

Abstract
The identification of native sources and vectors of introduced species informs their ecological and evolutionary history and may guide policies that seek to prevent future introductions. Population genetics provides a powerful set of tools to identify origins and vectors. However, these tools can mislead when the native range is poorly sampled or few molecular markers are used. Here, we traced the introduction of the Asian seaweed Gracilaria vermiculophylla (Rhodophyta) into estuaries in coastal western North America, the eastern United States, Europe, and northwestern Africa by genotyping more than 2,500 thalli from 37 native and 53 non‐native sites at mitochondrial cox1 and 10 nuclear microsatellite loci. Overall, greater than 90% of introduced thalli had a genetic signature similar to thalli sampled from the coastline of northeastern Japan, strongly indicating this region served as the principal source of the invasion. Notably, northeastern Japan exported the vast majority of the oyster Crassostrea gigas during the 20th century. The preponderance of evidence suggests G. vermiculophylla may have been inadvertently introduced with C. gigas shipments and that northeastern Japan is a common source region for estuarine invaders. Each invaded coastline reflected a complex mix of direct introductions from Japan and secondary introductions from other invaded coastlines. The spread of G. vermiculophylla along each coastline was likely facilitated by aquaculture, fishing, and boating activities. Our ability to document a source region was enabled by a robust sampling of locations and loci that previous studies lacked and strong phylogeographic structure along native coastlines.
Keywords: algae, biological invasion, Northwest Pacific, oysters, phylogeography, population genetics
1. Introduction
Non‐native species represent one of the greatest threats to native biodiversity by homogenizing the Earth's biota and altering community processes and ecosystem function. Predicting the environmental and evolutionary processes that facilitate invasions is one of the major goals of invasion biology (Kolar & Lodge, 2001). For a species with a broad distribution and strong population genetic structure in its native range, the pooling of all native populations with which to compare against introduced populations may provide an uninformative contrast. Nonsource populations in the native range may not share the same opportunities for invasion because the vector(s) may be absent (Miler et al., 2002) or the necessary ecological or evolutionary characteristics that enable successful colonization are missing (Colautti & Lau, 2015; Estoup & Guillemaud, 2010). Identifying population source(s) and elucidating invasion pathways are fundamental to the investigation of subsequent evolution in recipient regions (Bock et al., 2015), the identification of other invaders from the same region or as a result of the same vector (Brawley et al., 2009), and the development of effective management strategies (Dlugosch & Parker, 2008). For most invasions, our knowledge about pathways and vectors is largely based on historical and observational data.
In the marine environment, maritime shipping and aquaculture have long been implicated in the introduction of non‐native species, particularly in estuarine habitats where these activities are more intense (Ruiz et al., 1997, 2000; Ruesink et al., 2005; Preisler et al., 2009). Ballast water and hull fouling have received a great deal of attention (see, e.g., Carlton & Geller, 1993) and are often cited as the most likely vectors for many marine invasions (Ruiz et al., 2000, Seebens, Schwartz, Schupp, & Blasius, 2016). Population genetic tools and advances in statistical techniques have greatly facilitated our understanding of invasion pathways, in part because these data allow independent tests of predictions that stem from observational data (Estoup & Guillemaud, 2010; Geller, Darling, & Carlton, 2010). For example, shipping from the Gulf of Mexico and the eastern seaboard of the United States was the likely vector in the introductions of the ctenophore Mnemiopsis leidyi to the Black, Caspian, North, Baltic, and Mediterranean seas (Reusch, Bolte, Sparwel, Moss, & Javidpour, 2010). Voisin et al. (2005) found maritime traffic promoted recurrent invasions of the kelp Undaria pinnatifida to Australasia from its native range in the Northwest Pacific. For most studies, however, two limitations hamper genetic identification of source and pathway: (1) the lack of robust sampling of native and non‐native populations and (2) the lack of appropriately variable population genetic tools (Estoup & Guillemaud, 2010; Geller et al., 2010).
The Japanese oyster Crassostrea gigas has been proposed to be one of the main vectors facilitating the hitchhiking of estuarine invaders worldwide (Ruesink et al., 2005). For example, Bonnot (1935) documented the occurrence of invasive mollusks with imported Japanese oyster shipments to North American estuaries. Despite the recognized importance of C. gigas exports as a vector of invasion, to our knowledge, only one study has attempted to identify the source populations by genotyping native and non‐native populations of estuarine invaders associated with oysters. Based on genotypic diversity in Japan and the western coast of North America, the invasion of the Asian mud snail Batillaria attramentaria as well as one of its trematode parasites originated in the main region of Japanese C. gigas exports in the Miyagi Prefecture (i.e., Matsushima or Mangoku Bays; Miura, Torchin, Kuris, Hechinger, & Chiba, 2006).
We explored the native and non‐native population genetic structure of the haploid–diploid seaweed Gracilaria vermiculophylla (Ohmi) Papenfuss (Rhodophyta), which has spread from its native distribution in the northwestern Pacific Ocean to virtually every high‐salinity, temperate estuary in Europe and North America (Byers, Gribben, Yeager, & Sotka, 2012; Saunders, 2009; Weinberger, Buchholz, Karez, & Wahl, 2008). G. vermiculophylla can profoundly transform estuarine ecosystems (Byers et al., 2012; Thomsen, Stæhr, Nejrup, & Schiel, 2013) and result in negative economic impacts (Freshwater et al., 2006). G. vermiculophylla was first identified through molecular barcoding from samples collected in 1979 in Baja California (Bellorin, Oliveira, & Oliveira, 2004), 1994 in Elkhorn Slough (Bellorin et al., 2004), 1998 in the Chesapeake Bay (Thomsen, Gurgel, & McGlathery, 2006), and 1995 in France (Rueness, 2005), but these dates are unlikely the first dates of establishment due to poor taxonomic resolution in the Gracilariales (Gurgel & Fredericq, 2004).
Despite a flurry of studies assessing the impacts of the G. vermiculophylla invasion (e.g., Hammann, Buchholz, Karez, & Weinberger, 2013; Nyberg & Wallentinus, 2009; Thomsen et al., 2013), the source of the invasion is known only at a coarse spatial scale. Kim, Weinberger, and Boo (2010) sequenced a portion of the mitochondrial locus cytochrome oxidase subunit one and found the non‐native range was dominated by a single haplotype (Haplotype 6), which was found in the Sea of Japan/East Sea. Unfortunately, their conclusions were limited by population sampling, as they did not sequence any thalli from the Pacific coastline of northeastern Japan (Honshu and Hokkaido Islands), areas of important aquaculture activities not only for C. gigas (Byers, 1999; Ruesink et al., 2005), but also agar extraction of Gracilaria spp., including G. vermiculophylla (Okazaki, 1971). The investigation of the underlying evolutionary processes facilitating this widespread invasion is limited due to the uncertainty of the invasion source(s).
In a previous study describing the impacts of the invasion on genetic diversity and the haploid–diploid life cycle, we genotyped greater than 2,000 thalli sampled from 30 native sites, with an emphasis on the Japanese archipelago (23 of the 30 sites), and 35 non‐native sites along the coastlines of western and eastern North America and Europe (Krueger‐Hadfield et al., 2016). There was an ecological shift from hard to soft substratum during the invasion of North American and European estuaries by Gracilaria vermiculophylla that resulted in a shift from sexual to asexual reproduction as hard substratum is a necessity for algal spore recruitment (Krueger‐Hadfield et al., 2016). Non‐native sites were dominated by diploid tetrasporophytes (>80% diploid thalli, on average) as a result of asexual fragmentation. We also found comparable levels of genetic diversity between native and non‐native sites, suggesting highly genetically diverse inocula, multiple invasions or both (Krueger‐Hadfield et al., 2016).
In order to investigate the global pathways and genetic structure of the Gracilaria vermiculophylla invasion, we analyzed the spatial genetic structure of the thalli previously genotyped (Krueger‐Hadfield et al., 2016) as well as thalli from 25 new native and non‐native sites. Our results pinpoint the Pacific shorelines of northeastern Japan as the ultimate source of introduced populations. Based on ecological, genetic, and historical evidence, we further suggest that G. . vermiculophylla hitchhiked with the C. gigas exports from Japan during the 20th century.
2. Materials and Methods
2.1. Data generation
2.1.1. Sample collection
Algal thalli were sampled from 90 sites across the Northern Hemisphere from 2012 to 2016 (Table S1). Thirty‐seven sites were from the native range and sampled along the coastlines of China, South Korea, and Japan (Figure 1, Table S1, Figure S1). Fifty‐three sites were sampled along the coastlines of western North America (hereafter, WNA), the eastern United States (hereafter, EUSA), and northwestern Africa and Europe, including the British Isles (hereafter, EU).
Figure 1.
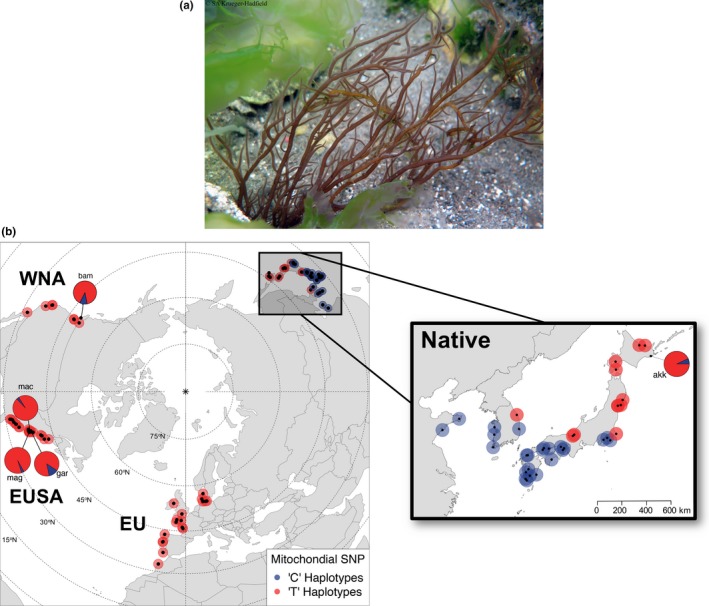
(a) Gracilaria vermiculophylla attached to hard substratum in a tide pool in the native range. Photographic credit: S.A. Krueger‐Hadfield. (b) Geographic distribution of the mitochondrial SNP frequency among sampled sites of G. vermiculophylla in the native range (Northwest Pacific) and non‐native range (WNA = western North America; EUSA = eastern United States; EU = British Isles, continental Europe, and northwestern Africa). At most sites, thalli were either “C” (blue) or “T” (red) at the 945th bp of the sequenced cox1 gene from Kim et al. (2010). At five sites, both “C” and “T” SNPs were detected and relative frequencies are shown by pie charts. Sampling information is provided in Table S1, and mitochondrial sequencing and genotyping data are provided in Figure S1 and Table S2
The species identity of all thalli was confirmed either using DNA barcoding (Kim et al., 2010) or the amplification of 10 species‐specific microsatellite markers (Kollars et al., 2015; Krueger‐Hadfield et al., 2016). Gracilaria vermiculophylla thalli from native sites were attached via holdfasts to hard substrata (i.e., bedrock, pebbles, gastropod shells), whereas, in the non‐native range, there was a spectrum of sites ranging from fully attached to fully free‐floating G. vermiculophylla thalli (Krueger‐Hadfield et al., 2016; Krueger‐Hadfield & Sotka, unpublished data). At all sites, samples were collected with at least one meter separating each putative genet to avoid sampling the same genet twice (Guillemin et al., 2008; Krueger‐Hadfield et al., 2016). Ploidy was determined by observing reproductive material under a binocular microscope (40×) and/or using microsatellite loci (following Krueger‐Hadfield et al., 2016).
2.1.2. DNA extraction, microsatellite and mitochondrial cox1 amplification and genotyping
Total genomic DNA was isolated and simplex PCRs of 10 microsatellite and fragment analysis were performed following Kollars et al. (2015) and Krueger‐Hadfield et al. (2016; see also Table S1). However, for sites sampled in 2015–2016 (Table S1), we only targeted and extracted phenotypically diploid thalli. In total, we genotyped 2,192 diploid and 785 haploid thalli across 90 native and non‐native sites. Only diploid thalli were used for subsequent microsatellite data analyses as the diploids dominated the non‐native range (Krueger‐Hadfield et al., 2016).
The mitochondrial gene cox1 was amplified using the primer sets 43F (Geraldino, Yang, & Boo, 2006) and 880R (Yang et al., 2008) and 622F (Yang et al., 2008) and 1549R (Geraldino et al., 2006) for 201 thalli sampled across both the native and introduced ranges (Table S2, Figure S1). We did not distinguish between haploid and diploid thalli for the mitochondrial sequencing. PCR amplification was performed on a total volume of 25 μl, containing 0.5 U of taq DNA polymerase, 2.5 mmol/L of each dNTP, 2 mmol/L MgCl2, 1× reaction buffer, 250 nmol/L of each primer, and 5 μl of DNA and PCR conditions previously described (Yang et al., 2008). Approximately, 5 μl of PCR product with 1 μl of Orange G loading dye was visualized on 1.5% agarose gels and 1 μl of ethidium bromide.
One microliter of ExoSAP‐It was added to 7 μl of PCR product (Affymetrix, Santa Clara, CA, USA) and incubated for 15 min at 37°C followed by 15 min at 80°C. Four microliters of 2 μmol/L primer was added to each product and sequenced in the forward direction commercially by Eurofins Genomics (Louisville, KY, USA).
We discovered a single nucleotide polymorphism (C/T) at the 945th base pair within the mitochondrial coxI fragment, which delineated among several haplotypes and could be genotyped using the restriction enzyme Af1III (New England Biolabs, Ipswich, MA, USA; ACGTG(T/C; Table 1, Table S2, Figure S1f). Restriction enzyme digestion was performed on thalli from each site using a total volume of 25 μl, containing 1× buffer, 10 U of AF1III and 5 μl of the cox1 PCR product amplified using the primers 622F (Yang et al., 2008) and 1549R (Geraldino et al., 2006) and under the following reaction conditions: 37°C for 2 h, 80°C for 20 min, and 20°C for 5 min. Restriction digest products were visualized on 1.5% agarose gels with 1 μl of ethidium bromide (Figure S1f).
Table 1.
Haplotypic diversity across the native and non‐native ranges of Gracilaria vermiculophylla. a) Sites south and north of approximately 35°N in China, South Korea, and Japan were delineated based on C:T frequencies (see also Figure 1). We used the haplotype numbers assigned by Kim et al. (2010) as these covered the ~1,200 bp of cox1. We have extended Kim et al. (2010) and generated haplotype numbers for six new haplotypes (haplotypes 26–31; see Table S2). The proportion of the “C” and “T” SNPs are given for each region (see Table S1 for sampling sizes are each site). b) The biogeographic province (Briggs & Bowen, 2011) and ecoregions (Spalding et al., 2007) in which each of the native range haplotypes were found
| (a) Region | “C” Haplotypes | “T” Haplotypes | C:T Ratio |
|---|---|---|---|
| South of ~35°N | 1–4, 8–15, 17, 27, 30–31 | None | 1.00: 0.00 |
| North of ~35°N | 26 | 5–7, 16, 18, 28 | 0.01: 0.99 |
| WNA | 15 | 6, 19, 29 | 0.02: 0.98 |
| EUSA | 15 | 6 | 0.01: 0.99 |
| EU | None | 6, 18 | 0.00: 1.00 |
| (b) Biogeographic province | Ecoregion | “C” Haplotypes | “T” Haplotypes |
|---|---|---|---|
| Cold temperate—Kurile | Sea of Okhotsk | None | 6 |
| Oyashio Current | 26 | 6 | |
| Northeastern Honshu | None | 6 | |
| Cold temperate—Oriental | Sea of Japan | None | 5–7 |
| Northeastern Honshu | None | 6, 18, 28 | |
| Yellow Sea | 1–2, 17 | None | |
| Warm temperate—Sino‐Japanese | Sea of Japan | 4 | None |
| East China Sea | 2–4; 8–13; 30–31 | None | |
| Central Kuroshio Current | 14–15; 27 | 16 |
2.2. Data Analyses
2.2.1. Mitochondrial analyses
Sequences were edited using 4Peaks (Nucleobytes, The Netherlands), aligned with the haplotypes from Kim et al. (2010) using Muscle (Edgar, 2004) in Seaview ver. 4.6 (Gouy, Guindon, & Gascuel, 2010) with default parameters. We assigned haplotype numbers using Kim et al. (2010), which sequenced a ~1200‐base pair fragment. We also defined six new ~1200‐bp haplotypes that were not previously sequenced (Kim et al., 2010) using DnaSP, ver. 5.10.1 (Librado & Rozas, 2009). We ignored the haplotype designations of Gulbransen, McGlathery, Marklund, Norris, & Gurgel (2012) because their haplotypes covered only a subset of this larger ~1200‐bp fragment.
A phylogeny of the haplotypes from Kim et al. (2010) and new haplotypes uncovered in this study was constructed using the Hasegawa–Kishino–Yano model (HKY) plus gamma (Hasegawa, Kishino, & Yano, 1985), one of the appropriate models detected using ModelTest (Darriba, Taboada, Doallo, & Posada, 2012; Guindon & Gascuel, 2003), as implemented in MEGA6 (Tamura et al. 2013). The resulting trees were edited using FigTree ver. 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/).
2.2.2. Ploidy and genet determination
The number of repeated identical multilocus microsatellite genotypes (MLGs) was computed using GenClone ver. 2.0 (Arnaud‐Haond & Belkhir, 2006) and a custom R, ver. 3.2.2 (R Core Team, 2016) routine following (Arnaud‐Haond, Duarte, Alberto, & Serrao, 2007; Parks & Werth, 1993), now implemented in poppr ver. 2.2.1 (Kamvar, Brooks, & Grünwald, 2015; Kamvar, Tabima, & Grünwald, 2014). P sex, or the probability for a given MLG to be observed in N samples as a consequence of different sexual reproductive events, was calculated for each repeated MLG. If P sex was greater than 0.05, duplicated multilocus genotypes were considered as different genets. If P sex was smaller than 0.05, the duplicated MLGs were considered as ramets (or clones) of the same genet.
Studies on the population genetics of clonal organisms remove repeated MLGs before calculating heterozygosity and other F‐statistics in order to avoid distorting these estimates (e.g., Halkett et al., 2005; Krueger‐Hadfield, Collen, Daguin‐Thiébaut, & Valero, 2011; Sunnucks, de Barro, Lushai, Maclean, & Hales, 1997). Following this convention, we chose the conservative approach of performing the following analyses for diploid MLGs for which P sex > 0.05 (i.e., one thallus per genotype based on P sex).
2.2.3. Within and between population genetic variation
Kollars et al. (2015) and Krueger‐Hadfield et al. (2016) provided locus information, the number of alleles, and basic summary statistics. For each site, the average expected heterozygosity (H E) was calculated using GenAlEx, ver. 6.5 (Peakall & Smouse, 2006, 2012). An estimate of the mean expected number of alleles (A E) was computed using rarefaction implemented in the program HP‐Rare, ver. 1.0 (Kalinowski, 2005) on the smallest sample size of 10 diploids (i.e., 20 alleles). Finally, the number of expected MLGs (eMLGs) at the smallest sample size (n = 10 diploid thalli) was estimated using rarefaction in the R package poppr, ver. 2.2.1 (Kamvar et al., 2014, 2015). For the latter two diversity metrics, sites with less than 10 diploid thalli were excluded from calculations.
We calculated pairwise genetic differentiation based on allele identity (F ST) and allele size (ρST) among sites along each of four coastlines: Japan, WNA, EUSA, and EU using genepop ver. 4.4 (Rousset, 2008). We measured geographic distance (km) following the contours of each coastlines between all site pairs using the measure distance function in Google® Maps. We performed Mantel tests in order to detect relationships between the genetic and geographic distance along each of the four coastlines using R (R Core Team, 2016).
2.2.4. Multivariate analyses of microsatellite data
To assess relationships among multilocus genotypes, we pursued a discriminant analysis of principal components (DAPC), a multivariate analysis that avoids making strong assumptions about the underlying genetic model (Jombart, Pontier, & Dufour, 2009). DAPC finds the principal components that best summarize the differences among clusters that are found, while also minimizing within‐cluster variation (Jombart, Devillard, & Balloux, 2010), and is implemented in the R package adegenet, ver. 2.0.1 (Jombart, 2008; Jombart & Ahmed, 2011). The procedure first generates a principal component analysis (PCA) on predefined groups (see below). These PCs were, then, used as variables for a discriminant analysis that maximizes the intergroup component of variation.
We performed the DAPC with increasing numbers of PCs on 90% of our data, and then the remaining 10% of the individuals were projected onto the discriminant axes constructed by the DAPC. It was possible to measure how accurately the remaining 10% of the individuals were placed in multidimensional space (i.e., how well their position corresponds to their group membership). Based on this cross‐validation with the xvalDapc function, we retained 88 principal components (PCs) that explained 87.8% of the total variance for subsequent DAPCs (Figure S2a).
We estimated how well‐supported the group membership was relative to five a priori subregions using the compoplot function in adegenet. Posterior group memberships were utilized in order to indicate admixture or the misclassification when prior groups are used to conduct the DAPC. Based on the mitochondrial SNP, we divided the native range into the “C” haplotype group and the “T” haplotype group corresponding to the break between ~35°N in Japan and South Korea in which the two SNPs dominated to the south and north, respectively (Figure 1, Table 1). For the introduced range, we treated each coastline as a different group: WNA, EUSA, and EU. The stability of a priori group membership probabilities, derived from proportions of successful reassignments based on retained discriminant functions of DAPC, was high (>92%) for the native “C” haplotype and EUSA and EU subregions, but lower for the native “T” haplotype and WNA subregions (85.3% and 76.4%, respectively; Figure S2). Based on these reassignment frequencies, we performed the DAPC with the aforementioned five subregions.
Next, we predicted the assignment of thalli from one region to another using supplementary individuals that were not used in model construction as implemented in adegenet. We performed this analysis with training data composed of (1) only the native sites and (2) sites from the native range and two of the three introduced coastlines. We transformed “new data” using the centering and scaling of training data (i.e., (1) the native range alone or (2) the native range and the two introduced coastlines). Using the same discriminant coefficients, we predicted the position of the new individuals (i.e., (1) all introduced sites and (2) the one introduced coastline not used in the training data).
Because both native and non‐native populations of Gracilaria vermiculophylla are out of Hardy–Weinberg equilibrium (Krueger‐Hadfield et al., 2016), we chose to use Bayesian model‐based clustering that relaxes the HWE assumption as implemented in the software instruct (Gao, Williamson, & Bustamante, 2007). Simulations were performed using instruct with a model including both biparental inbreeding and admixture, where each individual drew some fraction of its genome from each of the K populations. A burn‐in of 300,000 repetitions and a run length of 500,000 were used for K = 2 to K = 30, where 20 chains were run for each K.
To evaluate the values of K, we analyzed K = 2 to 30 clusters using clumpak (Kopelman, Mayzel, Jakobsson, Rosenberg, & Mayrose, 2015). clumpak identifies sets of highly similar runs across the 20 independent runs of each K generated with instruct and separates them into distinct major and minor modes. clumpak utilizes the software clumpp (Jakobsson & Rosenberg, 2007) in order to generate a consensus solution for each distinct mode using a Markov clustering algorithm that relies on a similarity matrix between replicate runs. Next, clumpak identifies an optimal alignment of inferred clusters and matches the clusters across the values of K tested.
We determined the optimal number of K using outputs from instruct and clumpak. First, we plotted the number of clusters against the values of DIC (± SE) and chose the value of K at the point at which the curve reached an asymptote. The lower the DIC value, the better fit of the model used (i.e., the number of K). Second, we used the number of independent chains out of 20 that generated the major mode and the highest mean similarity score from clumpak.
We constructed a neighbor‐joining (NJ) tree based on Jaccard distance using the R packages ape ver. 3.5 (Paradis, Claude, & Strimmer, 2004). The resulting trees were edited using FigTree.
3. Results
3.1. Mitochondrial structure across native and introduced ranges
Across 201 thalli sequenced from the native and non‐native ranges, we identified 10 distinct cox1 haplotypes (Figure S1, Table S2), four of which were described previously by Kim et al. (2010). Three groups of haplotypes were identified with a maximum‐likelihood (ML) analysis based on 19 previously described haplotypes (Kim et al., 2010) and the six new haplotypes from this study. The three clades were heterogeneously distributed across the native range (Figure S1a). Two clades were found south of ~35°N latitude: One consisted of 10 haplotypes including haplotypes 1, 2, 3, 9, 10, 11, 12, 17, 26, 30, 31 and the other clade of six haplotypes included 4, 8, 13, 14, 15, and 27 (Table 1, Figure S1a). The third clade of six haplotypes were only found sequenced north of ~35°N in the native range (Figure 1, Table 1, Figure S1). Seventy‐two of 77 thalli, or 94% of sequenced thalli, collected north of 35°N were haplotype 6, the haplotype that is known to dominate the introduced range (Kim et al., 2010).
Two single nucleotide polymorphisms, or SNPs (945th bp and 1040th bp from Kim et al., 2010; Table S2) delineated all haplotypes that occur north (“T”) from those that occurred south (“C”) of ~35°N in the native range (Figure S1, Table S2). Based on RFLP genotyping of 691 thalli collected south of ~35°N latitude, we found that all were “C” haplotypes (Figure 1, Table 1). An RFLP analysis of 375 thalli collected north of ~35°N revealed all were “T” haplotypes. The exceptions were three thalli from Akkeshi (akk) that belonged to the new “C” haplotype 26 that diverges from all other haplotypes by at least five base pairs (Figure S1a, Table S2).
In the non‐native range, 99.5% (1,597 of 1,605 thalli) of the thalli were “T” haplotypes and were dominated by haplotype 6 (82 of 101, or 81%, sequenced thalli; Figure 1, Table 1, Figure S1b‐d, Table S2). Only nine non‐native thalli were “C” haplotypes (three from Bamfield/bam, four from Gargatha/gar, one from Machipongo/mac, and one from Magotha/mag) and all belonged to haplotype 15. Haplotype 15 was originally sampled at Funabashi in Tokyo Bay (Kim et al., 2010), near the “C/T” haplotype break. The “T” haplotypes 19 (Elkhorn Slough/elk; Kim et al., 2010) and 29 (Bamfield/bam; this study) were detected in the non‐native range, but not in the native range, likely because of under‐sampling in northern Japan. For example, the “T” haplotype 18 was previously only sampled in France (Kim et al., 2010), but was found after sequencing thalli from Mangoku‐ura (mng; Figure S1, Table S2).
Taken together, mitochondrial data suggest the region north of ~35°N latitude was the source of the widespread Northern Hemisphere invasion (Figure 1). However, we could not resolve the sites that contributed to the invasion at finer spatial scales due to the lack of mitochondrial polymorphism within this northern native region.
3.2. Microsatellite structure across native and non‐native ranges
The ten microsatellite loci revealed profound genetic structure within the native range and identified cryptic genetic structure among and within non‐native coastlines that could not be detected with mitochondrial sequencing alone. We focus on each of these patterns in turn.
Within the native range, microsatellite genotypes distinguished northern sites dominated by “T” cox1 haplotypes and southern sites dominated by “C” haplotypes. This separation was evident along the first axis of a multivariate DAPC, which itself explained 55% of overall variation (Figure 2). “T” versus “C” differentiation was also confirmed by Bayesian clustering analyses in which sites were placed into subregions corresponding to genetic similarity (Figure 3, Figure S3, Table S3). At K = 5, the optimal K based on the similarity score using clumpak and the curve of DIC estimates (Figure S3) showed strong differentiation within and between the “T” and “C” regions in the native range (see Figure S3b–f for individual‐level analyses and across K's).
Figure 2.
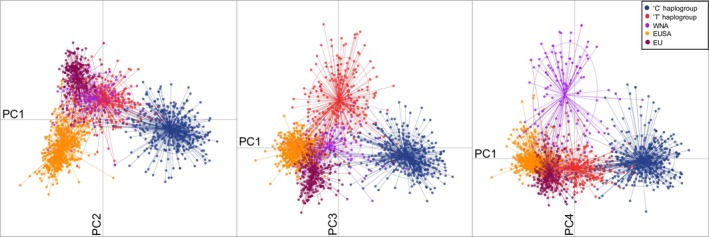
DAPC (discriminant analysis of principal components) relationships among microsatellite genotypes of Gracilaria vermiculophylla. We color‐coded individuals corresponding to five a priori groups with high reassignment frequencies (see Figure S2) as the “C” haplogroup (native sites dominated by the “C” mtSNP and south of ~35°N), the “T” haplogroup (native sites dominated by the “T” mtSNP and north of ~35°N), WNA (western North America), EUSA (eastern United States of America), and EU (Europe and northern Africa). The first four principal components are shown (PC1: 55.4%, PC2: 19.4%, PC3: 14.3%, PC4: 10.9%)
Figure 3.
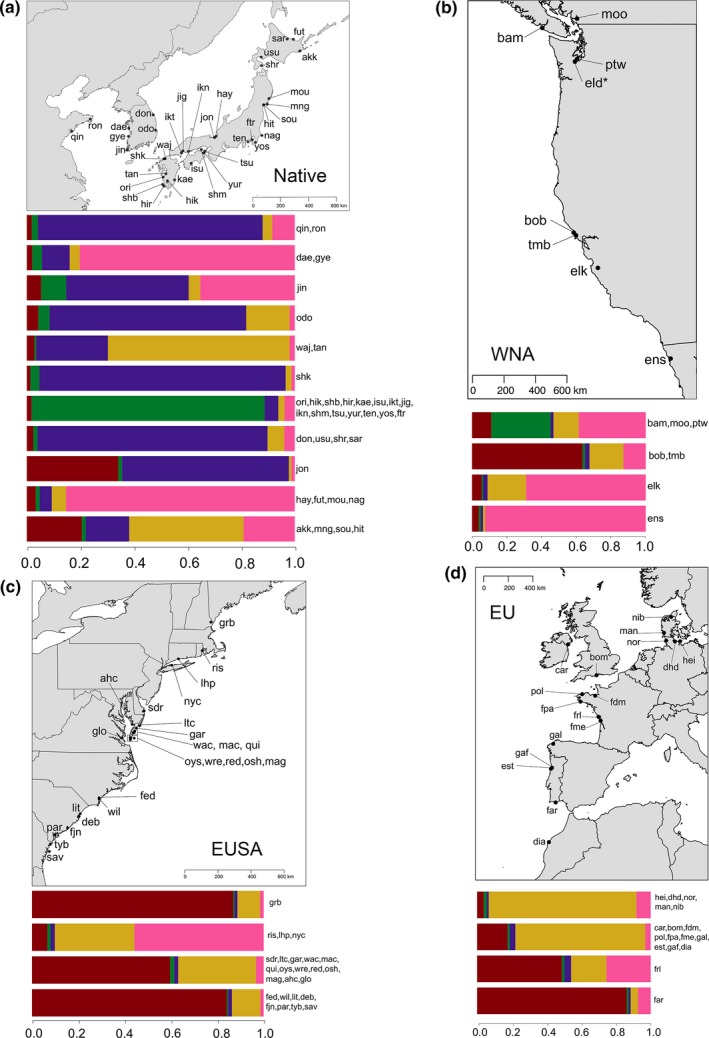
The mean assignment to five genetic clusters (colored as red, green, purple, gold, and pink) as generated using instruct (Gao et al., 2007) and clumpak (Kopelman et al., 2015). We grouped thalli across sites of similar genetic composition using a visual inspection of individual cluster assignment (see Figure S3e) and used barplots to display individual assignments averaged across sites for that group. As Eld Inlet (eld) had only one unique MLG based on P sex, it was excluded from Bayesian analyses, but is shown on the map
At finer geographic scales, there was a high degree of genetic differentiation among sites that were close in proximity (i.e., less than 100 km) in the native range. Genetic differentiation in the native range, as measured using allele identity (F ST) and allele size (ρST), was high on average (~0.47, Figure S4, Table S4). Genetic isolation was positively correlated with geographic distance when measured using allele identity (F ST), but not with allele size (ρST, Figure S4, Table S4).
In the non‐native range, the three continental shorelines were genetically differentiated and, thus, likely reflect different invasion histories. The DAPC analyses showed a separation of eastern United States (labeled as EUSA) thalli from the northern Japanese sites and other introduced shorelines along the second principal component axis, which itself explained ~19% of the variation (Figure 2). The British Isles, European and northwestern African (labeled as EU), and western North American (labeled as WNA) thalli were differentiated from other regions along the third (14.3% of variation) and fourth (10.9% of variation) principal component axes, respectively (Figure 2). With increasing numbers of genetic clusters using Bayesian analyses, introduced coastlines showed greater levels of genetic divergence from each other. At K = 3, WNA sites were differentiated from the majority of EUSA and EU sites (Figure S3c), while at K = 5, the EUSA and EU were genetically distinguishable (Figure S3e).
The strength of differentiation among populations along a shoreline differed across non‐native coastlines. Within WNA and EU shorelines, sites exhibited greater levels of genetic differentiation (average F ST ~ 0.33 and ρST ~ 0.20; Table S4) than was seen along the EUSA (average F ST ~ 0.15 and ρST ~ 0.18; Table S4). Significant patterns of isolation by distance (IBD) were detected along WNA and EU coastlines, but IBD was only marginally significant along the EUSA when measuring differentiation by allele identity (F ST; Figure S4, Table S4). In contrast, there were positive relationships between genetic differentiation and geographic distance along the EU and EUSA when measuring differentiation by allele size, but they were only marginally significant (ρST, Figure S4, Table S4).
It is likely that among‐site differentiation within the non‐native range reflected complex genetic origins that differ between coastlines. Along the WNA coastline, the Pacific Northwest (bam, pmo, and ptw), Californian (bob and tmb), Elkhorn Slough (elk), and Ensenada (ens) sites were composed of different genetic clusters (Figure 3; Figure S3). Similarly, a neighbor‐joining clustering analysis of site‐level genetic distances indicated that the WNA coastline has been separately invaded on two or three occasions, given that sites are in separate locations across the tree (Figure S5). However, the bootstrap support for the NJ tree was poor and, as a result, this analysis should be interpreted with caution.
Along the EUSA coastline, most sites were composed of a homogenous set of genetic constituents, regardless of the number of genetic clusters (Figure 3, Figure S3). The exceptions were the rocky‐shore sites of Long Island Sound (lhp and nyc) and Narragansett Bay (ris), which seemed to have genetic constituents more closely aligned with WNA and EU sites (Figure 3, Figure S3). In the NJ clustering analysis, the majority of the EUSA sites were also part of the same clade, with the exception of these three northeastern sites (ris, lhp, and nyc; Figure S5).
Along the EU coastline, Bayesian clustering revealed differences between sites sampled in Germany and Denmark (hei, dhd, nib, man, nor) versus a group of French (frl) and Portuguese populations (far), while the remaining populations in Ireland, the United Kingdom, France, Spain, Portugal, and Morocco reflected admixture and a similar set of genetic constituents (Figure 3, Figure S3). The NJ tree suggested a largely similar origin of all European populations, with the exception of the site in Dorset (bom) in the United Kingdom which clustered with a site in the Chesapeake Bay (ahc; Figure S5).
There was a significant relationship between different measures of genetic and genotypic diversity (H E , A E, and eMLG) versus latitude along the EUSA and for genetic diversity metrics (H E and A E) versus latitude (Figure 4, Table S5). Highest diversity was found in the mid‐latitudes along each coastline. Similar patterns were found along the EU coastline, but none of the patterns were significant (Table S5).
Figure 4.
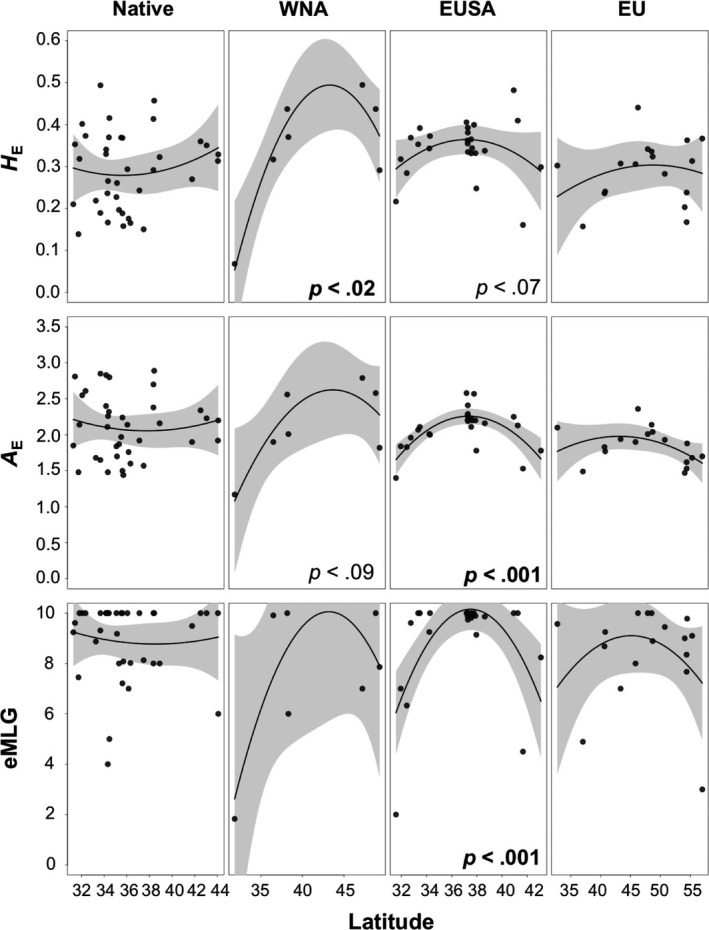
Genetic (H E and A E) and genotypic (eMLG) diversity as a function of latitude along the coastlines of the native range, WNA, EUSA, and EU. Significance values are shown in the plots and bold if less than p = .05
3.3. Population sources for primary and secondary introductions
Several analyses of microsatellite data indicated the Pacific shoreline of northeastern Japan was the ultimate genetic source of introduced Gracilaria vermiculophylla thalli throughout the Northern Hemisphere. Blind assignment of introduced populations onto multivariate DAPC space created by genotypes from native sites suggested most non‐native thalli originated in three northern Japanese sites (791 thalli; ~83%): Soukanzan (sou), Mangoku‐ura (mng), and Akkeshi (akk, Figure 1, Figure S6). Of the remaining 164 thalli, 101 (~11%) were assigned to other northern “T” haplotype sites, principally along northeastern Honshu and eastern Hokkaido Islands (Figure 1, Figure S6). Of the remaining 63 thalli, 14 WNA thalli (~2%) were assigned to Yoshio (yos), a site on the Chiba Peninsula at the “C/T” break, and 15 EU thalli (~2%) and 22 EUSA thalli (~2%) were assigned to Shikanoshima (shk), located in northern Kyushu Island (Figure 1, Figure S6).
Both Bayesian (Figure S3) and neighbor‐joining clustering methods (Figure S5) supported the notion that non‐native coastlines were most closely aligned with the Pacific shoreline of northeastern Japan. With increasing K's, the genetic clusters found in the northern “T” haplotype sites were the same clusters found predominately along each non‐native coastline (Figure 3, Figure S3). Similarly, NJ analyses also confirmed genetic similarity of Soukanzan (sou), Mangoku‐ura (mng), and Akkeshi (akk) with non‐native sites (Figure S5).
Finally, as briefly mentioned above, we found some evidence consistent with secondary introductions (i.e., movement of introduced thalli to other locations within the introduced range). The strongest evidence of secondary introduction came from the three sites from Long Island Sound and Narragansett Bay (nyc, lhp, ris). Bayesian clustering at K = 5 (Figure 3, Figure S3) and the NJ analyses (Figure S5) suggested these sites were more closely aligned with WNA sites (elk, ens) than with other EUSA sites. In addition, blind assignment of non‐native populations onto multivariate DAPC space created by genotypes from sites in the native range and two of the three non‐native coastlines suggested non‐native thalli may have more complex primary and secondary invasions (Figure S6). For example, EUSA and EU thalli were assigned to sites from the central Californian coast in addition to the northeastern coastline of Japan.
4. Discussion
Using a combination of mitochondrial and microsatellite genotyping, we discovered profound phylogeographic structure in the native range of Gracilaria vermiculophylla, reflecting low levels of contemporary gene flow. Introduced populations of North America and Europe exhibited mitochondrial and nuclear genotypes largely indistinguishable to those of the Pacific coastline of northeastern Japan (i.e., northern Honshu and southeastern Hokkaido Islands). Because this area historically served as the principal source for oyster exports worldwide, we suggest that oysters were the key vector for the G. vermiculophylla invasion. We discuss each of these findings in detail below.
4.1. Phylogeographic structure in the native range
In comparison with well‐studied biogeographic provinces, such as those in northeast (NE) Atlantic, less is known about the evolutionary history of the organisms that inhabit the northwest (NW) Pacific and it is not clear whether hypotheses drawn from patterns in other regions apply (Cheang et al., 2010). Phylogeographic studies in the NW Pacific have been biased toward commercially exploitable fish and mollusks and the majority have been underappreciated within English‐language journals because of language barriers (Ni, Li, Kong, & Yu, 2014). The existing studies reveal that phylogeographic patterns complex due to intricate topography and dynamic current systems (Wang, 1999).
The marginal seas of the South China Sea, the East China Sea, and the Sea of Japan/East Sea may have served as glacial refugia, although intraspecific variation in demographic histories is common across taxa (Ni et al., 2014). He, Mukai, Chung, and Zhang (2015) found the amphibious mudskipper Periophthalmus modestus colonized northward from the South China Sea through the East China Sea along the coasts of Korea and Japan. Other studies have found higher haplotypic diversity in southern South Korea and southern Japan (i.e., East China Sea) as compared to Honshu and Hokkaido populations (Ho, Kwan, Kim, & Won, 2014; Kim, Hoarau, & Boo, 2012), suggesting northward colonization. However, although studies have found southern clade haplotypes decrease to the north (Azuma & Chiba, 2015), no studies, to our knowledge, have found as sharp a break as observed in our study of Gracilaria vermiculophylla in the NW Pacific (Figure 1, Figure S1).
We found three shallow clades among mtDNA sequences, suggesting divergent demographic histories that were geographically correlated (Figure S1e). Two “C” groups were found south of ~35°N, corresponding to warm temperate biogeographic provinces, whereas the “T” group was found north of ~35°N corresponding to cold temperate biogeographic provinces (Briggs & Bowen, 2011; Spalding et al., 2007). The “C” clade with 10 haplotypes was found predominately in the East China Sea and Yellow Sea, whereas the other “C” clade was found further east from South Korea to the Chiba Peninsula in Central Honshu (Figure S1). The “T” haplotypes may have spread northward from the refugium in the Sea of Japan/East Sea, whereas the “C” haplotypes were restricted to warm‐water provinces. Present‐day maintenance of the “C”/“T” boundary may reflect current patterns, such as the barrier at Cape Inubo where the warm‐water Kuroshio Current meets the cold‐water Oyashio current. It is difficult to assess which clade is most basal given the lack of sequence information for closely related Gracilaria species.
Although the majority of studies have focused on broader scale patterns across the marginal seas of the NW Pacific from southern China to Japan (Ni et al., 2014), there is strong genetic differentiation between populations along the Sea of Japan/East Sea and the Pacific coast. For example, the eastern and western coastlines of Japan were genetically differentiated between populations of Pterogobus gobies (Akihito et al., 2008), the fucoids Sargassum hemiphyllum (Cheang & Chung, 2010) and S. horneri (Uwai, Kogame, Yoshida, Kawai, & Ajisaka, 2009) and the kelp Undaria pinnatifida (Uwai et al., 2006), but not in the gastropod Littorina brevicula (Azuma & Chiba, 2015). The “T” haplotype 6 was common on both coastlines of Japan at roughly 35°N and the eastern coastlines of South Korea and Russia, but haplotypes 5 and 7 and 16, 18, and 28 were only sampled in the Sea of Japan/East Sea and Pacific Ocean populations, respectively (Figure S1a).
Microsatellite genotypes, on the other hand, were characterized by genetic differentiation that corresponded to a large degree with extant current patterns (Figures 3 and S3). For example, the southern Japanese sites, from southeastern Kyushu to the Chiba Peninsula and likely isolated by the Kuroshio Current, formed a large cluster with minimal admixture (Figure S3). Likewise, Sea of Japan/East Sea and Hokkaido sites formed a group that was consistent with the flow of the Tsushima Current. In contrast, the thalli sampled at Hayase River (hay) and Futatsuiwa (fut) were genetically more similar to northeastern Honshu sites (mou and nag). It is possible that these reflect some movement of Gracilaria vermiculophylla thalli as a result of aquaculture practices (i.e., oysters or the alga itself). Finally, the similarity of thalli sampled at Shikanoshima (shk) to thalli sampled from putative source sites (akk, mng, sou) is likely due to the frequent fishery transport between Hokkaido and Kyushu (M. Nakaoka, personal communication), of which similar patterns have been found in the red seaweed Gelidium elegans (Kim et al., 2012).
4.2. Pathway of the oyster?
Gracilaria vermiculophylla invaded low‐energy, high‐salinity estuaries dominated by soft‐sediments on all continental margins of North America (Bellorin et al., 2004; Kim et al., 2010), habitats in which oysters were seeded and cultivated in huge quantities (Ruesink et al., 2005). However, the first records of G. vermiculophylla along the WNA, EUSA, and EU coastlines were 1979 (Bellorin et al., 2004), 1998 (Thomsen et al., 2006), and 1995 (Rueness, 2005), respectively, which are decades after deliberate oyster movement (Barrett, 1963; Ruesink et al., 2005), leading to speculation that hull fouling or ballast water vectors were more likely (Ruiz, Fofonoff, Steves, Foss, & Shiba, 2011; J. Carlton, personal communication). Although G. vermiculophylla thalli have been shown to survive desiccation (Nyberg & Wallentinus, 2009), they are negatively buoyant and are unlikely to be taken up in ballast water. Moreover, red algal spermatia and spores are only viable for several hours to a few days (see Destombe, Godin, & Remy, 1990 or reviewed in Krueger‐Hadfield, Roze, Correa, Destombe, & Valero, 2015) and would not likely survive an oceanic crossing. Moreover, G. vermiculophylla is not commonly found on boat hulls or pontoons in marinas (see Mineur, Johnson, & Maggs, 2008; S.A. Krueger‐Hadfield, personal observation).
Our results, however, point to oyster transport as the main vector for the spread of Gracilaria vermiculophylla. Specifically, our genetic analyses assigned nearly all introduced algal thalli to three prominent sites of oyster aquaculture (reviewed in Barrett, 1963; Byers, 1999; Ruesink et al., 2005) along the northeastern coastline of Japan: Soukanzan (sou, Matsushima Bay), Mangoku‐ura (mng, Mangoku Bay), and Akkeshi (akk, Lake Akkeshi). Anecdotal evidence suggests that oysters introduced to Puget Sound were from Akkeshi (Galtsoff, 1932) and that subsequent introductions to California (Barrett, 1963; Chew, 1979) and France (Goulletquer, 1998; Maurin & LeDantec, 1979) were mainly from Matsushima Bay (Chew, 1979). Moreover, sequencing of herbarium specimens has pre‐dated the first records of G. vermiculophylla thalli by several decades along the WNA and EUSA coastlines (S.A. Krueger‐Hadfield, E.E. Sotka & K.A. Miller, unpublished data). First records now align with the same decades in which Crassostrea gigas was introduced to WNA and EUSA estuaries. Mineur, Le Roux, Maggs, and Verlaque (2014) used the discovery rate of non‐native species in Europe from 1966 to 2012 and suggested continuous importation of oysters over this entire period. Thus, in EU estuaries, G. vermiculophylla introductions are possible much earlier than currently documented. While we cannot eliminate deliberate introductions for Gracilaria aquaculture, or incidental introductions from hull fouling or ballast water as playing a role, the simplest, most parsimonious explanation is oyster introductions.
Surprisingly, and despite the fact that dozens of species were introduced worldwide with Japanese Crossastrea gigas from northeastern Japan (Byers, 1999; Ruesink et al., 2005), we know only a single study in which this region was independently identified as an invasion source using genetic markers (i.e., the mud snail Batillaria attramentaria, Miura et al., 2006). The dearth of studies that identified this historically important source region is due either to poor resolution of markers, poor sampling of native range populations, or both. Our study and Miura et al. (2006) rigorously sampled the native and non‐native ranges enabling the identification of intensive oyster aquaculture as the source populations, while simultaneously excluding other native sites as candidate donor regions.
While Crassostrea gigas has been introduced to the Southern Hemisphere (Ruesink et al., 2005), there are no records of Gracilaria vermiculophylla from these areas. However, there are known free‐floating populations of Gracilaria and Gracilariopsis thalli from Argentina (Martín, Boraso de Ziaxso, & Leonardi, 2011) and South Africa (Govender, 2011; J. Bolton, personal communication). Thalli in other Northern Hemisphere bays and estuaries may have been previously misidentified as native congenerics or different genera (Tomales Bay, CA: Huntington & Boyer, 2008; Bodega Bay: Grosholz & Ruiz, 2009) until representative thalli were barcoded using molecular tools (e.g., Saunders, 2009). It is possible that G. vermiculophylla thalli are cryptogenic in the Southern Hemisphere and simply await documentation.
4.3. Complexity of introduction history
There were likely multiple Gracilaria vermiculophylla invasions along North American, European, and northwest African shorelines. Along the WNA coast, estuaries in Tomales Bay (tmb), Elkhorn Slough (elk), Puget Sound (ptw), and British Columbia (pmo, bam) were composed of different genetic cohorts of G. vermiculophylla. This is consistent with the anecdotal histories of oyster introduction and cultivation in these locations (Barrett, 1963). Most WNA thalli (~83%) had a genetic signature consistent with a northeastern Japanese source, but there were thalli from California and Puget Sound/British Columbia that appeared to be a mix of northern and southern Japanese genotypes (Figure 3, Figure S3). Similarly, some Bamfield thalli (bam) were the “C” mitochondrial genotypes. Together, this suggests some movement of southern thalli did occur historically (Byers, 1999), but the northeastern coastline of Japan was the source of the overwhelming majority of non‐native thalli.
Along the EU coast, we also found evidence of genetic structure consistent with multiple introductions. There is a long and complicated history of oyster introductions that dates back to the 1600s directly from the northwest Pacific (Goulletquer, 1998; Grizel & Héral, 1991; Haydar & Wolff, 2011; Lallias et al., 2015; Ó Foighil, Gaffney, Wilbur, & Hilbish, 1998). In addition to Crassostrea gigas introductions, C. virginica was introduced from the EUSA to hatcheries and aquaculture facilities throughout Western Europe (Ruesink et al., 2005). Other invaders, such as Sargassum muticum and Crepidula fornicata, are thought to have been accidentally introduced to Europe as a result of C. gigas oyster introductions from Japan or British Columbia (Farnham, Fletcher, & Irvine, 1973) or C. viriginica imported from EUSA (Riquet, Daguin‐Thiébaut, Ballenghien, Bierne, & Viard, 2013), respectively. The similarity of European sites to some WNA and EUSA sites suggests that the invasion of the European coastline may reflect primary invasions from Japan, secondary invasions from WNA and EUSA or both. With our current set of microsatellite genotypes, we cannot at present distinguish between these alternatives.
Along the EUSA coastline, there were at least two different invasions: one to Long Island Sound (nyc, lhp) and Narragansett Bay (ris) and the other to the outer coast of Virginia and the Chesapeake Bay (Figure 3). The first introduction stayed localized to Long Island Sound to the Cape Cod Peninsula and maintains a genetic constitution that is more similar to the EU and WNA than to the other sites along the EUSA. The second introduction rapidly spread along the eastern seaboard to New Hampshire (grb) and south to the Carolinas and Georgia and may have been introduced directly from Japan with oysters (Mann, 1979). However, as with the EU, we cannot distinguish among primary and secondary invasions with our current data.
Although the first records of EUSA Gracilaria vermiculophylla only date back to 1998, this alga likely occurred in the Chesapeake Bay and along New England coastlines for longer than currently thought and was misidentified as the native congeneric G. tikvahiae (Thomsen et al., 2006; Nettleton, Mathieson, Thornber, Neefus, & Yarish, 2013; S.A. Krueger‐Hadfield & K.A. Miller, unpublished data). Crassostrea gigas was introduced to Maine and Massachusetts in 1949 and the 1970s (Dean, 1979; Hickey, 1979), Connecticut in the 1940s (Loosanoff & Davis, 1963), New Jersey in the 1930s, Delaware in 1962, and Maryland in the 1970s (Andrews, 1980). Likewise, the arrival of an oyster pathogen in the Chesapeake Bay was linked to rogue plantings of C. gigas from Japan, California, the northwest Pacific, or some combination of these oyster cultivation regions (Burreson, Stokes, & Friedman, 2000). Taken together, our contemporary genetic data strongly point to the movement of oysters as the main vector and pathway for moving Gracilaria vermiculophylla across oceanic basins.
4.4. Secondary spread within coastlines
Our genetic data suggest different patterns of secondary spread once Gracilaria vermiculophylla invaded the three non‐native coastlines of the Northern Hemisphere. Along the WNA coastline, the genetic structure of G. vermiculophylla was the most profound, relative to other continental margins. This suggests that multiple primary invasions arrived from Japan and that secondary spread was likely less important than along the EUSA and EU coastlines. This is a tentative conclusion, however, as the geographic extent of the introduction of G. vermiculophylla is poorly characterized along the WNA coast. In 2015, for example, we found G. vermiculophylla in Bodega Bay (bob), Tomales Bay (tmb), and San Diego (E.E. Sotka and S.A. Krueger‐Hadfield, unpublished data). Grosholz and Ruiz (2009) documented the dramatic change in abundance of what they called Gracilariopsis sjoestedtii in Bodega Harbor as a result of the invasion of the European crab Carcinus maenas. They analyzed aerial photographs, but performed their study in the same area we sampled G. vermiculophylla in this study. An herbarium sample at the Bodega Marine Lab from the late 1960s from this same area was labeled Gracilaria cf. vermiculophylla (P.G. Connors, personal communication) and has subsequently been identified as G. vermiculophylla (S.A. Krueger‐Hadfield, E.E. Sotka & K.A. Miller, unpublished data).
Relative to the case along the WNA coastline, the magnitude of secondary spread along the EU and EUSA coastlines is less speculative. Along the EUSA, the high levels of diversity observed in the sites studied along the eastern shore of Virginia (Figure 4; see also Gulbransen et al., 2012) strongly suggest that the outer coast of Virginia and the Chesapeake Bay were the initial sites of introduction for the larger EUSA invasion. Rapid colonization southward has been documented, such as throughout the estuaries of South Carolina (Byers et al., 2012), possibly facilitated by the Intracoastal Waterway and movement of fishing vessels or leisure craft (Freshwater et al., 2006). Consistent with this southward invasion pathway, there was a decline in allelic diversity from Virginia southward to Georgia (Figure 4).
Along the coastlines of the British Isles, Europe, and northern Africa, our data are consistent with multiple invasion events into France and Portugal, followed by secondary spread to other estuaries. In the Marennes‐Oléron basin, for example, G. vermiculophylla thalli from Loix (frl) and Marennes (fme) belonged to different genetic cohorts (Figure S3). This basin is an area of intense aquaculture. The similarity of the sites sampled in Brittany (fdm, pol, fpa) to the thalli sampled at Marennes (fme) may be the result of secondary spread as oysters are commonly moved throughout French cultivation facilities in the English Channel, Bay of Biscay, and Mediterranean throughout their life cycle (Goulletquer, 1998). By contrast, sites sampled along the Jutland Peninsula (hei, dhd, nib, nor, man) were largely composed of a single genetic cluster from K = 2 to K = 23 (Figure 3, Figure S3) and exhibited lower estimates of diversity than other European sites (Figure 4). Gracilarioid seaweeds were not found in the Kiel fjord, for example, until the mid‐2000s, suggesting the invasion of Jutland estuaries is more recent (F. Weinberger, personal observation). These sites also exhibited genetic signatures of extensive vegetative fragmentation where we previously found few unique genotypes (Krueger‐Hadfield et al., 2016). On the other hand, oysters have repeatedly been introduced into the German Wadden Sea from Portugal (Mayer‐Waarden, 1964), which was reflected in the genetic similarity of the Jutland populations with those of the Ria d'Aveiro in Portugal (Gafanha/gaf and Estarreja/est; Figure S5a). We did not sample Mediterranean populations, which may help to identify pathways, both primary and secondary, that are cryptic in this current study, such as the introduction of the Manila clam (Venerupis philippinarum) to the Mediterranean (Sfriso, Maistro, Andreoli, & Moro, 2010).
The vector of secondary spread along the EU and EUSA coastlines is uncertain. Natural current patterns may contribute to the natural spread of thalli. Spread of Crassostrea gigas throughout the East Frisian Wadden Sea may be responsible for the encroachment of Gracilaria vermiculophylla along the Jutland Peninsula (Schmidt et al., 2008). Indeed, G. vermiculophylla is often found near oyster farms (Rueness, 2005). Other studies have documented the arrival of non‐native species as a result of leisure craft (e.g., Griffith et al., 2009); however, we have not found G. vermciulophylla on boat hulls or pontoons (S.A. Krueger‐Hadfield, personal observation). Nevertheless, some EU sites, such as Heiligenhafen (hei), were near marinas and could point to accidental within‐coastline transport by sailboats. Nyberg and Wallentinus (2009) suggested migrating seabirds may also be responsible for the transport of small fragments of G. vermiculophylla. Seabird sanctuaries in closed lagoons along the German Baltic Sea coast that are inaccessible to aquaculture, boat traffic, and other human activities often harbor isolated populations of G. vermiculophylla (F. Weinberger and M. Hammann, personal observation). Birds have also been known to use gracilarioid seaweeds in nests (Arnold, 1968), including G. vermiculophylla (S.A. Krueger‐Hadfield and S.J. Shainker, personal observation). Along the EUSA, spread of G. vermiculophylla may be via shrimp nets and crab pots, as G. vermiculophylla is known to be a nuisance in some estuaries to shrimpers (Freshwater et al., 2006).
5. Conclusions
Gracilaria vermiculophylla is now recognized as one of the most widespread and abundant marine invaders in the Northern Hemisphere (Kim et al., 2010; Krueger‐Hadfield et al., 2016; Saunders, 2009) and has transformed the ecosystems to which it has been introduced (Byers et al., 2012; Thomsen, Wernberg, Tuya, & Silliman, 2009). Nevertheless, this cryptic invader has likely been lurking in estuaries throughout the Northern Hemisphere for decades without recognition (S.A. Krueger‐Hadfield, E.E. Sotka and K.A. Miller, unpublished data). This oversight may reflect the fact that phycologists are more likely to study rocky shores than soft‐sediment estuaries where G. vermiculophylla has largely invaded, or the fact that cryptic species are common in seaweeds (Krueger‐Hadfield, Magill, Mieszkowska, Sotka, & Maggs, 2017). An analogous example comes from Provan, Booth, Todd, Beatty, and Maggs (2007), who used herbarium specimens coupled with molecular markers to conclude that the invasive strain of Codium fragile spp. tomentosoides colonized sites around the world for at least 100 years longer than previously reported from contemporary sampling.
Our identification of a source region has implications for understanding the evolutionary ecology of the Gracilaria vermiculophylla invasion, in particular. Previous studies have suggested introduced populations evolved greater tolerance for heat stress and resistance to herbivory during the invasion (Hammann, Wang, Boo, Aguilar‐Rosas, & Weinberger, 2016; Hammann, Wang, Rickert, Boo, & Weinberger, 2013). However, these studies compared introduced populations with South Korean and Chinese native populations that do not represent the source of the invasion. If there is geographic variation in the native range in these traits, then it is possible that the source populations had already evolved these traits before the invasion (Bossdorf, Lipowsky, & Prati, 2008; Colautti & Lau, 2015; Estoup & Guillemaud, 2010).
More broadly, Carlton (2009) argued convincingly that fewer assumptions should be made about the status of a given taxon (e.g., native or cryptogenic) without appropriate morphological and molecular analyses. This is particularly true for understudied groups in under‐explored habitats, such as macroalgae in estuarine habitats. We extend Carlton's (2009) suggestion to thoroughly explore not only the systematics of invaders, but also invasion vectors and pathways. Fewer assumptions should be made about the likelihood of a given vector without appropriate historical and contemporary genetic sampling and analyses.
Conflict of Interest
None declared.
Author Contributions
SAKH, NMK, and EES conceived the study; SAKH, NMK, JEB, MH, SJS, RT, FW, and EES collected samples; SAKH, NMK, and SJS extracted DNA; SAKH, NMK, SJS, TWG, and DM generated data; SAKH, AES, and EES analyzed data; JEB and AES contributed to discussions; SAKH and EES wrote the manuscript; all authors approved the final manuscript.
Supporting information
Acknowledgments
We are grateful for everyone who provided algal samples (see Table S1 for complete list), and in particular, R. Hadfield, C. Sotka, M. Nakaoka, M. Kamiya, and H. Endo. Thank you to J. Carlton, M. Valero, and C. Destombe for insightful discussions; L. Lees for help with mtDNA amplification; L. Burnett and K. Burnett for lab space for DNA extractions; B. Flanagan for help with DNA extractions; K. Hill‐Spanik for help with phylogenetic analyses; K. Holcombe at the Chincoteague National Wildlife Refuge (FWS Special Use Permit SUP 51570‐2014‐013), B. Hughes (Elkhorn Slough National Estuarine Research Reserve) for site access, and G. Saunders for field locations in British Columbia. This project was supported by NSF BIO‐OCE‐1057713, BIO‐OCE‐1057707, BIO‐OCE‐1357386; a College of Charleston Graduate Research Grant; the Phycological Society of America Grants‐in‐Aid‐of‐Research; Zostera Experimental Network Graduate Research Fellowship (NSF OCE‐1031061); and LLUR‐Schleswig‐Holstein.
The scientific results and conclusions, as well as any opinions expressed herein, are those of the author(s) and do not necessarily reflect the views of NOAA or the Department of Commerce. The mention of any commercial product is not meant as an endorsement by the Agency or Department.
Data Accessibility
Microsatellite primer sequences were deposited in GENBANK, accession numbers KT232089–KT232097 and KT232099 (Kollars et al., 2015). Ploidy designation for each thallus, GenAlEx files (diploid total, haploid total, diploid P sex), cox1 haplotypes (Kim et al., 2010; this study), cox1 sequences for the thalli from this study, and the cox1 SNP for each thallus were deposited in DRYAD, entry https://doi.org/10.5061/dryad.fn53k. Mitochondrial haplotypes were deposited in GENBANK (Acc. No. KY621338‐KY6211343).
Krueger‐Hadfield SA, Kollars NM, Strand AE, et al. Genetic identification of source and likely vector of a widespread marine invader. Ecol Evol. 2017;7:4432–4447. https://doi.org/10.1002/ece3.3001
Contributor Information
Stacy A. Krueger‐Hadfield, Email: sakh@uab.edu.
Erik E. Sotka, Email: eriksotka@gmail.com
References
- Akihito, F. A. , Ikeda, Y. , et al. (2008). Evolution of Pacific Ocean and the Sea of Japan populations of the gobiid species, Pterogobius elapoides and Pterogobius zonoleucus, based on molecular and morphological analyses. Gene, 427, 7–18. [DOI] [PubMed] [Google Scholar]
- Andrews, J. D. (1980). A review of introductions of exotic oysters and biological planning for new importations. Marine Fisheries Reviews, 42, 1–11. [Google Scholar]
- Arnaud‐Haond, S. , & Belkhir, K. (2006). GenClone: A computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Molecular Ecology Notes, 7, 15–17. [Google Scholar]
- Arnaud‐Haond, S. , Duarte, C. M. , Alberto, F. , & Serrao, E. A. (2007). Standardizing methods to address clonality in population studies. Molecular Ecology, 16, 5115–5139. [DOI] [PubMed] [Google Scholar]
- Arnold, A. F. (1968). The sea‐beach at ebb tide: A guide to the study of the seaweeds and the lower animal life found between tide‐marks. New York, NY: Dover Publications; 490 pp. [Google Scholar]
- Azuma, N. , & Chiba, S. (2015). Phylogeography of Littorina brevicula suggests postglacial colonization from south to north along the Japanese Archipelago. Journal of Molluscan Studies, 82, 259–267. [Google Scholar]
- Barrett, E. M. (1963). The California oyster industry. Fish Bulletin, 123, 1–53. [Google Scholar]
- Bellorin, A. M. , Oliveira, M. C. , & Oliveira, E. C. (2004). Gracilaria vermiculophylla: A western Pacific species of Gracilariaceae (Rhodophyta) first recorded from the eastern Pacific. Phycological Research, 52, 69–79. [Google Scholar]
- Bock, D. G. , Caseys, C. , Cousens, R. D. , et al. (2015). What we still don't know about invasion genetics. Molecular Ecology, 24, 2277–2297. [DOI] [PubMed] [Google Scholar]
- Bonnot, P. (1935). The California oyster industry. California Fish and Game, 21, 65–80. [Google Scholar]
- Bossdorf, O. , Lipowsky, A. , & Prati, D. (2008). Selection of preadapted populations allowed Senecio inaequidens to invade Central Europe. Diversity and Distributions, 14, 676–685. [Google Scholar]
- Brawley, S. H. , Coyer, J. A. , Blakeslee, A. M. H. , et al. (2009). Historical invasions of the intertidal zone of Atlantic North America associated with distinctive patterns of trade and emigration. Proceedings of the National Academy of Sciences of the United States of America, 106, 8239–8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs, J. C. , & Bowen, B. W. (2011). A realignment of marine biogeographic provinces with particular reference to fish distributions. Journal of Biogeography, 39, 12–30. [Google Scholar]
- Burreson, E. M. , Stokes, N. A. , & Friedman, C. S. (2000). Increased virulence in an introduced pathogen: Haplosporidium nelsoni (MSX) in the eastern Oyster Crassostrea virginica . Journal of Aquatic Animal Health, 12, 1–8. [DOI] [PubMed] [Google Scholar]
- Byers, J. E. (1999). The distribution of an introduced mollusc and its role in the long‐term demise of a native confamilial species. Biological Invasions, 1, 339–352. [Google Scholar]
- Byers, J. E. , Gribben, P. E. , Yeager, C. , & Sotka, E. E. (2012). Impacts of an abundant introduced ecosystem engineer within mudflats of the southeastern US coast. Biological Invasions, 149, 2587–2600. [Google Scholar]
- Carlton, J. T. , & Geller, J. B. (1993). Ecological roulette: The global transport of nonindigenous marine organisms. Science, 261, 78–82. [DOI] [PubMed] [Google Scholar]
- Carlton, J. T. (2009). Deep invasion ecology and the assembly of communities in historical time In Rilov G. & Crooks J. A. (Eds.), Biological invasions in marine ecosystems (pp. 13–56). Springer; Berlin Heidelberg. [Google Scholar]
- Cheang, C. C. , Chu, K. H. , & Ang, P. O. (2010). Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Molecular Ecology, 19, 2933–2948. [DOI] [PubMed] [Google Scholar]
- Chew, K. K. (1979). The Pacific Oyster (Crassostrea gigas) in the West Coast of the United States In Mann R. (Ed.), Exotic species in mariculture (pp. 54–82). MA: The MIT Press Cambridge, Cambridge. [Google Scholar]
- Colautti, R. I. , & Lau, J. A. (2015). Contemporary evolution during invasion: Evidence for differentiation, natural selection, and local adaptation. Molecular Ecology, 24, 1999–2017. [DOI] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2012). jModelTest 2: More models, new heuristics and parallel computing. Nature Methods, 9, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, D. (1979). Introduced species and the Maine situation In Mann R. (Ed.), Exotic species in mariculture (pp. 149–164). MA: The MIT Press Cambridge, Cambridge. [Google Scholar]
- Destombe, C. , Godin, J. , & Remy, J. M. (1990). Viability and dissemination of spermatia of Gracilaria verrucosa (Gracilariales, Rhodophyta). Hydrobiologia, 204/205, 219–233. [Google Scholar]
- Dlugosch, K. M. , & Parker, I. M. (2008). Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology, 17, 431–449. [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup, A. , & Guillemaud, T. (2010). Reconstructing routes of invasion using genetic data: Why, how and so what? Molecular Ecology, 19, 4113–4130. [DOI] [PubMed] [Google Scholar]
- Farnham, D. E. , Fletcher, R. L. , & Irvine, L. (1973). Attached Sargassum found in Britain. Nature, 243, 231–232.4706294 [Google Scholar]
- Freshwater, D. W. , Montgomery, F. , Greene, J. K. , et al. (2006). Distribution and Identification of an Invasive Gracilaria species that is hampering commercial fishing operations in Southeastern North Carolina, USA. Biological Invasions, 8, 631–637. [Google Scholar]
- Galtsoff, P. S. (1932). Introduction of Japanese oysters into the United States. United States Fishery Circular, 12, 1–16. [Google Scholar]
- Gao, H. , Williamson, S. , & Bustamante, C. D. (2007). A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics, 176, 1635–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller, J. B. , Darling, J. A. , & Carlton, J. T. (2010). Genetic perspectives on marine biological invasions. Annual Review of Marine Science, 2, 367–393. [DOI] [PubMed] [Google Scholar]
- Geraldino, P. J. L. , Yang, E. C. , & Boo, S. M. (2006). Morphology and molecular phylogeny of Hypnea flexicaulis (Gigartinales, Rhodophyta) from Korea. ALGAE, 21, 417–423. [Google Scholar]
- Goulletquer, P. (1998). Shellfish culture in France: Present status and new approaches to optimise production. Proceedings of the Twenty‐Ninth Annual Shellfish Conference, 69–80. [Google Scholar]
- Gouy, M. , Guindon, S. , & Gascuel, O. (2010). SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution, 27, 221–224. [DOI] [PubMed] [Google Scholar]
- Govender, K. (2001). Population genetic studies of economically important Gracilaria and Gracilariopsis (Rhodophyta) in the south western Cape. PhD Thesis, University of Cape Town. 155pp.
- Griffith, K. , Mowat, S. , Holt, R. H. F. , et al. (2009). First records in Great Britain of the invasive colonial ascidian Didemnum vexillum Kott, 2002. Aquatic Invasions, 4, 581–590. [Google Scholar]
- Grizel, H. , & Héral, M. (1991). Introduction in France of the Japanese Oyster (Crassostrea gigas). ICES Journal of Marine Science, 47, 399–403. [Google Scholar]
- Grosholz, E. D. , & Ruiz, G. M. (2009). Multitrophic effects of invasions in marine and estuarine ecosystems In Rilov G. & Crooks J. A. (Eds.). Biological invasions in marine ecosystems (pp. 305–324). Berlin Heidelberg: Springer. [Google Scholar]
- Guillemin, M.‐L. , Faugeron, S. , Destombe, C. , et al. (2008). Genetic variation in wild and cultivated populations of the haploid– diploid red alga Gracilaria chilensis: How farming practices favor asexual reproduction and heterozygosity. Evolution, 62, 1500–1519. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , & Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum‐likelihood”. Systematic Biology, 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Gulbransen, D. J. , McGlathery, K. J. , Marklund, M. , Norris, J. N. , & Gurgel, C. F. D. (2012). Gracilaria vermiculophylla (rhodophyta, gracilariales) in the Virginia coastal bays, USA: cox1 analysis reveals high genetic richness of an introduced macroalga. Journal of Phycology, 48, 1278–1283. [DOI] [PubMed] [Google Scholar]
- Gurgel, C. F. D. , & Fredericq, S. (2004). Systematics of the Gracilariaceae (Gracilariales, Rhodophyta): A critical assessment based on rbcl sequence analyses. Journal of Phycology, 40, 138–159. [Google Scholar]
- Halkett, F. , Plantegenest, M. , Prunier‐Leterme, N. , et al. (2005). Admixed sexual and facultatively asexual aphid lineages at mating sites. Molecular Ecology, 14, 325–336. [DOI] [PubMed] [Google Scholar]
- Hammann, M. , Buchholz, B. , Karez, R. , & Weinberger, F. (2013). Direct and indirect effects of Gracilaria vermiculophylla on native Fucus vesiculosus . Aquatic Invasions, 8, 121–132. [Google Scholar]
- Hammann, M. , Wang, G. , Boo, S. M. , Aguilar‐Rosas, L. E. , & Weinberger, F. (2016). Selection of heat‐shock resistance traits during the invasion of the seaweed Gracilaria vermiculophylla . Marine Biology, 163, 104. [Google Scholar]
- Hammann, M. , Wang, G. , Rickert, E. , Boo, S. M. , & Weinberger, F. (2013). Invasion success of the seaweed Gracilaria vermiculophylla correlates with low palatibility. Marine Ecology Progress Series, 486, 93–103. [Google Scholar]
- Hasegawa, M. , Kishino, H. , & Yano, T. (1985). Dating of human‐ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution, 22, 160–174. [DOI] [PubMed] [Google Scholar]
- Haydar, D. , & Wolff, W. J. (2011). Predicting invasion patterns in coastal ecosystems: Relationship between vector strength and vector tempo. Marine Ecology Progress Series, 431, 1–10. [Google Scholar]
- He, L. , Mukai, T. , Chung, M. Q. , & Zhang, J. (2015). Biogeographical role of the Kuroshio Current in the amphibious mudskipper. Scientific Reports, 5, 15645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey, J. M. (1979). Culture of the Pacific Oyster, Crassostrea gigas, in Massachusetts waters In Mann R. (Ed.), Exotic species in mariculture (pp. 129–148). MA: The MIT Press Cambridge. [Google Scholar]
- Ho, P.‐T. , Kwan, Y.‐S. , Kim, B. , & Won, Y.‐J. (2014). Postglacial range shift and demographic expansion of the marine intertidal snail Batillaria attramentaria . Ecology and Evolution, 5, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington, B. E. , & Boyer, K. E. (2008). Effect of red marcoalgal (Gracilariopsis sp.) abundance on eelgrass Zostera marina in Tomales Bay, California, USA. Marine Ecology Progress Series, 367, 133–142. [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics Applications Note, 23, 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics Applications Note, 24, 1403–1405. [DOI] [PubMed] [Google Scholar]
- Jombart, T. , & Ahmed, I. (2011). adegenet 1.3‐1: New tools for the analysis of genome‐wide SNP data. Bioinformatics, 27, 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , Devillard, S. , & Balloux, F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically. BMC Genetics, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , Pontier, D. , & Dufour, A.‐B. (2009). Genetic markers in the playground of multivariate analysis. Heredity, 102, 330–341. [DOI] [PubMed] [Google Scholar]
- Kalinowski, S. T. (2005). hp‐rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Molecular Ecology Notes, 5, 187–189. [Google Scholar]
- Kamvar, Z. N. , Brooks, J. C. , & Grünwald, N. J. (2015). Novel R tools for analysis of genome‐wide population genetic data with emphasis on clonality. Frontiers in Genetics, 6, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamvar, Z. N. , Tabima, J. F. , & Grünwald, N. J. (2014). Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ, 2, e281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. M. , Hoarau, G. G. , & Boo, S. M. (2012). Genetic structure and distribution of Gelidium elegans (Gelidiales, Rhodophyta) in Korea based on mitochondrial cox1 sequence data. Aquatic Botany, 98, 27–33. [Google Scholar]
- Kim, S. Y. , Weinberger, F. , & Boo, S. M. (2010). Genetic data hint at a common donor region for invasive atlantic and pacific populations of Gracilaria vermiculophylla (Gracilariales, Rhodophyta). Journal of Phycology, 46, 1346–1349. [Google Scholar]
- Kolar, C. S. , & Lodge, D. M. (2001). Progress in invasion biology: Predicting invaders. Trends in Ecology & Evolution, 16, 199–204. [DOI] [PubMed] [Google Scholar]
- Kollars, N. M. , Krueger‐Hadfield, S. A. , Byers, J. E. , et al. (2015). Development and characterization of microsatellite loci for the haploid–diploid red seaweed Gracilaria vermiculophylla . PeerJ, 3, e1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman, N. M. , Mayzel, J. , Jakobsson, M. , Rosenberg, N. A. , & Mayrose, I. (2015). Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources, 15, 1179–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger‐Hadfield, S. A. , Collen, J. , Daguin‐Thiébaut, C. , & Valero, M. (2011). Genetic population structure and mating system in Chondrus crispus (Rhodophyta). Journal of Phycology, 47, 440–450. [DOI] [PubMed] [Google Scholar]
- Krueger‐Hadfield, S. A. , Kollars, N. M. , Byers, J. E. , et al. (2016). Invasion of novel habitats uncouples haplo‐diplontic life cycles. Molecular Ecology, 25, 3801–3816. [DOI] [PubMed] [Google Scholar]
- Krueger‐Hadfield, S. A. , Magill, C. L. , Mieszkowska, N. , Sotka, E. E. , & Maggs, C. A. (2017). When invaders go unnoticed: The case of Gracilaria vermiculophylla in the British Isles. Cryptogamie Algologie (in press). [Google Scholar]
- Krueger‐Hadfield, S. A. , Roze, D. , Correa, J. A. , Destombe, C. , & Valero, M. (2015). O father where art thou? Paternity analyses in a natural population of the haploid‐diploid red seaweed Chondrus crispus . Heredity, 114, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallias, D. , Boudry, P. , Batista, F. M. , et al. (2015). Invasion genetics of the Pacific oyster Crassostrea gigas in the British Isles inferred from microsatellite and mitochondrial markers. Biological Invasions, 17, 2581–2595. [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics Applications Note, 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- Loosanoff, V. L. , & Davis, H. C. (1963). Rearing of bivalve mollusks. Advances in Marine Biology, 1, 1–136. [Google Scholar]
- Martín, L. A. , Boraso de Ziaxso, A. L. , & Leonardi, P. I. (2011). Biomass variation and reproductive phenology of Gracilaria gracilis in a Patagonian natural bed (Chubut, Argentina). Journal of Applied Phycology, 23, 643–654. [Google Scholar]
- Maurin, C. , & LeDantec, J. (1979). Experiments on the growth of the mangrove oyster, Crassostrea rhizophorae in France In Mann R. (Ed.), Exotic species in mariculture (pp. 123–128). MA: The MIT Press Cambridge. [Google Scholar]
- Mayer‐Waarden, P. F. (1964). Mastversuche mit portugiesischen Austern im deutschen Wattenmeer. Informationen für die Fischwirtschaft, 2, 77–78. [Google Scholar]
- Mineur, F. , Johnson, M. P. , & Maggs, C. A. (2008). Macroalgal introductions by hull fouling on recreational vessels: Seaweeds and sailors. Environmental Management, 42, 667–676. [DOI] [PubMed] [Google Scholar]
- Mineur, F. , Le Roux, A. , Maggs, C. A. , & Verlaque, M. (2014). Positive feedback loop between introductions of non‐native marine species and cultivation of oysters in Europe. Conservation Biology, 28, 1667–1676. [DOI] [PubMed] [Google Scholar]
- Miller, A. W. , Hewitt, C. L. , & Ruiz, G. M. (2002). Invasion success: Does size really matter? Ecology Letters, 5, 159–162. [Google Scholar]
- Miura, O. , Torchin, M. E. , Kuris, A. M. , Hechinger, R. F. , & Chiba, S. (2006). Introduced cryptic species of parasites exhibit different invasion pathways. Proceedings of the National Academy of Sciences of the United States of America, 103, 19818–19823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton, J. C. , Mathieson, A. C. , Thornber, C. S. , Neefus, C. D. , & Yarish, C. (2013). Introduction of Gracilaria vermiculophylla (Rhodophyta, Gracilariales) to New England, USA: Estimated arrival times and current distribution. Rhodora, 115, 28–41. [Google Scholar]
- Ni, G. , Li, Q. I. , Kong, L. , & Yu, H. (2014). Comparative phylogeography in marginal seas of the northwestern Pacific. Molecular Ecology, 23, 534–548. [DOI] [PubMed] [Google Scholar]
- Nyberg, C. D. , & Wallentinus, I. (2009). Long‐term survival of an introduced red alga in adverse conditions. Marine Biology Research, 5, 304–308. [Google Scholar]
- Ó Foighil, D. , Gaffney, P. M. , Wilbur, A. E. , & Hilbish, T. J. (1998). Mitochondrial cytochrome oxidase I gene sequences support an Asian origin for the Portuguese oyster Crassostrea angulata . Marine Biology, 131, 497–503. [Google Scholar]
- Okazaki, A. (1971). Seaweeds and their uses in Japan (165 pp). Tokyo: Tokai University Press. [Google Scholar]
- Paradis, E. , Claude, J. , & Strimmer, K. (2004). APE: Analyses of phylogenetics and evolution in R LANGUAGE. Bioinformatics Applications Note, 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Parks, J. C. , & Werth, C. R. (1993). A study of spatial features of clones in a population of bracken fern, Pteridium aquilinum (Dennstaedtiaceae). American Journal of Botany, 80, 537–544. [DOI] [PubMed] [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2006). GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics Applications Note, 28, 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisler, R. K. , Wasson, K. , Wolff, W. J. , & Tyrrell, M. C. (2009). Invasions of estuaries vs the adjacent open coast: A global perspective In Rilov G. & Crooks J. A. (Eds.,) Biological Invasions in Marine Ecosystems (pp 587–617). Springer Berlin Heidelberg. [Google Scholar]
- Provan, J. , Booth, D. , Todd, N. P. , Beatty, G. E. , & Maggs, C. A. (2007). Tracking biological invasions in space and time: Elucidating the invasive history of the green alga Codium fragile using old DNA. Diversity and Distributions, 14, 343–354. [Google Scholar]
- R Core Team . (2016). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from https://www.R-project.org/ [Google Scholar]
- Reusch, T. B. H. , Bolte, S. , Sparwel, M. , Moss, A. G. , & Javidpour, J. (2010). Microsatellites reveal origin and genetic diversity of Eurasian invasions by one of the world's most notorious marine invader, Mnemiopsis leidyi (Ctenophora). Molecular Ecology, 19, 2690–2699. [DOI] [PubMed] [Google Scholar]
- Riquet, F. , Daguin‐Thiébaut, C. , Ballenghien, M. , Bierne, N. , & Viard, F. (2013). Contrasting patterns of genome‐wide polymorphism in the native and invasive range of the marine mollusc Crepidula fornicata . Molecular Ecology, 22, 1003–1018. [DOI] [PubMed] [Google Scholar]
- Rousset, F. (2008). Genepop'007: A complete reimplementation of the Genepop software for Windows and Linux. Molecular Ecology Resources, 8, 103–106. [DOI] [PubMed] [Google Scholar]
- Rueness, J. (2005). Life history and molecular sequences of Gracilaria vermiculophylla (Gracilariales, Rhodophyta), a new introduction for European waters. Phycologia, 44, 120–128. [Google Scholar]
- Ruesink, J. L. , Lenihan, H. S. , Trimble, A. C. , et al. (2005). Introduction of non‐native oysters: Ecosystem Effects and Restoration Implications. Annual Review of Ecology, Evolution, and Systematics, 36, 643–689. [Google Scholar]
- Ruiz, G. M. , Carlton, J. T. , Grosholz, E. D. , & Hines, A. H. (1997). Global invasions of marine and estuarine habitats by non‐indigenous species: Mechanisms, extent, and consequences. Integrative and Comparative Biology, 37, 621–632. [Google Scholar]
- Ruiz, G. M. , Fofonoff, P. W. , Carlton, J. T. , Wonham, M. J. , & Hines, A. H. (2000). Invasion of coastal marine communities in North America: Apparent patterns, processes, and biases. Annual Review of Ecology and Systematics, 31, 481–531. [Google Scholar]
- Ruiz, G. M. , Fofonoff, P. W. , Steves, B. , Foss, S. F. , & Shiba, S. N. (2011). Marine invasion history and vector analysis of California: A hotspot for western North America. Diversity and Distributions, 17, 362–373. [Google Scholar]
- Saunders, G. W. (2009). Routine DNA barcoding of Canadian Gracilariales (Rhodophyta) reveals the invasive species Gracilaria vermiculophylla in British Columbia. Molecular Ecology Resources, 9, 140–150. [DOI] [PubMed] [Google Scholar]
- Schmidt, A. , Wehrmann, A. , & Dittmann, S. (2008). Population dynamics of the invasive Pacific oyster Crassostrea gigas during the early stages of an outbreak in the Wadden Sea (Germany). Helgoland Marine Research, 62, 367. [Google Scholar]
- Seebens, H. , Schwartz, N. , Schupp, P. J. , & Blasius, B. (2016). Predicting the spread of marine species introduced by global shipping. Proceedings of the National Academy of Sciences of the United States of America, 113, 5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfriso, A. , Maistro, S. , Andreoli, C. , & Moro, I. (2010). First record of Gracilaria vermiculophylla (Gracilariales, Rhodophyta) in the Po Delta Lagoons, Mediterranean Sea (Italy). Journal of Phycology, 46, 1024–1027. [Google Scholar]
- Spalding, M. D. , Fox, H. E. , Allen, G. R. , et al. (2007). Marine ecoregions of the world: A bioregionalization of coastal and shelf areas. BioScience, 57, 573–583. [Google Scholar]
- Sunnucks, P. , de Barro, P. J. , Lushai, G. , Maclean, N. , & Hales, D. (1997). Genetic structure of an aphid studied using microsatellites: Cyclic parthenogenesis, differentiated lineages and host specialization. Molecular Ecology, 6, 1059–1073. [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen, M. S. , Gurgel, C. F. D. , & McGlathery, K. J. (2006). Gracilaria vermiculophylla (Rhodophyta, Gracilariales) in Hog Island Bay, Virginia: A cryptic alien and invasive macroalga and taxonomic correction. Journal of Phycology, 42, 139–141. [DOI] [PubMed] [Google Scholar]
- Thomsen, M. , Stæhr, P. , Nejrup, L. , & Schiel, D. (2013). Effects of the invasive macroalgae Gracilaria vermiculophylla on two co‐occurring foundation species and associated invertebrates. Aquatic Invasions, 8, 133–145. [Google Scholar]
- Thomsen, M. S. , Wernberg, T. , Tuya, F. , & Silliman, B. R. (2009). Evidence for impacts of nonindigenous macroalgae: A meta‐analysis of experimental field studies. Journal of Phycology, 45, 812–819. [DOI] [PubMed] [Google Scholar]
- Uwai, S. , Kogame, K. , Yoshida, G. , Kawai, H. , & Ajisaka, T. (2009). Geographical genetic structure and phylogeography of the Sargassum horneri/filicinum complex in Japan, based on the mitochondrial cox3 haplotype. Marine Biology, 156, 901–911. [Google Scholar]
- Uwai, S. , Yotsukura, N. , Serisawa, Y. , et al. (2006). Intraspecific genetic diversity of Undaria pinnatifida in Japan, based on the mitochondrial cox3 gene and the ITS1 of nrDNA. Hydrobiologia, 553, 345–356. [Google Scholar]
- Voisin, M. , Engel, C. R. , & Viard, F. (2005). Differential shuffling of native genetic diversity across introduced regions in a brown alga: Aquaculture vs. maritime traffic effects. Proceedings of the National Academy of Sciences of the United States of America, 102, 5432–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. (1999). Response of Western Pacific marginal seas to glacial cycles: Paleoceanographic and sedimentological features. Marine Geology, 156, 5–39. [Google Scholar]
- Weinberger, F. , Buchholz, B. , Karez, R. , & Wahl, M. (2008). The invasive red alga Gracilaria vermiculophylla in the Baltic Sea: Adaptation to brackish water may compensate for light limitation. Aquatic Biology, 3, 251–264. [Google Scholar]
- Yang, E. C. , Kim, M. S. , Geraldino, P. J. L. , et al. (2008). Mitochondrial cox1 and plastid rbcL genes of Gracilaria vermiculophylla (Gracilariaceae, Rhodophyta). Journal of Applied Phycology, 20, 161–168. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials