

Abstract
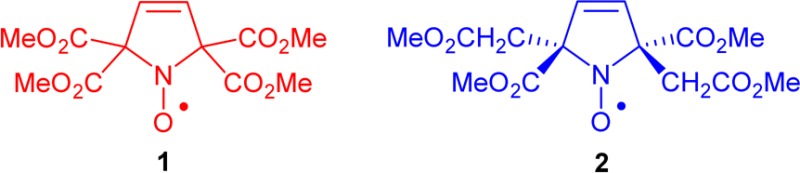
 We report the design,
synthesis, and electron spin relaxation properties
of hydrophilic tetracarboxylate ester pyrroline nitroxides 1 and 2, which serve as models in the search for new
spin labels for DEER distance measurement at room temperature. The
nitroxides are designed to have the methyl groups further away from
the N–O spin site to decrease the inequivalent couplings of
the unpaired electron to the methyl protons that shorten Tm at T > 70 K in currently used labels.
The key step in the synthesis of 1 and 2 is the reaction of the dianion of pyrrole-1,2,5-tricarboxylic acid tert-butyl ester dimethyl ester with electrophiles such
as methyl chloroformate and methyl bromoacetate. Structures of 1 and 2 are confirmed by X-ray crystallography.
Studies of electron spin relaxation rates in rigid trehalose/sucrose
matrices reveal approximately temperature independent values of 1/Tm for 1 and 2 up to
about 160 K and modest temperature dependence up to 295 K, demonstrating
that increasing the distance between the nitroxide moiety and methyl
groups is effective in lengthening Tm at T > 70 K.
We report the design,
synthesis, and electron spin relaxation properties
of hydrophilic tetracarboxylate ester pyrroline nitroxides 1 and 2, which serve as models in the search for new
spin labels for DEER distance measurement at room temperature. The
nitroxides are designed to have the methyl groups further away from
the N–O spin site to decrease the inequivalent couplings of
the unpaired electron to the methyl protons that shorten Tm at T > 70 K in currently used labels.
The key step in the synthesis of 1 and 2 is the reaction of the dianion of pyrrole-1,2,5-tricarboxylic acid tert-butyl ester dimethyl ester with electrophiles such
as methyl chloroformate and methyl bromoacetate. Structures of 1 and 2 are confirmed by X-ray crystallography.
Studies of electron spin relaxation rates in rigid trehalose/sucrose
matrices reveal approximately temperature independent values of 1/Tm for 1 and 2 up to
about 160 K and modest temperature dependence up to 295 K, demonstrating
that increasing the distance between the nitroxide moiety and methyl
groups is effective in lengthening Tm at T > 70 K.
Introduction
Nitroxides are stable organic radicals that have a wide range of applications in chemistry, biochemistry, biophysics, and biomedicine.1−5 One of the long-standing, remarkable applications is as spin labels, pioneered by McConnell and co-workers in 1965.6 More recently, the site-directed spin labeling (SDSL) technique coupled with pulsed electron paramagnetic resonance (EPR) dipolar spectroscopy has become one of the techniques of choice for investigating structure and dynamics of biomacromolecules, such as proteins, DNAs, and RNAs.3,7−19
Pulsed EPR dipolar spectroscopy, which includes double electron–electron resonance (DEER) and double quantum coherence (DQC), is among the best techniques for accurate measurement of the conformations of flexible regions of biomolecules. Advantages include high sensitivity, a wide range of distances, and absolute distance distributions. The most widely used nitroxide spin label is 1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-(methyl)methanethiosulfonate (MTSL) (Figure 1), which selectively binds to cysteine and can be attached at locations introduced by mutagenesis to provide doubly labeled proteins. Relatively small-size nitroxide spin labels typically cause minimal structural perturbation. Among the pulse EPR methods, DEER is the most widely used technique for the measurements of interspin distances of 1.5–8 nm in biomolecules. DEER experiments are typically carried out at temperatures between about 50 and 70 K, requiring use of liquid helium, or with lower sensitivity at 80 K with liquid nitrogen. This restriction to temperatures less than about 80 K is imposed by the rotation of the methyl group in the nitroxide at rates comparable to the inequivalent proton couplings to the unpaired electron. This process shortens the phase memory time, Tm, between about 80 and 300 K.20,21Tm is the time constant for the decay of the two-pulse spin echo as the time between pulses is increased. The length of Tm is important because the longer the Tm the stronger the echo at a particular pulse timing, the longer the distance that can be measured, and the more precisely the distribution of distances can be defined.13,14
Figure 1.
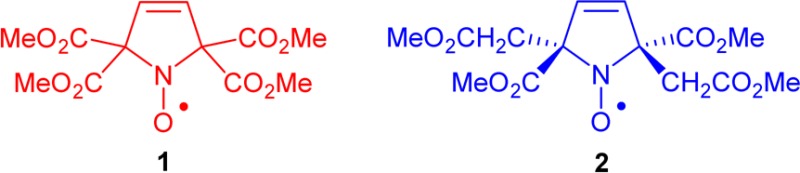
Spin labels and nitroxides 1 and 2.
We recently prepared a new class of nitroxide spin labels devoid of methyl groups, iodoacetamide (IA) spirocyclohexyl pyrimidine nitroxide, spiro-IA, and demonstrated DEER distance measurements at room temperature using T4 Lysozyme (T4L) that was doubly spin labeled with spiro-IA. The doubly labeled T4L possesses sufficiently long Tm at Q-band to allow measurement of 4 nm distances at 160 K in glycerol/water and 3 nm distances at room temperature in a trehalose matrix.22 Replacement of the gem-dimethyl groups by conformationally restrained spirocyclohexyl groups diminishes the dynamic averaging effects and lengthens Tm between 80 and 295 K in rigid matrices.22−25
The spin labels based on spirocyclohexyl nitroxides are however relatively hydrophobic, which presumably lowers the yield for incorporation of the label at certain sites in proteins, e.g., Cys-8 in T4L.26 We found that an analogous spirocyclohexyl spin label, i.e., spiro-TOAC (as an Fmoc-derivative),23 could not be efficiently incorporated in phospholamban (PLB).27
In the search for a hydrophilic spin label with adequately long Tm for distance measurement at room temperature, we are designing nitroxides with longer Tm compared to the gem-dimethyl analogues. Our approach is to decrease the inequivalent couplings of the unpaired electron to the methyl protons that shorten Tm at T > 70 K. This can be achieved if the methyl groups are further away from the N–O spin site. Examples of the prototype structure are tetracarboxylate ester pyrroline nitroxides 1 and 2 (Figure 1). Notably, according to the calculated partition coefficients (cLog P) and the Connolly solvent excluded volumes (Vol), nitroxides 1 and 2 with four methyl ester groups are much more polar, though somewhat larger, compared to gem-dimethyl and spirocyclohexyl analogues (Table 1). Thus, 1 and 2 are excellent models for the novel design of new spin labels.
Table 1. Calculated Partition Coefficients (cLog P) and the Connolly Solvent Excluded Volumes (Vol) for Selected Nitroxidesa.
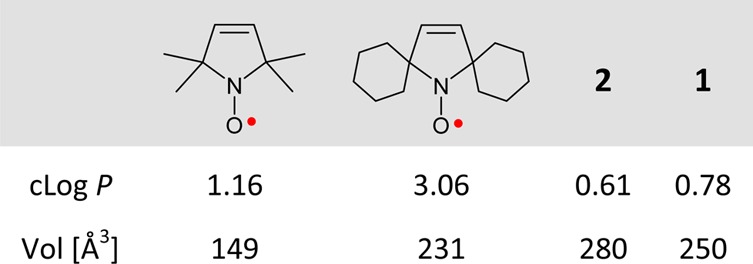
Geometries of 1 and 2 are optimized at the UB3LYP/6-311G(d,p)+ZPVE level; see: Tables S5–S7, SI.
Here we report the synthesis of the tetracarboxylate ester pyrroline nitroxides 1 and 2 and their electron spin relaxation rates in rigid trehalose/sucrose matrices.
Results and Discussion
The synthesis of nitroxides 1 and 2 starts from commercially available N-Boc-pyrrole which is then converted to pyrrole 3 in one step according to the procedures for the synthesis of pyrrolizidine alkaloids (Scheme 1, Supporting Information (SI)).28,29 Subsequently, the dianion of 3 in THF is generated using the conditions reported for preparation of the cis-2,5-tetrasubstituted pyrroline, which was formed by quenching the dianion with 2,6-di-tert-butylphenol. Because our target is the trans-isomer, we quench the dianion of 3 with electrophiles, ClCO2Me and BrCH2CO2Me, to provide products 4 and 6, respectively. In analogy to the reported reaction of the dianion of 3 with an acid,28 we expect a bulky electrophile such as BrCH2CO2Me to provide a similar outcome. That is, after the first C–C bond formation, the resulting monoenolate anion reacts, from the least hindered face, with the second molecule of BrCH2CO2Me, to produce trans-6. Subsequent deprotections of the N-Boc groups with trifluoroacetic acid (TFA) in dichloromethane (DCM) provided amines 5 and 7, which are then oxidized to the corresponding nitroxides 1 and 2, using meta-chloroperbenzoic acid (m-CPBA) in DCM.
Scheme 1. Synthesis of Nitroxides 1 and 2.
Structures of nitroxides 1 and 2 are confirmed by single-crystal X-ray analysis (Figure 2). In particular, the trans-stereochemistry for 2, which is C2-symmetric in the crystal, is unequivocally confirmed.
Figure 2.

X-ray structures for nitroxide radicals 1 (left) and 2 (right). Carbon, nitrogen, and oxygen atoms are depicted with thermal ellipsoids set at the 50% probability level.
Purity of nitroxides 1 and 2 was confirmed by paramagnetic 1H NMR spectra and EPR spectroscopic spin concentrations. In addition, nitroxides 1 and 2 are reduced, using an excess of ascorbate and reduced glutathione (GSH), to the corresponding diamagnetic hydroxylamines 1-H and 2-H. Lastly, structures of 1-H and 2-H are confirmed by 1H and 13C NMR spectroscopy, IR spectroscopy, and mass spectrometry.
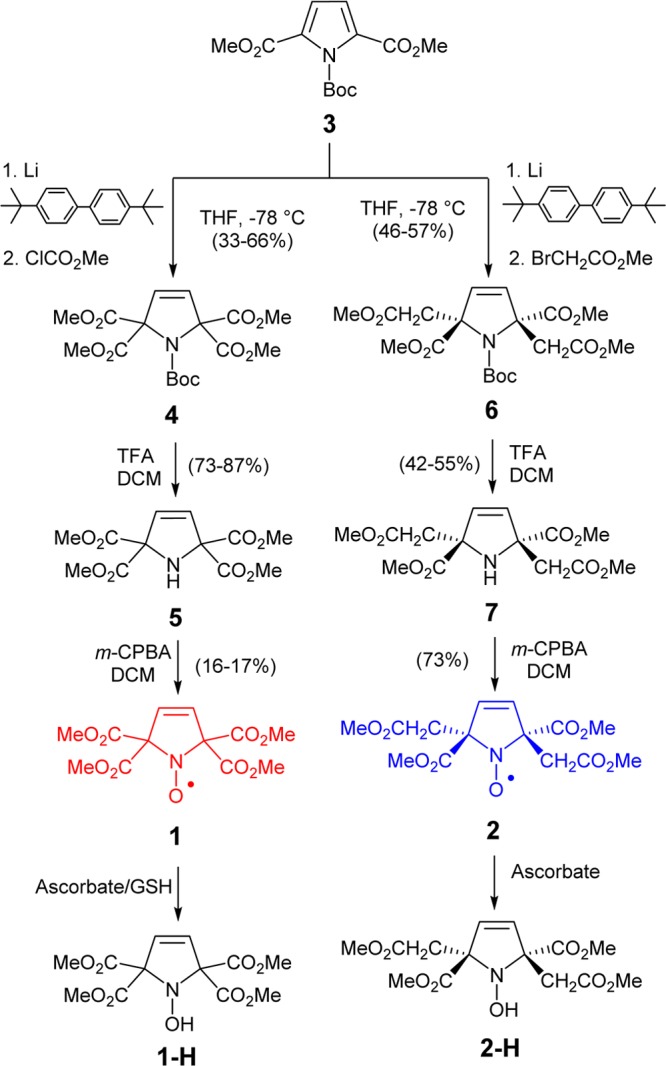
EPR spectra of nitroxides 1 and 2 in chloroform show triplet patterns due to isotropic 14N hyperfine coupling with A(14N) = 34.2 and 37.7 MHz, respectively; g-values are near 2.006 (Figure 3). For 1, each of the triplet peaks is resolved into triplets, corresponding to the coupling of two vinylic protons with isotropic 1H hyperfine coupling, A(1H) = 1.10 MHz (top panel in Figure 3).
Figure 3.
EPR (v = 9.6505 GHz) spectrum of 0.1 mM nitroxide 1 in chloroform. (Top panel) Complete spectrum; (bottom panel) expansion plot near the center peak showing 13C satellite peaks. Simulation: g = 2.0062, A(14N) = 34.2 MHz (n = 1), A(1H) = 1.10 MHz (n = 2), A(15N) = 48.1 MHz (n = 1), A(13C) = 25.3 MHz (n = 4), A(13C) = 15.5 MHz (n = 2), Lorentzian/Gaussian = 0.4, LW = 0.020 mT (all nuclei are at natural abundance).
Two well resolved side bands originating from 13C hyperfine couplings of two quaternary carbons and four carbonyl (C=O) carbons with A(13C) = 15.5 and 25.3 MHz, respectively, are observed (bottom panel in Figure 3, Table 2). Similarly, for 2, multiplet patterns are observed, corresponding to the hyperfine coupling of three pairs of protons, i.e., protons of the vinylic group and diastereotopic protons within the methylene (CH2) groups, with A(1H) = 1.86, 1.43, and 1.09 MHz (Figure S9, SI).
Table 2. Experimental and DFT-Computed Isotropic 1H Hyperfine Couplings (A(1H) in MHz) for Nitroxides 1 and 2a.
| (Me) |
(vinylic) |
(CH2)b |
||||||
|---|---|---|---|---|---|---|---|---|
| EPR | NMRc | EPRd | NMRc | DFT | EPRd | NMRc | DFT | |
| 1e | – | +0.034 | 1.10 | –1.08 | –1.68 | – | – | – |
| 2 | – | +0.058 | 1.43 | –1.26 | –1.80 | 1.09 | –1.6 | –1.37 |
| +0.017 | 1.86 | –1.94 | ||||||
DFT computations at the UB3LYP/EPR-III//UB3LYP/6-311G(d,p)+ZPVE level; because no conformational analysis of the methyl groups was carried out, the computed values of A(1H) for the methyl groups are not reported.
Protons in the CH2 moiety are diastereotopic.
For paramagnetic 1H NMR spectra of 1 and 2, see: Figures S15, S16, S24, and S25, SI.
For fluid solution CW EPR spectra of 1 and 2 with spectral simulations, see: Figures S7–S9, SI.
Computed values of A(13C) (in MHz) in C2-symmetric 1 are −16.9 for C1 and C4 (quaternary carbons), +30.5 and +22.2 (avg. = +26.4) for C9–C13 (carbonyl carbons); all other carbons have relatively small values of A(13C) ≤ 0.5 MHz, and thus their sidebands are not resolved in the experimental spectrum.
Notably, the EPR spectral line widths are rather narrow, ≤ 0.02 mT, indicating that the unresolved isotropic hyperfine couplings from protons of the methyl groups are very small, i.e., A(1H) < 0.1 MHz. This finding is confirmed by paramagnetic 1H NMR spectroscopy.30−32 For 1, the spectrum consists of a singlet peak at δ = 4.72 ppm and the other, much broader singlet at δ = −22.9 ppm (Figure 4).
Figure 4.

Paramagnetic 1H NMR (400 MHz) spectrum for 0.9 M nitroxide 1 in chloroform-d.
Based on the corresponding paramagnetic shifts (vs diamagnetic reference such as 1-H), the values of isotropic 1H hyperfine couplings are A(1H) = +0.034 MHz and A(1H) = −1.08 MHz for the methyl and vinylic protons, respectively. Similar analyses for 2 (and 2-H) provide A(1H) = +0.058 MHz and A(1H) = +0.017 MHz for the two types of methyl protons (Table 2).33 For comparison, an ∼1 order of magnitude larger value of |A(1H)| = 0.90 MHz for the methyl group protons was measured in a typical five-membered ring gem-dimethyl nitroxide such as Proxyl.34 Values of isotropic A(14N) and of the anisotropic Azz(14N) (Figures S5 and S6, SI) increase in the order 1 < 2 < MTSL. The smaller hyperfine couplings for the carboxy-substituted nitroxides are attributed to the electron-withdrawing and electron delocalization effects of the carboxylate groups.35
Using the lowest energy C2-symmetric conformational minima for 1 and 2, identified at the UB3LYP/6-311G(d,p)+ZPVE level of theory, the UB3LYP-computed values of isotropic A(1H) using the EPR-III basis set are in qualitative agreement with experiment (Table 2).36 For 1, the computed values of isotropic A(13C) for quaternary and carbonyl carbons well reproduce experimental values (Footnote in Table 2 and Figure 3).
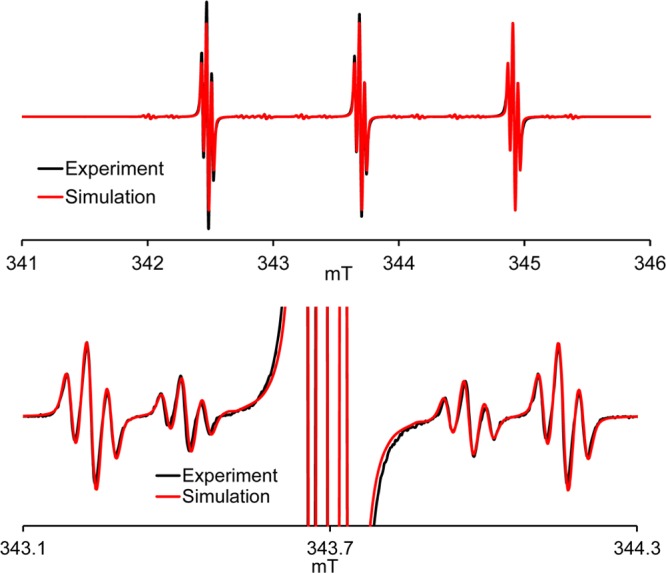
Electron spin relaxation times were measured on samples immobilized in a glassy sugar matrix consisting of 90% trehalose and 10% sucrose. Trehalose has been shown to cryoprotect biomaterials better than other sugars.37 Trehalose also maintains protein function and enzyme activity better than dehydrated sucrose.38,39 Glasses containing 10% sucrose were observed to be more stable during drying than 100% trehalose glasses. If the glass crystallizes, the solute is excluded from the lattice which results in locally high concentrations of solute that enhance relaxation rates (Figure S4, SI). The temperature dependence of 1/Tm for 1 and 2 at the X- and Q-band is summarized in Figure 5. Rates at both frequencies were measured at the field that corresponds to the maximum amplitude of the absorption spectrum. Relaxation rates are similar for the two radicals. We observe that 1/Tm is approximately independent of temperature below about 160 K. Notably, the 1/Tm values for 1 and 2 in trehalose/sucrose at T < 160 K are similar to those observed for spirocyclohexyl nitroxides in 1:1 water/glycerol at T < 70 K,22,23 except that we observe a small enhancement in 1/Tm at about 60 to 70 K. We attribute this enhancement to the rotation of the carboxy methyl groups at rates comparable to the electron proton dipolar coupling. This effect on 1/Tm is much smaller than in the case of the traditional gem-dimethyl groups of MTSL because the electron–proton couplings are much smaller, as suggested by the relatively small values of isotropic A(1H) (Table 2). It occurs at lower temperatures than those for gem-dimethyl groups of MTSL because the barrier to rotation is smaller. This enhancement would be a small disadvantage for DEER experiments at 40 to 70 K but not a factor for experiments at higher temperatures, with optimum conditions, i.e., long Tm and short T1, at about 120 K. At temperatures above about 160 K, the 1/Tm relaxation rates increase, which is attributed to increasing motion of the radicals in the lattice and shorter T1. The 1/Tm values for the two radicals remain similar at the X- and Q-band.
Figure 5.
Temperature dependence of 1/T1 at X-band (9.44 GHz) in the perpendicular plane (blue circles) 1, (red squares) 2, and at Q-band (33.90 GHz) in the perpendicular plane (red triangles) 2, along the z-axis (red inverted triangles) 2. Temperature dependence of 1/Tm at X-band in the perpendicular plane (blue triangles) 1, (red plus) 2. Lines connect the data points. Samples were in 9:1 trehalose/sucrose.
The spin–lattice relaxation rate, 1/T1, is important for the DEER experiments because it determines how quickly the signal averaging can be repeated. The temperature dependence of 1/T1 for 1 and 2 in the perpendicular plane of the molecule is similar at X- and Q-band and similar for the two radicals (Figure 5). This result is consistent with the expectation that the Raman process dominates spin–lattice relaxation at lower temperatures and a local mode process dominates at higher temperatures.40 The 1/T1 relaxation rates for both of these processes are independent of the microwave frequency. Previous studies demonstrated that 1/T1 is slower near the parallel axis of the g-matrix (along the p-π orbital of the nitrogen) than in the perpendicular plane.41 At Q-band, anisotropy is more highly resolved than at X-band, and the anisotropy in 1/T1 is readily observed (Figure 5). At temperatures up to about 200 K, 1/T1 for these radicals in trehalose/sucrose is similar to values for Spiro-TOAC in 1:1 water/glycerol.23 The water/glycerol glass softens and melts at about 200 K, whereas the trehalose/sucrose glass remains relatively rigid at ambient temperature. In the relatively rigid trehalose/sucrose glass, there is negligible contribution to 1/T1 from modulation of g and A-anisotropy, so relaxation rates at temperatures above about 200 K are slower in this environment than in water/glycerol.40
Minimal dependence of 1/Tm on the position in the spectrum is important for the DEER experiments, because the amplitude of the echo that is used to monitor the electron–electron dipolar interaction depends on Tm. If Tm were much shorter for some orientations of the molecule in the magnetic field than for others, those orientations would be under-represented in the DEER response. Orientation dependence of 1/Tm was examined in detail at Q-band. Field-swept echo detected spectra of 2 are shown in Figure 6. The spectrum at 80 K is typical for immobilized nitroxides. The amplitude at each position in the spectrum depends on the fraction of radicals on resonance at that field and on the value of 1/Tm. The extremum of the absorption spectrum at about 1213 mT (position 3 in Figure 6) corresponds to molecules with nitrogen mI = −1 that are aligned with the magnetic field along the molecular z-axis. The region of the spectrum near 1212 mT corresponds to molecules with the magnetic field at an orientation that is intermediate between the z-axis and the xy plane, and is particularly sensitive to motions that alter the orientation of the molecular z-axis relative to the external field on the time scale of the spin echo experiment.42
Figure 6.
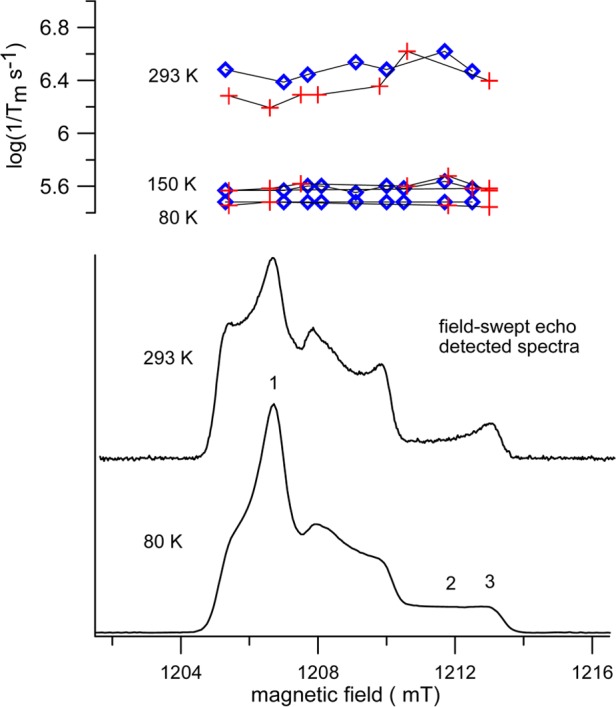
Lower panel: Field-swept echo-detected spectra for 2 in 9:1 trehalose/sucrose at 33.90 GHz at 80 and 293 K. Positions labeled as 1, 2, and 3 correspond, respectively, to resonance for molecules with the magnetic field in the xy plane, an orientation intermediate between the z-axis and the xy plane, and near the z-axis. The upper panel displays 1/Tm as a function of position in the spectrum for 1 (blue diamonds) and 2 (red plus) at 80, 150, and 293 K.
At 293 K the decreased amplitude of the field-swept echo-detected signal at about 1212 mT (position 2) relative to that at about 1213 mT (position 3) reflects the increase in 1/Tm at the intermediate orientation of the molecule that arises from motion on the time scale of the spin–echo experiment. Values of 1/Tm for 1 and 2 as a function of position in the spectrum are shown in the upper panel of Figure 6. Between 80 and 150 K there is little temperature dependence of 1/Tm, and values show little dependence on position in the spectrum, as is expected when the radical is well immobilized. Relaxation rates at 293 K are faster than at lower temperature and more dependent on position in the spectrum. The slowest rates are observed at position 1, which is in the perpendicular plane. At this position in the spectrum, low amplitude motions cause little change in resonance field.42 The fastest relaxation rates are near position 2, where the resonance field is more sensitive to motion. However, even at 293 K, the variation of 1/Tm through the spectrum of 1 or 2 is less than a factor of 2.5 for the positions at which values were recorded.
Electron spin relaxation rates at room temperature for nitroxides immobilized in trehalose matrices have been compared for nitroxides with a range of structures.25 The concern was expressed that structures and substituents that made Tm longer also made T1 longer. The signal-to-noise in the DEER experiment is much more strongly dependent on Tm than on T1. The intensity of an echo at a particular time between pulses depends exponentially on Tm. Thus, a factor of 2 decrease in Tm, decreases the echo intensity by a factor of e–2 = 0.14, which means that the echo is only 14% of what it would have been at the longer Tm. A factor of 2 increase in T1 only increases the data acquisition time by a factor of 2. Furthermore, near room temperature T1 of nitroxides is short enough that the duty cycle of the high power amplifiers may limit the pulse repetition time, rather than the nitroxide relaxation. Thus, modest increases in T1 that accompany lengthening of Tm are not a major concern. It is also expected that rigid attachment of a spin label to a macromolecule will enhance immobilization and lengthen Tm beyond what is observed for the small molecule nitroxides.
Conclusion
The approximately temperature independent value of 1/Tm for 1 and 2 up to about 160 K and modest temperature dependence up to 295 K demonstrate that increasing the distance between the nitroxide moiety and methyl groups is effective in increasing Tm at T > 70 K. Rigid attachment to a biomolecule is expected to further limit motion at room temperature and thereby decrease 1/Tm and the orientation dependence of 1/Tm.
Experimental Section
Synthesis of 1 and 2
Chemicals and per-deuterated solvents for NMR spectroscopy were obtained from commercial sources and used as received unless otherwise indicated. Purification and titration of commercial m-CPBA (70%) were carried out as previously described.43 1-(tert-Butyl)-2,5-dimethyl 1H-pyrrole-1,2,5-tricarboxylate (3) was prepared according to the literature method.28,29 Column chromatography was carried out on flash grade silica gel, using 0–20 psig pressure. Preparative TLC (PTLC) was carried out using tapered silica plates with a preadsorbent zone.
NMR spectra were obtained using commercial spectrometers (1H, 300, 400, and 700 MHz) using chloroform-d (CDCl3) as solvent. The 700 MHz instrument was equipped with a cryoprobe. The chemical shift references were as follows: (1H) chloroform, 7.260 ppm and (13C) chloroform-d, 77.2 ppm. Typical 1D FID was subjected to exponential multiplication with an exponent of 0.3 Hz (1H) or 1 Hz (13C). IR spectra were obtained using a commercial instrument, equipped with an ATR sampling accessory. MS analyses were carried out at the local mass spectrometry facility.
Detailed procedures for all new compounds may be found in the SI.
1-(tert-butyl)-2,2,5,5-Tetramethyl-1H-pyrrole-1,2,2,5,5-pentacarboxylate (4) and 1-(tert-Butyl)-2,5-dimethyl-trans-2,5-bis(2-methoxy-2-oxoethyl)-2,5-dihydro-1H-pyrrole-1,2,5-tricarboxylate (6)
4,4′-Di-tert-butylbiphenyl (1 equiv) in THF was added to Li metal (20+ equiv) in THF at 0 °C. After stirring for 3 h, the reaction mixture was cooled to −78 °C and a solution of 3 (2 equiv) in THF (0.2–0.4 mM) was added dropwise.28 After 16 h, the dark brown liquid part of the reaction mixture (i.e., dianion of 3) was cannulated to mostly frozen methyl chloroformate (2 equiv) or methyl bromoacetate (2 equiv) and then stirred for 1 d at −78 °C. After addition of saturated aqueous ammonium chloride the reaction mixture was allowed to reach room temperature. Subsequent aqueous workup with diethyl ether provided crude products, which were purified by column chromatography (silica gel, pentane/ethyl acetate, 1:1): 4, Rf 0.32, 1.86 g (66%) from 2.0 g of 3, and for 6, Rf 0.52, 0.71 g (47%) from 1.0 g of 3. In the 1H and 13C NMR spectra of 4 and 6, some of the peaks are doubled due to the presence of two conformers of the Boc group.28,29 Characterization for compound 4. Light yellow solid, mp 105–107 °C. 1H NMR (700 MHz, CDCl3): δ 6.139 (d, J = 5.9 Hz, 1H), 6.108 (d, J = 5.9 Hz, 1H), 3.816 (s, 6H), 3.811 (s, 6H), 1.440 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 166.8, 151.8, 130.5, 130.1, 82.4, 78.85, 78.75, 53.4, 53.2, 28.1. HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C17H23NO10Na 424.1220; found 424.1216. IR (diamond, cm–1): 3101, 2996, 2976, 2957, 1772, 1746, 1717, 1433, 1368, 1231, 1203, 1166, 1131, 1091, 1045, 1012, 970, 906, 845, 810, 766, 738, 719, 624. Characterization for compound 6. Light yellow oil. 1H NMR (300 MHz, CDCl3): δ 6.43 (d, J = 6 Hz, 1H), 6.40 (d, J = 6 Hz, 1H), 3.723 (s, 6H), 3.690 (s, 6H), 3.71 (d, J ≈ 10 Hz, 1H), 3.490 (d, J = 15 Hz, 1H), 2.910 (dd, J = 15 Hz, J = 6.0 Hz, 2H), 1.432 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 170.89, 170.75, 170.60, 170.54, 151.5, 131.7, 131.5, 82.3, 74.4, 73.7, 53.2, 53.1, 51.90, 51.80, 40.7, 39.4, 28.3. HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C19H27NO10Na 452.1533; found 452.1543. IR (diamond, cm–1): 2984, 2954, 1738, 1703, 1436, 1349, 1329, 1289, 1220, 1160, 1110, 1068, 1001, 899, 864, 845, 801, 771, 716, 670.
Tetramethyl-1H-pyrrole-2,2,5,5-tetracarboxylate (5) and Dimethyl-trans-2,5-bis(2-methoxy-2-oxoethyl)-2,5-dihydro-1H-pyrrole-2,5-dicarboxylate (7)
To compound 4 (2.64 g, 6.57 mmol, 1 equiv) or 6 (0.400 g, 0.932 mmol, 1 equiv) in dichloromethane (DCM, 0.02–0.1 mM) trifluoroacetic acid (TFA, 30–300 equiv) was added at 0 °C. Following the addition, the reaction mixture was kept for 5 min at 0 °C, and then it was stirred at room temperature for 1–2 h, until TLC analyses indicated the absence of the starting material. DCM and TFA were rapidly removed under vacuo; the resultant crude product was treated with methanol (3 times), which was then removed under vacuo. Purification by column chromatography (silica gel) gave the products: compound 5, methanol/DCM, 3:97, Rf 0.48, 1.720 g (87%); compound 7, pentane/ethyl acetate, 3:1, Rf 0.15, 0.127 g (42%). Characterization of compound 5. Sand colored solid, mp 46–52 °C. 1H NMR (600 MHz, CDCl3): δ 6.156 (s, 2H), 3.801 (s, 12H). 13C NMR (176 MHz, CDCl3): δ 169.4, 130.5, 79.4, 53.5. HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C12H15NO8Na 324.0695; found 324.0685. IR (diamond, cm–1): 3318, 3011, 2958, 2844, 1735, 1435, 1267, 1226, 1153, 1073, 1034, 965, 928, 860, 804, 765, 734, 699. Characterization for compound 7. Brown solid, mp 80–81 °C. 1H NMR (300 MHz, CDCl3): δ 5.908 (s, 2H), 3.688 (d, J = 18 Hz, 12H), 2.929 (d, J = 16 Hz, 4H). 13C NMR (75 MHz, CDCl3): δ 173.9, 171.2, 132.7, 73.9, 53.0, 52.0, 43.7. HRMS (ESI/TOF-Q) m/z: [M + H]+ Calcd for C14H20NO8 330.1189; Found 330.1177. IR (diamond, cm–1): 3398, 3091, 2957, 1731, 1713, 1433, 1410, 1354, 1320, 1285, 1274, 1244, 1202, 1174, 1079, 1059, 1047, 994, 890, 850, 808, 797, 758, 708.
Nitroxides 1 and 2
0.1–0.4 mM purified m-CPBA43 (3.8 equiv) in DCM was added dropwise to 0.3–0.4 mM 5 (0.849 g, 2.82 mmol) or 7 (250 mg, 0.76 mmol) in DCM in an ice bath (0 °C). Following the addition, the resultant mixture was stirred for 5 min at 0 °C, and then for 2–3 h at room temperature. The solvents were removed under vacuum, and the residue was dissolved in diethyl ether, washed with aqueous saturated sodium bicarbonate and brine, dried over sodium sulfate, filtered, and evaporated, to provide a crude product. The crude was purified with column chromatography (silica gel, chloroform/ethyl acetate, 2:1, Rf 0.45) and then treated with diethyl ether to provide nitroxide 1 (145 mg, 16%) or purified by column chromatography (silica gel, pentane/ethyl acetate, 3:1, Rf = 0.31) to provide nitroxide 2 (190 mg, 73%). Characterization of nitroxide 1. Orange powder, mp 127–129 °C. EPR (X-band, 0.1 mM in C6H6): spin concentration 99%. Paramagnetic 1H NMR (400 MHz, CDCl3): δ 4.72 (s), −22.87 (bs). HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C12H14NO9Na 339.0566; found 339.0578. IR (diamond, cm–1): 3121, 2961, 1759, 1737, 1441, 1340, 1244, 1168, 1115, 1047, 960, 918, 890, 801, 769, 728, 659, 640. Characterization for nitroxide 2. Light yellow solid, mp 119–120 °C. EPR (X-band, 0.1 mM in chloroform): spin concentration ∼100%. Paramagnetic 1H NMR (400 MHz, CDCl3): δ 5.31 (s), 4.13 (s), −27.72 (bs), −40 (very bs). HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C14H18NO9Na 367.0879; found 367.0870. IR (diamond, cm–1): 2965, 2916, 2848, 2160, 1978, 1745, 1725, 1432, 1414, 1359, 1336, 1267, 1226, 1209, 1169, 1128, 1062, 1003, 977, 965, 922, 901, 853, 797, 766, 716, 674, 626.
Hydroxylamines: Tetramethyl-1-hydroxy-1H-pyrrole-2,2,5,5-tetracarboxylate (1-H) and Dimethyl-trans-1-hydroxy-2,5-bis(2-methoxy-2-oxoethyl)-2,5-dihydro-1H-pyrrole-2,5-dicarboxylate (2-H)
Nitroxide 1 (5.1 mg) or nitroxide 2 (10.6 mg) was dissolved in a pH 7.4 buffered ascorbate solution: for 1, 2 mL, 20 mM ascorbate, 20 mM reduced glutathione, and 125 mM phosphate; for 2, 2.5 mL, 0.062 mM ascorbate, 12.4 mM phosphate. For 1 and 2, the reaction mixtures were stirred at room temperature under the exclusion of light for 15 min and 3 h, respectively. Subsequent extraction with ethyl acetate, drying over sodium sulfate, and removal of solvents provided crude products 1-H and 2-H. Characterization for crude 1-H. 1H NMR (700 MHz, CDCl3): δ 6.605 (bs, 1H), 6.180 (s, 2H), 3.811 (s, 12H). 13C NMR (176 MHz, CDCl3): δ 167.5, 130.3, 82.2, 53.3. HRMS (ESI/TOF-Q) m/z: [M + Na]+ calcd for C12H15NO9Na 340.0645; found 340.0642. IR (diamond, cm–1): 3440 (br), 2958, 1733, 1434, 1255, 1155, 1039, 801. Characterization for crude 2-H. 1H NMR (700 MHz, CDCl3): δ 6.105 (s, 2H), 5.79 (bs, 1H), 3.755 (s, 6H), 3.665 (s, 6H), 3.294 (d, J = 17 Hz, 2H), 2.893 (d, J = 17 Hz, 2H). 13C NMR (176 MHz, CDCl3): δ 171.7, 171.3, 131.7, 78.1, 52.8, 51.9, 39.3. HRMS (ESI/TOF-Q) m/z: [M]+ calcd for C14H19NO9 345.1060; found 345.1069. IR (diamond, cm–1): 3442 (br), 2955, 1733, 1437, 1348, 1207, 1079, 1005, 785.
X-ray Crystallography of 1 and 2
For 1 and 2, crystals were obtained by slow evaporation of a DCM/heptane (1:1) solution and vapor diffusion of pentane into a chloroform solution, respectively. Data were collected at low temperature using a commercial diffractometer for 1 and synchrotron radiation for 2. Data reduction and absorption corrections were carried out using standard procedures. Structure solutions were obtained by intrinsic methods.44 Further details are provided in the SI PDF and CIF files.
Electron Spin Relaxation Studies
Samples for relaxation time measurements were prepared as follows: Solutions of 0.1 mM nitroxide in 1:1 water/ethanol were mixed with an aqueous solution containing 9:1 trehalose/sucrose. The mole ratio of sugars to nitroxide was 2000:1. The solutions were placed in a thin layer on a watch glass. The solvent evaporated overnight, forming a glassy film. The film was scraped off the water glass and transferred to a 4 mm quartz EPR tube. EPR tubes were evacuated on a vacuum line overnight to remove oxygen and residual water and then flame-sealed. Samples for the Q-band were aliquots of materials that had been dried in vacuum overnight and then transferred to 1.8 mm quartz capillaries, without further evacuation.
Relaxation times at the X- and Q-band were measured. Tm was measured by two-pulse echo decay using a π/2 – τ – π – τ – echo sequence and initial τ = 200 or 300 ns. T1 was measured by inversion recovery using a π – T – π/2 – τ – π – τ – echo sequence with a constant τ = 800 ns at Q-band or 600 ns at X-band. The lengths of the π/2 pulses were 40 ns. The temperature dependence of relaxation was monitored at the field that corresponds to the maximum amplitude in the absorption spectrum. At the X-band this position is near the center of the spectrum, and at the Q-band this position is near the low-field end of the spectrum. At both frequencies these positions correspond predominantly to molecules for which the magnetic field is in the molecular xy plane. Echo decays and inversion recovery curves were fitted with single exponentials.
Acknowledgments
We thank the National Science Foundation and National Institutes of Health for support of this work through Grants CHE-1362454 (A.R.), NIBIB R01 EB019950-02 (A.R. and S.S.E.), and NIGMS U54GM087519-06 (A.R.). ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under Grant Number NSF/CHE-1346572. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We thank Dr. Xinming Liu (UNL) for his help with the synthesis and Dr. Nolan Gallagher (UNL) for his help with the EasySpin spectral simulations.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b02737.
Author Contributions
∥ S.H. and J.T.P. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Tebben L.; Studer A. Angew. Chem., Int. Ed. 2011, 50, 5034–5068. 10.1002/anie.201002547. [DOI] [PubMed] [Google Scholar]
- Krishna M. C.; Russo A.; Mitchell J. B.; Goldstein S.; Dafni H.; Samuni A. J. Biol. Chem. 1996, 271, 26026–26031. 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- Hubbell W. L.; Lopez C. J.; Altenbach C.; Yang Z. Curr. Opin. Struct. Biol. 2013, 23, 725–733. 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J.; Burks S. R.; Liu K. J.; Kao J. P. Y.; Rosen G. M. J. Magn. Reson. 2016, 271, 68–74. 10.1016/j.jmr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rajca A.; Wang Y.; Boska M.; Paletta J. T.; Olankitwanit A.; Swanson M. A.; Mitchell D. G.; Eaton S. S.; Eaton G. R.; Rajca S. J. Am. Chem. Soc. 2012, 134, 15724–15727. 10.1021/ja3079829. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sowers M. A.; McCombs J. R.; Wang Y.; Paletta J. T.; Morton S. W.; Dreaden E. C.; Boska M. D.; Ottaviani M. F.; Hammond P. T.; Rajca A.; Johnson J. A. Nat. Commun. 2014, 5, 5460. 10.1038/ncomms6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone T. J.; Buckman T.; Nordio P. L.; McConnell H. M. Proc. Natl. Acad. Sci. U. S. A. 1965, 54, 1010–1017. 10.1073/pnas.54.4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C.; Flitsch S. L.; Khorana H. G.; Hubbell W. L. Biochemistry 1989, 28, 7806–7812. 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- Altenbach C.; Marti T.; Khorana H. G.; Hubbell W. L. Science 1990, 248, 1088–1092. 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Sajid M.; Schulte M.; Ramezanian N.; Volkov A.; Zimmermann H.; Godt A. J. Am. Chem. Soc. 2010, 132, 10107–10117. 10.1021/ja102983b. [DOI] [PubMed] [Google Scholar]
- Akyuz N.; Georgieva E. R.; Zhou Z.; Stolzenberg S.; Cuendet M. A.; Khelashvili G.; Altman R. B.; Terry D. S.; Freed J. H.; Weinstein H.; Boudker O.; Blanchard S. C. Nature 2015, 518, 68–73. 10.1038/nature14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B.; Sikora A.; Cafiso D. S. J. Am. Chem. Soc. 2016, 138, 1844–1847. 10.1021/jacs.5b13382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser P.; Schmidt M. J.; Drescher M.; Summerer D. Org. Biomol. Chem. 2016, 14, 5468–5476. 10.1039/C6OB00473C. [DOI] [PubMed] [Google Scholar]
- Jeschke G. Annu. Rev. Phys. Chem. 2012, 63, 419–446. 10.1146/annurev-physchem-032511-143716. [DOI] [PubMed] [Google Scholar]
- Baber J. L.; Louis J. M.; Clore G. M. Angew. Chem., Int. Ed. 2015, 54, 5336–5339. 10.1002/anie.201500640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbat P. P.; Mchaourab H. S.; Freed J. H. J. Am. Chem. Soc. 2002, 124, 5304–5353. 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- Borbat P. P.; Davis J. H.; Butcher S. E.; Freed J. H. J. Am. Chem. Soc. 2004, 126, 7746–7747. 10.1021/ja049372o. [DOI] [PubMed] [Google Scholar]
- Yang Z. Y.; Liu Y. P.; Borbat P.; Zweier J. L.; Freed J. H.; Hubbell W. L. J. Am. Chem. Soc. 2012, 134, 9950–9952. 10.1021/ja303791p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarkh M.; Singh V.; Okle O.; Seemann I. T.; Dietrich D. R.; Hartig J. S.; Drescher M. Nat. Protoc. 2012, 8, 131–147. 10.1038/nprot.2012.136. [DOI] [PubMed] [Google Scholar]
- Shevelev G. Y.; Krumkacheva O. A.; Lomzov A. A.; Kuzhelev A. A.; Rogozhnikova O. Y.; Trukhin D. V.; Troitskaya T. I.; Tormyshev V. M.; Fedin M. V.; Pyshnyi D. V.; Bagryanskaya E. G. J. Am. Chem. Soc. 2014, 136, 9874–9877. 10.1021/ja505122n. [DOI] [PubMed] [Google Scholar]
- Dzuba S. A.; Maryasov A. G.; Salikhov K. M.; Tsvetkov Yu. D. J. Magn. Reson. 1984, 58, 95–117. 10.1016/0022-2364(84)90010-6. [DOI] [Google Scholar]
- Nakagawa K.; Candelaria M. B.; Chik W.W. C.; Eaton S. S.; Eaton G. R. J. Magn. Reson. 1992, 98, 81–91. 10.1016/0022-2364(92)90111-J. [DOI] [Google Scholar]
- Meyer V.; Swanson M. A.; Clouston L. J.; Boratyński P. J.; Stein R. A.; Mchaourab H. S.; Rajca A.; Eaton S. S.; Eaton G. R. Biophys. J. 2015, 108, 1213–1219. 10.1016/j.bpj.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajca A.; Kathirvelu V.; Roy S. K.; Pink M.; Rajca S.; Sarkar S.; Eaton S. S.; Eaton G. R. Chem. - Eur. J. 2010, 16, 5778–5782. 10.1002/chem.200903102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirilyuk I. A.; Polienko Y. F.; Krumkacheva O. A.; Strizhakov R. K.; Gatilov Y. V.; Grigor’ev I. A.; Bagryanskaya E. G. J. Org. Chem. 2012, 77, 8016–8027. 10.1021/jo301235j. [DOI] [PubMed] [Google Scholar]
- Kuzhelev A. A.; Strizhakov R. K.; Krumkacheva O. A.; Polienko Y. F.; Morozov D. A.; Shevelev G. Y.; Pyshnyi D. V.; Kirilyuk I. A.; Fedin M. V.; Bagryanskaya E. G. J. Magn. Reson. 2016, 266, 1–7. 10.1016/j.jmr.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Personal communication from Drs. Elka R. Georgieva, Peter P. Borbat, and Jack H. Freed (Cornell University). Approximately 8% efficiency in labelling of Cys-8 in T4L with Spiro-IA was determined; the reason for this low efficiency was not established. [Google Scholar]
- Personal communication from Dr. Christine B. Karim (University of Minnesota). [Google Scholar]
- Donohoe T. J.; Headley C. E.; Cousins R. P. C.; Cowley A. Org. Lett. 2003, 5, 999–1002. 10.1021/ol027504h. [DOI] [PubMed] [Google Scholar]
- a Donohoe T. J.; Thomas R. E.; Cheeseman M. D.; Rigby C. L.; Bhalay G.; Linney I. D. Org. Lett. 2008, 10, 3615–3618. (b) NMR data for compound 3 from refs (28) and (29a): 1H NMR (400 MHz, CDCl3) δ 6.84 (2 H, s), 3.87 (6 H, s), 1.67 (9 H, s); 13C NMR (101 MHz, CDCl3) δ 159.9, 148.9, 126.7, 115.8, 86.3, 52.0, 27.3. (c) NMR data for compound 3: 1H NMR (300 MHz, CDCl3): δ 6.83 (s, 2H), 3.86 (s, 6H), 1.66 (s, 9H). 13C NMR (176 MHz, CDCl3): δ 160.0, 149.0, 126.8, 115.9, 86.4, 52.1, 27.4 10.1021/ol801415d. [DOI] [PubMed] [Google Scholar]
- la Mar G. N., Ed. Nuclear Magnetic Resonance of Paramagnetic Macromolecules; Springer: Netherlands, 1995; pp 1 – 391. [Google Scholar]
- Olankitwanit A.; Kathirvelu V.; Rajca S.; Eaton G. R.; Eaton S. S.; Rajca A. Chem. Commun. 2011, 47, 6443–6445. 10.1039/c1cc11172h. [DOI] [PubMed] [Google Scholar]
- Gallagher N. M.; Bauer J. J.; Pink M.; Rajca S.; Rajca A. J. Am. Chem. Soc. 2016, 138, 9377–9380. 10.1021/jacs.6b05080. [DOI] [PubMed] [Google Scholar]
- These two values of A(1H) = +0.058 and +0.017 MHz in nitroxide 2 are the higher and lower bounds, due to uncertainty in the assignment of the resonances for the methyl groups in 2 and in 2-H.
- a Biller J. R.; Meyer V.; Elajaili H.; Rosen G. M.; Kao J. P. Y.; Eaton S. S.; Eaton G. R. J. Magn. Reson. 2011, 212, 370–377. 10.1016/j.jmr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rockenbauer A.; Gyor M.; Hankovszky H. O.; Hideg K. Electron Spin Resonance 1988, 11, 145–182. 10.1039/9781847553454-00145. [DOI] [Google Scholar]
- Rassat A.Molecular Spectroscopy; Institute of Petroleum, distributed by Elsevier Publishing, Brighton, U.K., 1968; pp 145–155. [Google Scholar]
- Frisch M. J., et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Crowe J. H.; Carpenter J. F.; Crowe L. M. Annu. Rev. Physiol. 1998, 60, 73–103. 10.1146/annurev.physiol.60.1.73. [DOI] [PubMed] [Google Scholar]
- Martinez L. M.; Videa M.; Mederos F.; deMoral Y. J. Mex. Chem. Soc. 2011, 55, 185–189. [Google Scholar]
- Möbius K.; Savitsky A.; Nalepa A.; Malferrari M.; Francia F.; Lubitz W.; Venturoli G. Appl. Magn. Reson. 2015, 46, 435–464. 10.1007/s00723-014-0633-4. [DOI] [Google Scholar]
- Yu Z.; Quine R. W.; Rinard G. A.; Tseitlin M.; Elajaili H.; Kathirvelu V.; Clouston L. J.; Boratyński P. J.; Rajca A.; Stein R. A.; Mchaourab H. S.; Eaton S. S.; Eaton G. R. J. Magn. Reson. 2014, 247, 67–71. 10.1016/j.jmr.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.-L.; Eaton G. R.; Eaton S. S. J. Magn. Reson., Ser. A 1995, 115, 213–221. 10.1006/jmra.1995.1169. [DOI] [Google Scholar]
- Du J.-L.; More K. M.; Eaton S. S.; Eaton G. R. Isr. J. Chem. 1992, 32, 351–355. 10.1002/ijch.199200041. [DOI] [Google Scholar]
- Rajca A.; Shiraishi K.; Rajca S. Chem. Commun. 2009, 4372–4374. 10.1039/b909741d. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.