

Abstract
Purpose
The mutation R345W in EFEMP1 (fibulin-3) causes macular degeneration. This study sought to determine whether proteoglycan content and diffusion across Bruch's membrane are altered in Efemp1ki/ki mice carrying this mutation or in Efemp1−/− mice.
Methods
Proteoglycans in mouse Bruch's membranes were stained with Cupromeronic Blue (CB). Heparan sulfated proteoglycan (HSPG) and chondroitin/dermatan sulfate proteoglycan (C/DSPG) distributions were visualized following treatments with chondroitinase ABC (C-ABC) or nitrous acid. Total sulfated glycosaminoglycans (sGAGs) in Bruch's membrane/choroid (BrM/Ch) were measured with dimethylmethylene blue (DMMB). Matrix metalloprotease (MMP)-2, MMP-9, and tissue inhibitor of metalloproteinase (TIMP)-3 were examined by immunofluorescence and quantified using Image J. Molecules with different Stokes radius (Rs) were allowed simultaneously to diffuse through mouse BrM/Ch mounted in a modified Ussing chamber. Samples were quantified using gel exclusion chromatography.
Results
HSPGs and C/DSPGs were markedly increased in Efemp1ki/ki Bruch's membrane, and MMP-2 and MMP-9 were decreased, but TIMP-3 was increased. Diffusion across Efemp1ki/ki Bruch's membrane was impaired. In contrast, the proteoglycan amount in Efemp1−/− Bruch's membrane was not significantly different, but the size of proteoglycans was much larger. MMP-2, MMP-3, and TIMP-3 levels were similar to that of Efemp1+/+ mice, but they were localized diffusely in retinal pigment epithelium (RPE) cells instead of Bruch's membrane. Diffusion across Efemp1−/− Bruch's membrane was enhanced.
Conclusions
Mutant fibulin-3 causes proteoglycan accumulation, reduction of MMP-2 and MMP-9, but increase of TIMP-3, and impairs diffusion across Bruch's membrane. Fibulin-3 ablation results in altered sizes of proteoglycans, altered distributions of MMP-2, MMP-9, and TIMP-3, and enhances diffusion across Bruch's membrane.
Keywords: age-related macular degeneration, Malattia Leventinese, EFEMP1 (fibulin-3), proteoglycans, diffusion
Bruch's membrane is a thin layer of connective tissue interposed between the RPE and the choriocapillaris.1 It serves as a semipermeable filtration barrier for bidirectional diffusion of nutrients and metabolites between the outer retina and the choriocapillaris.1,2 Bruch's membrane contains five parts distinguishable by electron microscopy: the basement membrane of the RPE, inner collagenous zone, elastic fiber zone, outer collagenous zone, and the basement membrane of the endothelium of the choriocapillaris.2 The ground substance in Bruch's membrane is composed largely of sulfated proteoglycans.3 These anionic proteoglycans contribute to charge-selective properties of Bruch's membrane associated with diffusion.1,2 It has been suggested that the sulfated glycosaminoglycan (sGAG) side chains of proteoglycans provide an electrolytic barrier to diffusion. Thus, alterations in the proteoglycan content of Bruch's membrane could influence its diffusion properties, cause retention of materials in Bruch's membrane, and ultimately disrupt the function of the RPE and outer retina leading to disease. Sub-RPE deposits between the RPE and Bruch's membrane (basal laminar deposit [BLamD]) or within Bruch's membrane (basal linear deposits and drusen) are a hallmark of age-related macular degeneration (AMD),4,5 the most common cause of incurable blindness in developed countries.6 Although it is still not clear how these deposits develop, one hypothesis is that the sub-RPE deposits may result from a change in the ability of materials to diffuse across Bruch's membrane.7
Similar to AMD, an autosomal dominant macular degeneration Malattia Leventinese/Doyne's honeycomb retinal dystrophy (ML/DHRD) is also characterized by the presence of sub-RPE deposits.8–12 ML/DHRD is caused by the mutation R345W in fibulin-3, a basement membrane glycoprotein encoded by the gene EGF containing fibulin-like extracellular matrix protein 1 (EFEMP1).13 Fibulin-3 is a member of the fibulin family.14 Fibulins are a family of extracellular matrix (ECM) proteins that share an elongated structure containing tandem arrays of calcium-binding epidermal growth factor (EGF)-like domains and a carboxy-terminal fibulin-type module.14,15 The functions of fibulins are not completely understood yet, but a common feature among them is that they have a high affinity for proteoglycans.15,16 In addition, fibulin-1 is a co-factor for proteoglycanase ADAMTS-1 (a disintegrin-like and metalloproteinase with thrombospondin type-1 motifs 1),17 and an antagonist for ADAMTS-4 and 9.18,19 Fibulin-3 up-regulates tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-3, physically interacts with TIMP-3, and down-regulates matrix metalloprotease (MMP)-2, MMP-3, and MMP-9.20–25 These MMPs have known proteoglycanase activities.26 TIMP-3 is a broad inhibitor for ADAMTSs and MMPs.27 Mutations in TIMP-3 cause Sorsby's fundus dystrophy,28 another inherited macular degenerative disease characterized by sub-RPE deposits.29 The activities of MMP-2 and MMP-9 of Bruch's membrane are significantly reduced in AMD.30 Both fibulin-3 and TIMP-3 are components of sub-RPE deposits in AMD and ML/DHRD.20,31 In normal eyes, fibulin-3 is localized to Bruch's membrane.32 It is possible that fibulin-3 functions to regulate the turnover of proteoglycans in Bruch's membrane through modifying the activities of MMPs and TIMPs. The mutation in fibulin-3 may alter proteoglycan content, affect diffusion across Bruch's membrane, and lead to retention of materials and sub-RPE deposit formation.
In Efemp1ki/ki mice that carry the R345W mutation, basement membrane-like materials accumulate between the plasma and basement membranes of the RPE to form BLamDs.33,34 Lipid-rich debris is retained within continuous sheets of BLamDs. There is also an accumulation of heterogeneous materials in a thickened Bruch's membrane.33 These mice recapitulate the important histopathology of ML/DHRD. In contrast, BLamD is not observed in Efemp1−/− mice that lack fibulin-3.35,36 Thus, Efemp1ki/ki and Efemp1−/− mice can serve as useful tools for studying the role of fibulin-3 in Bruch's membrane and the underlining mechanism by which mutant fibulin-3 causes sub-RPE deposit formation.
The cationic dye Cupromeronic Blue (CB) binds glycosaminoglycan side chains of sulfated proteoglycans.37 Different types of proteoglycans bound to CB can be visualized under electron microscopy as filaments with different sizes and electron density.3,38 CB staining coupled with treatments that eliminate selective groups of proteoglycans can reveal proteoglycan distribution patterns in tissues.3 Bruch's membrane mainly contains Heparan sulfated proteoglycans (HSPGs) in the basement membranes of the RPE and endothelium of choriocapillaris, and chondroitin/dermatan sulfate proteoglycans (C/DSPGs) in the fibrous layers.3 Treatment with nitric acid eliminates HSPGs, and treatment with chondroitinase ABC (C-ABC) removes C/DSPGs.3 Content of sGAGs in tissues can be measured by a colorimetric assay with the metachromatic dye dimethylmethylene blue (DMMB).39 In this study, we investigated whether distribution and content of sulfated proteoglycans are altered in Bruch's membrane of Efemp1ki/ki or Efemp1−/− mice by these methods.
Studies of the permeability of Bruch's membrane using randomly coiled linear polymers (dextrans) have shown that there is an age-related decline in the diffusion of linear polymers.40 We have previously established a method to study globular protein and small molecule diffusion across Bruch's membrane by simultaneously measuring the flux of multiple molecules with different Rs using quantitative gel exclusion chromatography.41 Coupling this method with a modified Ussing chamber has allowed us to study diffusion across very small pieces (1.8 mm2) of isolated Bruch's membrane/choroid (BrM/Ch).41 In this study, we used this system to examine Bruch's membrane's diffusion properties in Efemp1ki/ki and Efemp1−/− mice.
Methods
Mice
Efemp1ki/ki and Efemp1−/− mice were generated previously.33,35 Mice were handled in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, using protocols approved by the Institutional Animal Care and Use Committee of the University of Arizona or Mayo Clinic. Animals were housed under standard conditions and maintained on a 12-hour light/dark cycle with free access to food and water.
Proteoglycan Distribution in Bruch's Membrane
Eyes from 9-month-old wild-type (Efemp1+/+), Efemp1ki/ki, and Efemp1−/− mice were fixed at 4°C in 1% formaldehyde, 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Chorioretinal tissues were dissected from fixed eyes, cut into strips, and divided into four groups for enzymatic or nitrous acid treatment: (group 1) control group without enzyme or nitrous acid treatment; (group 2) C-ABC (Sigma-Aldrich Corp., St. Louis, MO, USA) treatment; (group 3) nitrous acid (Sigma-Aldrich Corp.) treatment; and (group 4) combinational treatment with both C-ABC and nitrous acid. For C-ABC treatment, tissue strips were incubated with 1 unit/mL C-ABC in 0.25 M Tris buffer containing 0.05% BSA, 5 mM benzamidine-HCl, and 0.1 M 6-amino-caproic acid, pH 8.0, at 37°C for 24 hours. For nitrous acid treatment, tissue strips were incubated in a nitrous acid solution containing 5% sodium nitrite, 33% acetic acid, 5 mM benzamidine-HCl, and 0.1 M 6-amino-caproic acid for 90 minutes at room temperature. Each group of tissue strips was then stained with 0.05% CB (Sigma-Aldrich Corp.) in 25 mM sodium acetate, 0.2 M MgCl2 and 2.5% glutaraldehyde, pH 5.7 overnight as described.3 After staining, tissue strips were processed for transmission electron microscopy. Thin sections were cut with a Reichert Ultracut microtome, stained with uranyl acetate and lead citrate, and imaged with a Philips CM-12 electron microscope equipped with an AMT CCD camera (Advanced Microscopy Techniques Corp., Danvers, MA, USA).
CB-stained proteoglycan filaments were quantified by counting the filaments from different view fields at the same magnification. Specimens from three individual mice for each genotype were included in the filament quantification. Three sections from each specimen were used, and 10 different fields from each section were counted. The numbers of filaments from Efemp1ki/ki or Efemp1−/− mice were compared with those in Efemp1+/+ mice using a Student's t-test. Proteoglycan filament lengths were measured using the Adobe Photoshop ruler tool. Ten different filaments per field for each category (HSPG or C/DSPG) were measured. Due to their small size, filament lengths were measured to the nearest 5 nm.
sGAG Quantification Assay
After mice were killed with with CO2 asphyxiation, eyes from 9-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice were enucleated, and the lens and anterior segments were removed. The retina was detached using forceps, the eyecup filled with PBS buffer, and the RPE was removed using a fine camel's hair brush. The BrM/Ch was detached from the sclera, weighed, and minced. Five sets of BrM/Ch samples from five mice per genotype (Efemp1+/+, Efemp1ki/ki, or Efemp1−/−) were analyzed. Samples were digested with 300 μg papain (Sigma-Aldrich Corp.) in 20 mM sodium phosphate buffer (pH 6.8) containing 1 mM EDTA and 2 mM dithiothreitol at 60°C for 2 hours. Samples were diluted with 10 mM iodoacetic acid and 50 mM Tris/HCl (pH 8.0). The sGAG content in papain-digested samples was measured by colorimetric assay with DMMB using an assay kit (Amsbio, Cambridge, MA, USA) according to the manufacturer's instructions. sGAG measurements from Efemp1ki/ki or Efemp1−/− mice were compared with those in Efemp1+/+ mice using a Student's t-test.
Immunofluorescence
Eyes from 9-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice were fixed in 4% paraformaldehyde. Immunofluorescence staining of 10-μm frozen sections of mouse eyes was performed as previously described33 using a rabbit polyclonal antibody against MMP-2 (Abcam, Cambridge, MA, USA), MMP-9 (Abcam), TIMP-3 (Abcam), or fibulin-3.33 A goat anti-rabbit IgG Alexa Fluor 488 conjugate was used as a secondary antibody (Invitrogen, Carlsbad, CA, USA). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Sections were examined and photographed using a Nikon E600 microscope (Melville, NY, USA) equipped with a CCD camera.
Quantification of immunofluorescence was performed using Image J software (National Institutes of Health, Bethesda, MD, USA). Sections from three mice (n = 3) per genotype (Efemp1+/+, Efemp1ki/ki, or Efemp1−/−) were used. Three images taken from three randomly selected areas in each stained section were measured. In each image, the outline of the RPE layer and Bruch's membrane was drawn, and the area, mean fluorescence, and background readings were measured. The total fluorescence was calculated as integrated density subtracting (area of selected sections multiplying mean fluorescence of background readings). Fluorescence from Efemp1ki/ki or Efemp1−/− mice was compared with that in Efemp1+/+ mice using a Student's t-test.
Diffusion Assay
The BrM/Ch prepared from 9- or 18-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice was mounted between two pieces of X-ray film, which had corresponding 1.0-mm diameter drilled holes and were coated with a thin film of silicon grease. Diffusion of molecules through mouse BrM/Ch was studied using a dual Ussing chamber model U2500 (Warner Instruments, Hamden, CT, USA). The BrM/Ch-X-ray film “sandwich” was mounted, separating the two chambers with only the 1.0-mm diameter circle of BrM/Ch allowing communication between them. Four molecules with different Rs were selected for their ability to separate cleanly by gel exclusion chromatography41: cytosine (molecular weight [MW] = 110.1, Rs < 1.0 nm; Sigma-Aldrich Corp.), RNase A (MW = 13,700, Rs = 1.72 nm; Teknova, Hollister, CA, USA), human serum albumin (MW = 66,382, Rs = 3.55 nm; EMD Chemicals, Gibbstown, NJ, USA), and horse spleen ferritin (MW = 450,000, Rs = 6.15 nm; EMD Chemicals). A mixture of these molecules dissolved in PBS with 1 mM MgCl, and 0.1 mM CaCl2 (PBS-CM) was added to the reservoir on the BrM side of the tissue with only PBS-CM on the Ch side.
Flux and Permeability Coefficients
Flux and permeability coefficients were calculated as previously described.41 Molecules were quantified after 48 hours using gel exclusion chromatography with a 1.6 × 60 cm Hi-Prep Sephacryl S-300HR (Amersham Biosciences, Piscataway, NJ, USA) column equilibrated with PBS-CM. The column was developed at a linear flow rate of 15 mL cm−1 hr−2 using an AKTA prime chromatography system controlled by PrimeView software (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Peaks were detected by their absorbance at 280 nm, and peak areas were determined by integration using PrimeView software. Solute concentrations were quantified by linear regression analysis using peak areas derived from known quantities of each test molecule. Flux (J) was calculated using the equation J = (C)/(A·t) where C = accumulated molar concentration, A = the area (cm2) of the exposed tissue (0.0.00785 cm2), and t = 48 hours. Permeability coefficients (P) were calculated using Fick's first law P = (J·L)/ΔC where L = the thickness of the BrM/Ch preparation and ΔC describes the difference in moles of molecules between the two compartments of the Ussing chamber.
Results
Rod-like filaments representing sulfated proteoglycans were observed at high magnifications under electron microscopy in Bruch's membrane of 9-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice after CB staining (Fig. 1). The size and number of filaments were different in mice with different Efemp1 genotypes (Fig. 1). In Efemp1+/+ Bruch's membrane, filaments with an average length of 50 nm were observed in the collagenous/elastic fibrous layers, and filaments with an average length of 40 nm were observed in the basement membranes of the RPE and endothelium of choriocapillaris (Figs. 1A, 1a1, 1a2; Table 1). Smaller sizes of filaments may represent the same type of filaments cut shorter during sectioning or only partially visible in any given microscopic viewing field. Filaments in the fibrous layers were eliminated by C-ABC treatment (Fig. 2), indicating that they were C/DSPGs. Filaments in the basement membranes were removed by nitric acid treatment (Fig. 3), indicating that those filaments were HSPGs. Combined treatment of both C-ABC and nitrous acid eliminated all the filaments from Bruch's membrane (Fig. 4). This indicates that all the filaments observed were either HSPGs or C/DSPGs.
Figure 1.

Electron micrographs of Bruch's membrane of 9-month-old mice stained by CB. (A) Efemp1+/+ (+/+). a1 and a2 are higher magnification of box “a1” and “a2” areas of (A). (B) Efemp1ki/ki (ki/ki). b1 and b2 are higher magnification of box “b1” and “b2” areas of (B). (C) Efemp1−/− (−/−). c1 and c2 are higher magnification of box “c1” and “c2” areas of (C). Arrows indicate filaments representing sulfated proteoglycans in Bruch's membrane. OS, photoreceptor outer segment; RPE BI, RPE basal infolding; BrM, Bruch's membrane. Scale bar: 2 μm for A, B, and C; 200 nm for a1 to c2.
Table 1.
Proteoglycan Sizes in Efemp1+/+, Efemp1ki/ki, or Efemp1−/− Bruch's Membrane

Figure 2.

Electron micrographs of mouse Bruch's membrane of 9-months-old Efemp1+/+ (+/+), Efemp1ki/ki (ki/ki), and Efemp1−/− (−/−) mice treated with C-ABC and stained by CB. Note that the filaments representing C/DSPGs in the fibrous layers of Bruch's membrane were eliminated by C-ABC treatment, but that filaments (arrows) representing HSPGs in the basement membranes of the RPE and the endothelium of choriocapillaris were preserved. BI, basal infolding; BM, basement membrane; EN, endothelium. Scale bar: 500 nm.
Figure 3.

Electron micrographs of mouse Bruch's membrane of 9-month-old Efemp1+/+ (+/+), Efemp1ki/ki (ki/ki), and Efemp1−/− (−/−) mice treated with nitrous acid and stained by CB. Note that filaments representing HSPGs in the basement membranes of the RPE and the endothelium of choriocapillaris were eliminated, but that filaments (arrowheads) representing C/DSPGs in the fibrous layers of Bruch's membrane were preserved. BI, basal infolding; BM, basement membrane; EN, endothelium. Scale bar: 500 nm.
Figure 4.

Electron micrographs of mouse Bruch's membrane of 9-month-old Efemp1+/+ (+/+), Efemp1ki/ki (ki/ki), and Efemp1−/− (−/−) mice treated with C-ABC and nitrous acid coupled by CB staining. Note that all the filaments in Bruch's membrane were eliminated. Black dots distinct from the filaments representing proteoglycans in +/+ and ki/ki samples were likely background staining. BI, basal infolding; BM, basement membrane; EN, endothelium. Scale bar: 500 nm.
Proteoglycan Accumulation in Bruch's Membrane of Efemp1ki/ki Mice
Markedly increased filaments were observed in Bruch's membrane of Efemp1ki/ki mice (Figs. 1B, 1b1, 1b2). By counting the filaments from different view fields at the same magnification, we determined that there were on average three times as many filaments in Efemp1ki/ki Bruch's membrane as those in Efemp1+/+ Bruch's membrane (Table 2).
Table 2.
Proteoglycan Distributions in Efemp1+/+, Efemp1ki/ki, and Efemp1−/− Bruch's Membrane

After C-ABC treatment, filaments with an average length of 40 nm (Table 1) representing HSPGs remained in the basement membranes of the RPE and endothelium of choriocapillaris in Efemp1ki/ki mice (Fig. 2). There were 2.35 times as many filaments in Efemp1ki/ki basement membranes as those in Efemp1+/+ mice (Table 2). After nitrous acid treatment, filaments with an average length of 50 nm (Table 1) representing C/DSPGs remained in the fibrous layers of Efemp1ki/ki Bruch's membrane (Fig. 3). There were 4.15 times as many filaments in Efemp1ki/ki fibrous layers as those in Efemp1+/+ fibrous layers (Table 2). Combined treatment of C-ABC and nitrous acid eliminated all the filaments from Efemp1ki/ki Bruch's membrane (Fig. 4), indicating that all the filaments observed were either HSPGs or C/DSPGs.
Biochemical analysis by DMMB colorimetric assays revealed that total sGAG content in 9-month-old Efemp1ki/ki BrM/Ch was significantly higher than that of Efemp1+/+ mice (Fig. 5). The average amount of sGAGs in Efemp1ki/ki BrM/Ch was 15.89 μg/mg wet tissue weight, comparing to 10.15 μg/mg in Efemp1+/+ mice.
Figure 5.
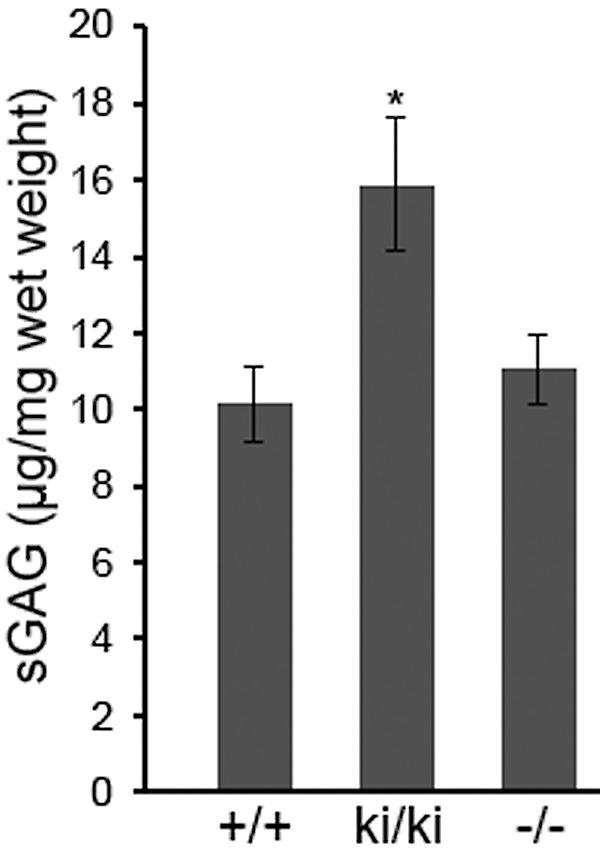
Total amount of sGAG of BrM/Ch samples from 9-month-old mice. Error bars indicate the mean ± SD. n = 5 mice per genotype. *P < 0.05 comparing to the value of Efemp1+/+ mice. +/+, Efemp1+/+; ki/ki, Efemp1ki/ki; −/−, Efemp1−/−.
Different Sizes of Proteoglycans in Bruch's Membrane of Efemp1−/− Mice
In Efemp1−/− mice, there were slightly more filaments observed in Bruch's membrane than in Efemp1+/+ mice, but the difference was not statistically significant (Table 2). However, the size of filaments in Efemp1−/− Bruch's membrane was strikingly larger (Figs. 1C, 1c1, 1c2). After C-ABC treatment, filaments with an average length of 120 nm (Table 1) representing HSPGs were observed in Efemp1−/− basement membranes of the RPE and endothelium of choriocapillaris (Fig. 2). After nitrous acid treatment, filaments with an average length of 105 nm (Table 1) representing C/DSPGs were observed in the fibrous layers of Efemp1−/− Bruch's membrane (Fig. 3). Despite the different sizes, combined treatment of C-ABC and nitrous acid eliminated all the filaments from Efemp1−/− Bruch's membrane (Fig. 4), indicating that all the filaments were either HSPGs or C/DSPGs.
Results of DMMB assays showed that total sGAG content in BrM/Ch from 9-month-old Efemp1−/− mice was similar to that of Efemp1+/+ mice (Fig. 5). The average amount of sGAGs in Efemp1−/− BrM/Ch was 11.04 μg/mg.
Altered Levels and Distributions of MMP-2, MMP-9, and TIMP-3 in the RPE and Bruch's Membrane of Efemp1ki/ki and Efemp1−/− Mice
Immunofluorescence revealed that MMP-2, MMP-9, and TIMP-3 were localized to the basal side of the RPE along Bruch's membrane in Efemp1+/+ and Efemp1ki/ki mice (Fig. 6A). TIMP-3 and fibulin-3 were co-localized together and had a complete overlap-staining pattern in these mice (Fig. 6A). However, quantification of immunofluorescence showed that the levels of both MMP-2 and MMP-9 were significantly reduced in Efemp1ki/ki mice (Fig. 6B). In contrast, TIMP-3′s level was significantly increased in Efemp1ki/ki mice (Fig. 6B). Without fibibulin-3 in Efemp1−/− mice, MMP-2, MMP-9, and TIMP-3 had a drastically different pattern of distribution. All three proteins had a diffuse distribution in RPE cells (Fig. 6A). This suggests that fibulin-3 may normally function to anchor these proteins to Bruch's membrane. Quantification of immunofluorescence showed that the levels of all three proteins in the Efemp1−/− RPE and Bruch's membrane were slightly lower than that in Efemp1+/+ mice, but the difference was not statistically significant (Fig. 6B).
Figure 6.

Immunofluorescence for MMP-2, MMP-9, TIMP-3, and fibulin-3 in the RPE and Bruch's membrane of 9-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice. (A) Frozen sections were stained with antibodies (red signal) against MMP-2, MMP-9, TIMP-3 (green signal) or fibulin-3. The nuclei were stained with DAPI (blue signal). Note the more defined linear staining along Bruch's membrane for MMP-2, MMP-9, TIMP-3, and fibulin-3 in Efemp1+/+ and Efemp1ki/ki mice, but a diffuse pattern of staining in RPE cells in Efemp1−/− mice. TIMP-3 and fibulin-3 signals overlapped in the RPE and Bruch's membrane of Efemp1+/+ and Efemp1ki/ki mice. BrM, Bruch's membrane. (B) Immunofluorescence of MMP-2, MMP-9, TIMP-3, and fibuin-3 in the RPE and Bruch's membrane was quantified using Image J software. n = 3 mice per genotype. Error bars indicate the mean ± SD. n = 3 mice per genotype. *P < 0.05 comparing to the values of wild-type controls for each protein in a Student's t-test.
Different Diffusion Rates Across Mouse Bruch's Membrane of Efemp1+/+, Efemp1ki/ki, and Efemp1−/− Mice
We previously established a system using a modified Ussing chamber coupled with quantitative gel exclusion chromatography to study the diffusion of multiple molecules with different Rs through small samples (<2 mm2) of Bruch's membrane.41 Here we used this system to measure differences in the diffusion of four molecules through Bruch's membrane of 9- and 18-month-old Efemp1+/+, Efemp1ki/ki, and Efemp1−/− mice in the BrM to Ch direction. We established a concentration gradient of all four molecules across BrM/Ch and applied Fick's first law to calculate the flux and permeability coefficient of each molecule as described previously.41 The flux of each molecule remained constant, and the concentration gradient was not significantly influenced by the diffusion of molecules throughout the time course of each experiment. Flux and permeability coefficient decreased with increasing Rs (Fig. 7).
Figure 7.

Total diffusion flux (A) and permeability coefficient (B) of molecules through mouse Bruch's membrane plotted as a function of Rs. Molecules include cytosine (Rs < 1.0 nm), RNase A (Rs = 1.72 nm), albumin (Rs = 3.55 nm), and ferritin (Rs = 6.15 nm). Data points are given as mean ± SD, n ≥ 5 mice per genotype. +/+, Efemp1+/+; ki/ki, Efemp1ki/ki; −/−, Efemp1−/−.
For Efemp1+/+ tissue (n = 5 mice per age), cytosine had a flux of 5.33 × 104 (9 months) and 2.99 × 104 nmol cm−2 h−1 (18 months), and a permeability coefficient of 0.94 (9 months) and 0.47 cm−2 h−1 (18 months). Ferritin had a flux of 1.32 (9 months) and 0.86 nmol cm−2 h−1 (18 months), and a permeability coefficient of 9.25 × 10−5 (9 months) and 2.38 × 10−5 cm−2 h−1 (18 months). Flux and permeability coefficient of each molecule were higher for 9-month-old than 18-month-old tissue (Fig. 7).
We found both flux and permeability coefficients of each molecule were significantly (P < 0.05) decreased for BrM/Ch samples from Efemp1ki/ki mice (n = 5 for 9 months old, and n = 6 for 18 months old) (Fig. 7). Cytosine had a flux of 8.54 (9 months) and 6.71 nmol cm−2 h−1 (18 months), and a permeability coefficient of 6.92 × 10−4 (9 months) and 2.24 × 10−4 cm−2 h−1 (18 months). Ferritin had a flux of 4.80 × 10−2 (9 months) and 1.47 × 10−2 nmol cm−2 h−1 (18 months), and a permeability coefficient of 1.01 × 10−6 (9 months) and 3.13 × 10−7 cm−2 h−1 (18 months). This indicates that diffusion across Bruch's membrane of Efemp1ki/ki mice was markedly decreased, with ferritin virtually impermeable at both 9 and 18 months of ages.
In contrast to Efemp1ki/ki mice, we found increased flux and permeability coefficient of each molecule for BrM/Ch samples from Efemp1−/− mice (n = 6 for 9 months old, and n = 8 for 18 months old) (Fig. 7). Cytosine had a flux of 1.13 × 105 (9 months) and 4.79 × 104 nmol cm−2 h−1 (18 months), and a permeability coefficient of 5.30 (9 months) and 2.93 cm−2 h−1 (18 months). Ferritin had a flux of 3.98 nmol (9 months) and 2.19 nmol cm−2 h−1 (18 months), and a permeability coefficient of 5.56 × 10−4 (9 months) and 1.12 × 10−4 cm−2 h−1 (18 months). This indicates that diffusion across Bruch's membrane of Efemp1−/− mice was increased at both 9 and 18 months of ages. The permeability coefficient was similar for albumin and ferritin for BrM/Ch samples from either Efemp1+/+ or Efemp1−/− mice (Fig. 7B), suggesting that the ferritin is near the physical size exclusion limit of Bruch's membrane for these mice.
Discussion
The R345W mutation in fibulin-3 causes sub-RPE deposit formation both in human and mouse,13,33,34 but the pathogenic mechanism behind this remains unknown. In this study, we have found that Efemp1ki/ki mice carrying this mutation have markedly increased sulfated proteoglycans in their Bruch's membrane, although the type and size of proteoglycans are similar to those found in the wild-type mice. Both HSPGs and C/DSPGs are substantially increased. In Efemp1−/− mice, which do not express fibulin-3, we have found that the sizes of HSPGs and C/DSPGs are significantly larger than those observed in wild-type mice, but their amount is not significantly different. These results indicate that fibulin-3 regulates the level and type of proteoglycans in Bruch's membrane.
The accumulated proteoglycans in Efemp1ki/ki mice can be due to either over-synthesis or deficient degradation. Our data suggest that deficient degradation may be a more important factor. MMP-2 and MMP-9, two ECM enzymes with known proteoglycanase activities, are significantly decreased in Efemp1ki/ki Bruch's membrane, but TIMP-3, an inhibitor of a broad range of proteoglycanases, is significantly increased. Without fibulin-3 in Efemp1−/− mice, MMP-2, MMP-9, and TIMP-3 are diffusely localized in RPE cells instead of Bruch's membrane, suggesting that fibulin-3 may normally be an anchor for these proteins to Bruch's membrane. Fibulin-3 has ECM enzyme inhibitory activity,22,25,42 we hypothesize that it regulates proteoglycan turnover in Bruch's membrane by controlling proteoglycanase activities. In the absence of fibulin-3, proteoglycans normally present in Bruch's membrane may be degraded. The different sizes of proteoglycans in Efemp1−/− mice may be derived from a compensatory effect induced by the lack of normal proteoglycans. They may be different types of HSPGs or C/DSPGs or same core proteoglycans with different levels of sulfation. The R345W mutation does not impair fibulin-3′s activity but makes the protein more resistant to degradation leading to its accumulation.32 In Efemp1ki/ki mice, fibulin-3 is accumulated in Bruch's membrane. This accumulation could provide a high level of localized inhibitory activity on proteoglycanase(s) and cause proteoglycan accumulation in Bruch's membrane. Fibulin-3 is overexpressed by senescent cells in aging tissues.43,44 It accumulates in Bruch's membrane of both ML/DHRD and AMD donor eyes.32 There is no fibulin-3 mutation found in AMD patients.13,32 This supports the idea that elevated fibulin-3 activity from fibulin-3 accumulation due to mutation or aging causes pathologic changes associated with sub-RPE deposits.
Studies on the composition of sub-RPE deposits have found that lipoproteins, cholesterol, and products of the complement system are common components of sub-RPE deposits.45–52 This leads to an ocular “response-to-retention” hypothesis for sub-RPE deposit formation reminiscent of that for atherosclerosis,45 which proposes that lipoproteins are retained on proteoglycans in the arterial intima, become modified, and then elicit an inflammatory response leading to atherosclerotic plaque formation.53 We have previously found that lipid-containing particles (previously referred to as membranous debris) accumulate in linear tracks within BLamDs in Efemp1ki/ki mice.33 Our findings here suggest that the lipid-containing particles may be trapped by the proteoglycans accumulated in Bruch's membrane, thus, leading to the exposure of retained lipids to the high oxidative stress environment at the RPE/Bruch's membrane interface. Oxidative modification of lipids triggers complement activation to remove the oxidatively damaged molecules.54 The sustained supply of lipids trapped in proteoglycans can turn complement activation into chronic inflammation with the accumulation of inflammatory products to form sub-RPE deposits.
Proteoglycans affect the diffusion properties of Bruch's membrane. Consistent with the changes in proteoglycans in Efemp1ki/ki and Efemp1−/− mice, we have found that diffusion across Bruch's membrane is impaired in Efemp1ki/ki mice, but enhanced in Efemp1−/− mice. Both flux and permeability coefficient decreased exponentially for all four molecules with Rs ranging from <1.0 to 6.15 nm for Efemp1ki/ki mice comparing to those for the wild-type mice. This suggests that the porosity of Bruch's membrane is greatly reduced in Efemp1ki/ki mice. The increased amount of proteoglycans in these mice may structurally obscure pores of Bruch's membrane, or may increase the viscosity of the fibrous layers. Impaired diffusion would hinder the removal of metabolic wastes and other molecules secreted from the RPE, and accelerate the deposition of heterogeneous materials in Bruch's membrane of Efemp1ki/ki mice. For Efemp1−/− mice, the flux and permeability coefficient across Bruch's membrane are more in line with those of the wild-type mice, but the increase is significant. The substantially larger sizes of proteoglycans in Efemp1−/− mice may structurally expand pore sizes of Bruch's membrane resulting in enhanced diffusion as we observed.
In summary, our study has found that mutant fibulin-3 causes proteoglycan accumulation and impaired diffusion across Bruch's membrane. Fibulin-3 may have specific proteoglycanase substrate(s). It would be worthwhile to investigate this in future studies. The impairment of diffusion across Bruch's membrane would block nutrient and waste flow to and from the retina, contribute to the initiation and/or growth of sub-RPE deposits, and induce degeneration of the RPE and photoreceptors. The enhanced diffusion across Bruch's membrane of Efemp1−/− mice suggests that reduction or elimination of fibulin-3 could be a valuable strategy in developing treatments for macular degeneration.
Acknowledgments
The authors thank Taylor Berent for technical assistance.
Supported by National Institutes of Health Grants EY13847 (LYM), EY13160 (ADM), and EY21153 (ADM), The Edward N. & Della L. Thome Memorial Foundation (LYM), Mayo Foundation, and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology at the Mayo Clinic in Rochester, MI, USA.
Disclosure: A. Zayas-Santiago, None; S.D. Cross, None; J.B. Stanton, None; A.D. Marmorstein, None; L.Y. Marmorstein, None
References
- 1. Booij JC,, Baas DC,, Beisekeeva J,, Gorgels TG,, Bergen AA. The dynamic nature of Bruch's membrane. Prog Retin Eye Res. 2010; 29: 1–18. [DOI] [PubMed] [Google Scholar]
- 2. Guyer DR,, Schachat AP,, Green WR. The choroids: structural consideration. : Ryan SJ, Retina. St. Louis, MO: Mosby; 2001: 29. [Google Scholar]
- 3. Call TW,, Hollyfield JG. Sulfated proteoglycans in Bruch's membrane of the human eye: localization and characterization using cupromeronic blue. Exp Eye Res. 1990; 51: 451–462. [DOI] [PubMed] [Google Scholar]
- 4. Sarks SH,, Sarks JP. Age-related maculopathy: nonneovascular age-related macular degeneration and the evolution of geographic atrophy. : Ryan SJ, Retina. St. Louis, MO: Mosby; 2001: 1064. [Google Scholar]
- 5. Sarks S,, Cherepanoff S,, Killingsworth M,, Sarks J. Relationship of Basal laminar deposit and membranous debris to the clinical presentation of early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007; 48: 968–977. [DOI] [PubMed] [Google Scholar]
- 6. de Jong PT. Age-related macular degeneration. N Engl J Med. 2006; 355: 1474–1485. [DOI] [PubMed] [Google Scholar]
- 7. Guyer DR,, Bird AC. Age changes in Bruch's membrane and related structures. : Ryan SJ, Retina. St. Louis, MO: Mosby; 2001: 1051–1063. [Google Scholar]
- 8. Piguet B,, Haimovici R,, Bird AC. Dominantly inherited drusen represent more than one disorder: a historical review. Eye. 1995; 9 pt 1: 34–41. [DOI] [PubMed] [Google Scholar]
- 9. Marmorstein L. Association of EFEMP1 with malattia leventinese and age-related macular degeneration: a mini-review. Ophthalmic Genet. 2004; 25: 219–226. [DOI] [PubMed] [Google Scholar]
- 10. Streicher T,, Schmidt K,, Dusek J. Hereditary drusen of Bruch's membrane. I. Clinical and light microscopical study [in German]. Klin Monatsbl Augenheilkd. 1982; 181: 27–31. [DOI] [PubMed] [Google Scholar]
- 11. Dusek J,, Streicher T,, Schmidt K. Hereditary drusen of Bruch's membrane. II: Studies of semi-thin sections and electron microscopy results [in German]. Klin Monatsbl Augenheilkd. 1982; 181: 79–83. [PubMed] [Google Scholar]
- 12. Collins T. A pathological report on a case of Doyne's choroiditis (“honeycomb” or “family choroiditis”). Ophthalmoscope. 1913; 11: 537–538. [Google Scholar]
- 13. Stone EM,, Lotery AJ,, Munier FL,, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999; 22: 199–202. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y,, Marmorstein LY. Focus on molecules: fibulin-3 (EFEMP1). Exp Eye Res. 2010; 90: 374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Timpl R,, Sasaki T,, Kostka G,, Chu ML. Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol. 2003; 4: 479–489. [DOI] [PubMed] [Google Scholar]
- 16. Djokic J,, Fagotto-Kaufmann C,, Bartels R,, Nelea V,, Reinhardt DP. Fibulin-3, -4, and -5 are highly susceptible to proteolysis, interact with cells and heparin, and form multimers. J Biol Chem. 2013; 288: 22821–22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee NV,, Rodriguez-Manzaneque JC,, Thai SN,, et al. Fibulin-1 acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005; 280: 34796–34804. [DOI] [PubMed] [Google Scholar]
- 18. Hesselson D,, Newman C,, Kim KW,, Kimble J. GON-1 and fibulin have antagonistic roles in control of organ shape. Curr Biol. 2004; 14: 2005–2010. [DOI] [PubMed] [Google Scholar]
- 19. Kubota Y,, Kuroki R,, Nishiwaki K. A fibulin-1 homolog interacts with an ADAM protease that controls cell migration in C. elegans. Curr Biol. 2004; 14: 2011–2018. [DOI] [PubMed] [Google Scholar]
- 20. Klenotic PA,, Munier FL,, Marmorstein LY,, Anand-Apte B. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. J Biol Chem. 2004; 279: 30469–30473. [DOI] [PubMed] [Google Scholar]
- 21. Albig AR,, Neil JR,, Schiemann WP. Fibulins 3 and 5 antagonize tumor angiogenesis in vivo. Cancer Res. 2006; 66: 2621–2629. [DOI] [PubMed] [Google Scholar]
- 22. Rahn DD,, Acevedo JF,, Roshanravan S,, et al. Failure of pelvic organ support in mice deficient in fibulin-3. Am J Pathol. 2009; 174: 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim EJ,, Lee SY,, Woo MK,, et al. Fibulin-3 promoter methylation alters the invasive behavior of non-small cell lung cancer cell lines via MMP-7 and MMP-2 regulation. Int J Oncol. 2012; 40: 402–408. [DOI] [PubMed] [Google Scholar]
- 24. Wang Z,, Cao CJ,, Huang LL,, et al. EFEMP1 promotes the migration and invasion of osteosarcoma via MMP-2 with induction by AEG-1 via NF-kappaB signaling pathway. Oncotarget. 2015; 6: 14191–14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fernandez-Godino R,, Garland DL,, Pierce EA. A local complement response by RPE causes early-stage macular degeneration. Human Mol Genet. 2015; 24: 5555–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein T,, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011; 41: 271–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Woessner JF., Jr. That impish TIMP: the tissue inhibitor of metalloproteinases-3. J Clin Invest. 2001; 108: 799–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weber BH,, Vogt G,, Pruett RC,, Stohr H,, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby's fundus dystrophy. Nat Genet. 1994; 8: 352–356. [DOI] [PubMed] [Google Scholar]
- 29. Capon M,, Marshall J,, Krafft JI,, Alexander RA,, Hiscott PS,, Bird AC. Sorsby's fundus dystrophy: a light and electron microscopic study. Ophthalmology. 1989; 95: 1769–1777. [DOI] [PubMed] [Google Scholar]
- 30. Hussain AA,, Lee Y,, Zhang JJ,, Marshall J. Disturbed matrix metalloproteinase activity of Bruch's membrane in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011; 52: 4459–4466. [DOI] [PubMed] [Google Scholar]
- 31. Crabb J,, Miyagi M,, Gu X,, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marmorstein LY,, Munier FL,, Arsenijevic Y,, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 13067–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marmorstein LY,, McLaughlin PJ,, Peachey NS,, Sasaki T,, Marmorstein AD. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: a model for the early pathogenic course of macular degeneration. Human Mol Genet. 2007; 16: 2423–2432. [DOI] [PubMed] [Google Scholar]
- 34. Fu L,, Garland D,, Yang Z,, et al. The R345W mutation in EFEMP1 is pathogenic and causes AMD-like deposits in mice. Hum Mol Genet. 2007; 16: 2411–2422. [DOI] [PubMed] [Google Scholar]
- 35. McLaughlin PJ,, Bakall B,, Choi J,, et al. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Human Mol Genet. 2007; 16: 3059–3070. [DOI] [PubMed] [Google Scholar]
- 36. Stanton JB,, Marmorstein AD,, Zhang Y,, Marmorstein LY. Deletion of Efemp1 is protective against the development of sub-RPE deposits in mouse eyes. Invest Ophthalmol Vis Sci. 2017; 58: 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott JE. Morphometry of cupromeronic blue-stained proteoglycan molecules in animal corneas, versus that of purified proteoglycans stained in vitro, implies that tertiary structures contribute to corneal ultrastructure. J Anatomy. 1992; 180 pt 1: 155–164. [PMC free article] [PubMed] [Google Scholar]
- 38. Scott JE. Collagen--proteoglycan interactions. Localization of proteoglycans in tendon by electron microscopy. Biochem J. 1980; 187: 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ho LT,, Harris AM,, Tanioka H,, et al. A comparison of glycosaminoglycan distributions, keratan sulphate sulphation patterns and collagen fibril architecture from central to peripheral regions of the bovine cornea. Matrix Biol. 2014; 38: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hussain AA,, Starita C,, Hodgetts A,, Marshall J. Macromolecular diffusion characteristics of ageing human Bruch's membrane: implications for age-related macular degeneration (AMD). Exp Eye Res. 2010; 90: 703–710. [DOI] [PubMed] [Google Scholar]
- 41. Zayas-Santiago A,, Marmorstein AD,, Marmorstein LY. Relationship of stokes radius to the rate of diffusion across Bruch's membrane. Invest Ophthalmol Vis Sci. 2011; 52: 4907–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin Z,, Wang Z,, Li G,, Li B,, Xie W,, Xiang D. Fibulin-3 may improve vascular health through inhibition of MMP-2/9 and oxidative stress in spontaneously hypertensive rats. Mol Med Rep. 2016; 13: 3805–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun BS,, Zhu X,, Clayton MM,, Pan J,, Feitelson MA. Identification of a protein isolated from senescent human cells that binds to hepatitis B virus X antigen. Hepatology. 1998; 27: 228–239. [DOI] [PubMed] [Google Scholar]
- 44. Lecka-Czernik B,, Lumpkin CK, Jr,, Goldstein S. An overexpressed gene transcript in senescent and quiescent human fibroblasts encoding a novel protein in the epidermal growth factor-like repeat family stimulates DNA synthesis. Mol Cell Biol. 1995; 15: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Curcio CA,, Johnson M,, Huang JD,, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009; 28: 393–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rudolf M,, Curcio CA. Esterified cholesterol is highly localized to Bruch's membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem. 2009; 57: 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Curcio CA,, Johnson M,, Huang JD,, Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res. 2010; 51: 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Curcio CA,, Johnson M,, Rudolf M,, Huang JD. The oil spill in ageing Bruch membrane. Br J Ophthalmol. 2011; 95: 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rudolf M,, Seckerdieck K,, Grisanti S,, Curcio CA. Internal structure consistent with remodelling in very small drusen, revealed by filipin histochemistry for esterified cholesterol. Br J Ophthalmol. 2014; 98: 698–702. [DOI] [PubMed] [Google Scholar]
- 50. Kunchithapautham K,, Atkinson C,, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem. 2014; 289: 14534–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Johnson LV,, Forest DL,, Banna CD,, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2011; 108: 18277–18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garland DL,, Fernandez-Godino R,, Kaur I,, et al. Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Human Mol Genet. 2014; 23: 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Williams KJ,, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995; 15: 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Handa JT,, Cano M,, Wang L,, Datta S,, Liu T. Lipids, oxidized lipids, oxidation-specific epitopes, and age-related macular degeneration. Biochim Biophys Acta. 2017; 1862: 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]