 1. Delagoutte E, von Hippel PH (2002) Helicase mechanisms and the coupling of helicases within macromolecular machines Part 1: Structures and properties of isolated helicases. Quart Rev Biophys 35:431–478. [DOI] [PubMed] [Google Scholar]
- 2. Delagoutte E, von Hippel PH (2003) Helicase mechanisms and the coupling of helicases within macromolecular machines ‐ Part II: Integration of helicases into cellular processes. Quart Rev Biophys 36:1–69. [DOI] [PubMed] [Google Scholar]
- 3. Lohman TM, Bjornson KP (1996) Mechanisms of helicase‐catalyzed unwinding. Annu Rev Biochem 65:169–214. [DOI] [PubMed] [Google Scholar]
- 4. West SC (1996) DNA helicases: New breeds of translocating motors and molecular pumps. Cell 86:177–180. [DOI] [PubMed] [Google Scholar]
- 5. Soultanas P, Wigley DB (2001) Unwinding the ‘Gordian knot’ of helicase action. Trends Biochem Sci 26:47–54. [DOI] [PubMed] [Google Scholar]
- 6. Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2004) Molecular biology of the gene. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 7. Patel SS, Picha KM (2000) Structure and function of hexameric helicases. Ann Rev Biochem 69:651. [DOI] [PubMed] [Google Scholar]
- 8.DNA Helicases and DNA Motor Proteins, Maria Spies, Volume 973 of the series Advances in Experimental Medicine and Biology; Springer (2012)
- 9. Jankowsky E (2012) Methods in enzymology. Preface. Methods Enzymol 511:xix–xxx. doi:10.1016/B978-0-12-396546-2.00029-2. [DOI] [PubMed] [Google Scholar]
- 10. Gorbalenya AE, Koonin EV (1993) Helicase amino acid comparisons and structure function relationship. Curr Opin Struct Biol 3:419–429. [Google Scholar]
- 11. Singleton MR, Dillingham MS, Wigley DB (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. [DOI] [PubMed] [Google Scholar]
- 12. Fairman‐Williams ME, Guenther U‐P, Jankowsky E (2010) SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benkovic SJ, Raney KD (2011) Mechanisms: molecular machines. Curr Opin Chem Biol 15:577–579. [DOI] [PubMed] [Google Scholar]
- 14. Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97:75–84. [DOI] [PubMed] [Google Scholar]
- 15. Wigley DB (2007) RecBCD: the supercar of DNA repair. Cell 131:651–653. [DOI] [PubMed] [Google Scholar]
- 16. Nanduri B, Byrd AK, Eoff RL, Tackett AJ, Raney KD (2002) Pre‐steady‐state DNA unwinding by bacteriophage T4 Dda helicase reveals a monomeric molecular motor. Proc Natl Acad Sci USA 99:14722–14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raney KD, Byrd AK, Aarattuthodiyil S (2013) Structure and mechanisms of SF1 DNA helicases. Adv Exp Med Biol 767:17–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee JY, Yang W (2006) UvrD helicase unwinds DNA one base pair at a time by a two‐part power stroke. Cell 127:1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dillingham MS, Kowalczykowski SC (2008) RecBCD enzyme and the repair of double‐stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang W (2010) Lessons learned from UvrD helicase: mechanism for directional movement. Annu Rev Biophys 39:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jia H, Korolev S, Niedziela‐Majka A, Maluf NK, Gauss GH, Myong S, Ha T, Waksman G, Lohman TM (2011) Rotations of the 2B sub‐domain of E. coli UvrD helicase/translocase coupled to nucleotide and DNA binding. J Mol Biol 411:633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manthei KA, Hill MC, Burke JE, Butcher SE, Keck JL (2015) Structural mechanisms of DNA bnding and unwinding in bacterial RecQ helicases. Proc Natl Acad Sci USA 114:4292–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. von Hippel PH, Delagoutte E (2001) “A general model for nucleic acid helicases and their coupling” within macromolecular machines. Cell 104:177–190. [DOI] [PubMed] [Google Scholar]
- 24. Liu B, Baskin RJ, Kowalczykowski SC (2013) DNA unwinding heterogeneity by RecBCD results from static molecules able to equilibrate. Nature 500:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu CG, Bradford C, Lohman TM (2010) Escherichia coli RecBC helicase has two translocase activities controlled by a single ATPase motor. Nat Struct Mol Biol 17:1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roman LJ, Kowalczykowski SC (1989) Characterization of the helicase activity of the E. coli RecBCD enzyme using a novel helicase assay. Biochemistry 28:2863–2873. [DOI] [PubMed] [Google Scholar]
- 27. Roman LJ, Eggleston AK, Kowalczykowski SC (1992) Processivity of the DNA helicase activity of Escherichia‐coli RecBCD enzymze. J Biol Chem 267:4207–4214. [PubMed] [Google Scholar]
- 28. Roman LJ, Kowalczykowski SC (1989) Characterization of the ATPase activity of the E. coli RecBCD enzyme: relationship of ATP hydrolysis to the unwinding of duplex DNA. Biochemistry 28:2873–2881. [DOI] [PubMed] [Google Scholar]
- 29. Lucius AL, Vindigni A, Gregorian R, Ali JA, Taylor AF, Smith GR, Lohman TM (2002) DNA unwinding step‐size of E. coli RecBCD helicase determined from single turnover chemical quenched‐flow kinetic studies. J Mol Biol 324:409–428. [DOI] [PubMed] [Google Scholar]
- 30. Lucius AL, Jason Wong C, Lohman TM (2004) Fluorescence stopped‐flow studies of single turnover kinetics of E. coli RecBCD helicase‐catalyzed DNA unwinding. J Mol Biol 339:731–750. [DOI] [PubMed] [Google Scholar]
- 31. Lucius AL, Vindigni A, Gregorian R, Ali JA, Taylor AF, Smith GR, Lohman TM (1989) DNA unwinding step‐size of E. coli RecBCD helicase determined from single turnover chemical quenched‐flow kinetic studies. Biochemistry 28:2863–2873. [DOI] [PubMed] [Google Scholar]
- 32. Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, Baskin RJ (2001) Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature 409:374–378. [DOI] [PubMed] [Google Scholar]
- 33. Handa N, Bianco PR, Baskin RJ, Kowalczykowski SC (2005) Direct visualization of RecBCD movement reveals cotranslocation of the RecD motor after Chi recognition. Mol Cell 17:771–750. [DOI] [PubMed] [Google Scholar]
- 34. Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, Kowalczykowski SC (2003) A molecular throttle: The recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell 114:647–654. [DOI] [PubMed] [Google Scholar]
- 35. Strick T, Allemand JF, Bensimon D, Bensimon A, Croquette V (1996) The elasticity of a single supercoiled DNA molecule. Science 271:835–1837. [DOI] [PubMed] [Google Scholar]
- 36. Lansdorp BM, Tabrizi SJ, Dittmore A, Saleh OA (2013) A high‐speed magnetic tweezer beyond 10,000 frames per second. Rev Sci Instrum 84:044301. [DOI] [PubMed] [Google Scholar]
- 37. Lee KS, Balci H, Jia H, Lohman TM, Ha T (2013) Direct imaging of single UvrD helicase dynamics on long single‐stranded DNA. Nat Commun 4:1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dulin D, Cui T, Cnossen J, Docter MW, Lipfert J, Dekker NH (2015) High spatiotemporal resolution magnetic tweezers for single‐molecule force spectroscopy: Calibration and applications to DNA dynamics. Biophys J 109:2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Einstein A (1956) Investigation of the Brownian theory of movement. Mineola, New York: Dover Publication. [Google Scholar]
- 40. Reif F (1965) Fundamentals of statistical and thermal physics. New York: McGraw‐Hill. [Google Scholar]
- 41. Lionnet T, Allemand JF, Revyakin A, Strick TR, Saleh OA, Bensimon D, Croquette V, Single‐molecule studies using magnetic traps In: Ha T, Selvin PR, Eds. (2008) Single‐molecule techniques: A laboratory manual. Cold Spring Harbor, New York: CSHL Press. [DOI] [PubMed] [Google Scholar]
- 42. Allemand JF (1997) Micromanipulation de molécules d'ADN isolées, Thèse Université Pierre et Marie Curie.
- 43. Velthuis AJ, Kerssemakers JW, Lipfert J, Dekker NH (2010) Quantitative guidelines for force calibration through spectral analysis of magnetic tweezers data. Biophys J 99:1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lansdorp BM, Saleh OA (2014) Power spectrum and Allan variance methods for calibrating single‐molecule video‐tracking instruments. Rev Sci Instrum 83:025115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Vlaminck W, Henighan T, van Loenhout MTJ, Pfeiffer I, Huijts J, Kerssemakers JWJ, Katan AJ, van Langen‐Suurling A, van der Drift E, Wyman C, Dekker C (2011) Highly parallel magnetic tweezers by targeted DNA tethering. Nano Lett 11:5489–5493. [DOI] [PubMed] [Google Scholar]
- 46. Plénat T, Tardin C, Rousseau P, Salomé L (2012) High‐throughput single‐molecule analysis of DNA–protein interactions by tethered particle motion. Nucleic Acids Res 40:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kemmerich FE, Kasaciunaite K, Seidel R (2016) Modular magnetic tweezers for single‐molecule characterizations of helicases. Methods 108:4–13. [DOI] [PubMed] [Google Scholar]
- 48. Matson SW (1986) Escherichia coli helicase ii (uvrd gene product) translocates unidirectionnaly in a 3' to 5' direction. J Biol Chem 261:10169–10175. [PubMed] [Google Scholar]
- 49. Runyon GT, Lohman TM (1989) Escherichia coli helicase ii (UvrD) protein can completely unwind fully duplex linear and nicked circular DNA. J Biol Chem 264:17502–17512. [PubMed] [Google Scholar]
- 50. Dessinges M‐N, Lionnet T, Xi X, Bensimon D, Croquette V (2004) Single molecule assay reveals strand switching and enhanced processivity of UvrD. Proc Natl Acad SCI USA 101:6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ali JA, Lohman TM (1997) Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science 275:377–380. [DOI] [PubMed] [Google Scholar]
- 52. Manosas M, Xi X, Bensimon D, Croquette V (2010) Active and passive mechanisms of helicases. Nucleic Acids Res 38:5518–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xie P (2016) Dynamics of monomeric and hexameric helicases. Biophys Chem 211:49–58. [DOI] [PubMed] [Google Scholar]
- 54. Sun B, Johnson DS, Patel G, Smith BY, Pandey M, Patel SS, Wang MD (2011) ATP‐induced helicase slippage reveals highly coordinated subunits. Nature 478:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morris PD, Byrd AK, Tackett AJ, Cameron CE, Tanega P, Ott R, Fanning E, Raney KD (2002) Hepatitis C virus NS3 and simian virus 40 T antigen helicases displace streptavidin from 5′‐biotinylated oligonucleotides but not from 3′‐biotinylated oligonucleotides: evidence for directional bias in translocation on single‐stranded DNA. Biochemistry 7:2372–2378. [DOI] [PubMed] [Google Scholar]
- 56. Lionnet T, Spiering MM, Benkovic SC, Bensimon D, Croquette V (2007) Real‐time observation of t4 gp41 helicase reveals a passive unwinding mechanism. Proc Natl Acad Sci USA 104:19790–19795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Betterton MD, Jülicher F (2005) Opening of nucleic‐acid double strands by helicases: active versus passive opening. Phys Rev E Stat Nonlin Soft Matter Phys 71:011904. Erratum in: Phys Rev E Stat Nonlin Soft Matter Phys 72:029906. [DOI] [PubMed] [Google Scholar]
- 58. Klaue D, Kobbe D, Kemmerich F, Kozikowska A, Puchta H, Seidel R (2013) Fork sensing and strand switching control antagonistic activities of RecQ helicases. Nat Commun 4:2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qi Z, Pugh RA, Spies M, Chemla YR (2013) Sequence‐dependent base pair stepping dynamics in XPD helicase unwinding. eLife 2:e00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cheng W, Dumont S, Tinoco I, Bustamante C (2007) NS3 helicase actively separates RNA strands and senses sequence barriers ahead of the opening fork. Proc Natl Acad Sci USA 104:13954–13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Syed S, Pandey M, Patel SS, Ha T (2014) Single‐molecule fluorescence reveals the unwinding stepping mechanism of replicative helicase. Cell Rep 6:1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Byrd AK, Matlock DL, Bagchi B, Aarattuthodiy S, Harrison D, Croquette V, Raney KD (2012) Dda helicase tightly couples translocation on single‐stranded DNA to unwinding of duplex DNA: Dda is an optimally active helices. J Mol Biol 420:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ribeck N, Kaplan DL, Bruck I, Saleh OA (2010) DnaB helicase activity is modulated by DNA geometry and force. Biophys J 99:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ribeck N, Saleh OA (2013) DNA unwinding by ring‐shaped T4 helicase gp41 is hindered by tension on the occluded strand. PLoS One 8:e79237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee S‐J, Syedd S, Enemark EJ, Schuckc S, Stenlundc A, Ha T, Joshua‐Tor L (2014) Dynamic look at DNA unwinding by a replicative helicase. Proc Natl Acad Sci USA 111:E827–E835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Johnson DS, Bai L, Smith BY, Patel SS, Wang MD (2007) Single molecule studies reveal dynamics of DNA unwinding by the ring‐shaped T7 helicase. Cell 129:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ha T, Kozlov AG, Lohman TM (2012) Single‐molecule views of protein movement on single‐stranded DNA. Annu Rev Biophys 41:295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, Croquette V (2009) Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat Chem Biol 5:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen D, Yue H, Spiering MM, Benkovic SJ (2013) Insights into okazaki fragment synthesis by the T4 replisome the fate of lagging‐strand holoenzyme components and their influence on Okazaki fragment size. J Biol Chem 288:20807–20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Noble E, Spiering MM, Benkovic SJ (2015) Coordinated DNA replication by the bacteriophage T4 peplisome. Viruses 7:3186–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Patel SS, Pandey M, Nandakumar D (2011) Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Curr Opin Chem Biol 15:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee S‐J, Richardson CC (2011) Choreography of bacteriophage T7 DNA replication. Curr Opin Chem Biol 15:580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pandey M, Patel SS (2014) Helicase and polymerase move together close to the fork junction and copy DNA in one‐nucleotide steps. Cell Rep 6:1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Geertsema HJ, van Oijen AM (2013) A single‐molecule view of DNA replication: The dynamic nature of multi‐protein complexes revealed. Curr Opin Struct Biol 23:788–793. [DOI] [PubMed] [Google Scholar]
- 75. Manosas M, Spiering MM, Ding F, Bensimon D, Allemand JF, Benkovic SJ, Croquette V (2012) Mechanism of strand displacement synthesis by DNA replicative polymerases. Nucleic Acids Res 40:6174–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Manosas M, Spiering MM, Ding F, Croquette V, Benkovic SJ (2012) Collaborative coupling between polymerase and helicase for leading‐strand synthesis. Nucleic Acids Res 40:6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nandakumar D, Pandey M, Patel SS (2015) Cooperative base pair melting by helicase and polymerase positioned one nucleotide from each other. eLife 4:e06562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nandakumar D, Patel SS (2016) Methods to study the coupling between replicative helicase and leading strand DNA polymerase at the replication fork. Methods 108:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Manosas M, Perumal SK, Croquette V, Benkovic SJ (2012) Direct observation of stalled fork restart via fork regression in the T4 replication system. Science 338:1217–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Manosas M, Perumal SK, Bianco PR, Ritort F, Benkovic SJ, Croquette V (2014) RecG and UvWcatalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun 4:2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bétous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D (2013) Substrate‐selective repair and restart of replication forks by DNA translocases. Cell Rep 3:1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jankowsky E (2011) RNA helicases at work: binding and rearranging. Trends Biochem Sci 36:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bourgeois CF, Mortreux F, Auboeuf D (2016) The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol 17:426–438. [DOI] [PubMed] [Google Scholar]
- 84. García‐García C, Frieda KL, Feoktistova K, Fraser CS, Block SM (2015) Factor‐dependent processivity in human eIF4A DEAD‐box helicase. Science 348:1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pyle AM (2008) Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys 37:317–336. [DOI] [PubMed] [Google Scholar]
- 86. Vilfan ID, Kamping W, van den Hout M, Candelli A, Hage S, Dekker NH (2007) An RNA toolbox for single‐molecule force spectroscopy studies. Nucleic Acids Res 35:6625–6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fiorini F, Bagchi D, Le Hir H, Croquette V (2015) Human Upf1 is a highly processive RNA helicase and translocase with RNP remodeling activities. Nat Commun 6:7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hug N, Longman D, Cáceres JF (2016) Mechanism and regulation of the nonsense‐mediated decay pathway. Nucleic Acids Res 44:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E (2011) Molecular mechanisms for the RNA‐dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41:693–703. [DOI] [PubMed] [Google Scholar]
- 90. Myong S, Ha T (2010) Stepwise translocation of nucleic acid motors. Curr Opin Struct Biol 20:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Arslan S, Khafizov R, Thomas CD, Chemla YR, Ha T (2015) Engineering of a superhelicase through conformational control. Science 348:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Duderstadt KE, Geertsema HJ, Stratmann SA, Punter CM, Kulczyk AW, Richardson CC, van Oijen AM (2016) Simultaneous real‐time imaging of leading and lagging strand synthesis reveals the coordination dynamics of single replisomes. Mol Cell 64:1035–1047. [DOI] [PubMed] [Google Scholar]
- 93. Elshenawy MM, Jergic S, Xu ZQ, Sobhy MA, Takahashi M, Oakley AJ, Dixon NE, Hamdan SM (2015) Replisome speed determines the efficiency of the Tus‐Ter replication termination barrier. Nature 525:394. [DOI] [PubMed] [Google Scholar]
- 94. Sun B, Pandey M, Inman JT, Yang Y, Kashlev M, Patel SS, Wang MD (2015) T7 replisome directly overcomes DNA damage. Nat Commun 6:10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fan J, Leroux‐Coyau M, Savery NJ, Strick TR (2016) Reconstruction of bacterial transcription‐coupled repair at single‐molecule resolution. Nature 536:234–237. [DOI] [PubMed] [Google Scholar]
- 96. Paeschke K, Bochman ML, Garcia PD, Cejka P, Friedman KL, Kowalczykowski SC, Zakian VA (2013) Pif1 family helicases suppress genome instability at G‐quadruplex motifs. Nature 497:718–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mendoza O, Bourdoncle A, Boulé JB, Brosh RM, Jr , Mergny JL (2016) G‐quadruplexes and helicases. Nucleic Acids Res 44:1989–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cnossen JP, Dulin D, Dekker NH (2014) An optimized software framework for real‐time, high‐throughput tracking of spherical beads. Rev Sci Instrum 85:103712. [DOI] [PubMed] [Google Scholar]