

Abstract
Patient: Male, newborn
Final Diagnosis: Simpson-Golabi-Behmel syndrome
Symptoms: Dyspnea
Medication: —
Clinical Procedure: —
Specialty: Pediatrics and Neonatology
Objective:
Congenital defects/diseases
Background:
Simpson-Golabi-Behmel syndrome (SGBS) is a rare X-linked recessive syndrome characterized by fetal overgrowth.
Case report:
We present a case of a male infant with SGBS. Abnormal prenatal ultrasound (including congenital diaphragmatic hernia) prompted microarray testing of amniotic fluid cells, which showed deletion on chromosome Xq26.2 affecting the glypican-3 gene consistent with SGBS type I. The infant died six hours after birth and at autopsy showed features of SGBS, including macrosomia, organomegaly, diaphragmatic hernia with consequent pulmonary hypoplasia, cleft palate, large tongue with a midline groove, a supernumerary nipple, Meckel’s diverticulum, and abnormal phalanges. Additionally, we observed features that have previously not been described in SGBS, including testes with hyperplastic seminiferous tubules and Mullerian remnants, and placenta with incipient fetal thrombotic vasculopathy.
Conclusions:
While most patients with SGBS type I survive into childhood or even adulthood, the severe course in our patient was ascribed to pulmonary hypoplasia secondary to the bilateral diaphragmatic hernia.
MeSH Keywords: Congenital Abnormalities; Hernia, Diaphragmatic; Testicular Diseases; Genetic Diseases, X-Linked
Background
Multiple neonatal syndromes are characterized by overgrowth. While some result in generalized overgrowth (including Perlman syndrome, Simpson-Golabi-Behmel, Sotos, PTEN hamartoma tumor syndrome and Gorlin syndrome), others show mosaic overgrowth (including hemihyperplasia, Beckwith-Wiedemann and Proteus syndrome). Many of these syndromes are associated with abnormal growth factor signaling, and thus include alterations of glucose metabolism and propensity for development of tumors.
Simpson-Golabi-Behmel syndrome (SGBS, OMIM312870; ORPHA373) is a rare, X-linked recessive genetic syndrome [1]. The majority of cases are inherited from the mother, and ~25% are due to de novo mutations. The incidence is not known, with only approximately 250 cases described in the literature. The first cases were described in 1975 and 1984 [2–4], and a case series which named the syndrome was published in 1988 [5]. While primarily occurring in males, carrier females may have mild features; a few case reports with full disease expression in females have also been described, caused by skewed X inactivation, homozygosity, compound heterozygosity, or other mechanisms [1,6]. Genetically, two types of SGBS have been identified: type I with an alteration (mutation or deletion) on chromosome Xq26 involving genes for glypican-3 [7] and glypican-4, and type II (OMIM 300209; ORPHA79022) with mutation in the CXORF5 gene on chromosome Xp22. The latter is more severe and presents with hydrops [8]. Until recently, SGBS was diagnosed primarily postnatally and on clinical grounds. However, significant overlap exists between features of SGBS and other overgrowth syndromes, especially Beckwith-Wiedemann syndrome, resulting in common misdiagnosis. Recently, microarray testing and whole exome sequencing [9,10] have enabled prenatal diagnosis, which enables proactive management of these patients.
Typical features of SGBS include macrosomia, facies, macroglossia, diaphragmatic hernia [11], supernumerary nipples, genitourinary and gastrointestinal anomalies, skeletal anomalies, neonatal hypoglycemia, and increased risk for embryonal tumors. Glypicans are heparan sulfate proteoglycans involved in control of cell growth and cell division, and thus their mutation leads to overgrowth, and increased risk of developing embryonal tumors, mostly Wilms and liver tumors [12]. However, clinical features vary significantly, from very mild forms occasionally seen in carrier females, to severe disease. Due to lyonization, some female carriers have high stature, supernumerary nipples, coarse face, abnormal hands and midline defects, while others are completely asymptomatic. In males, the disease features are variable including physical characteristics, and intellectual disability which may or may not be present. Tenorio et al. provide a review of SGBS features [1]. Furthermore, studies to date have not identified specific genotype-phenotype correlations [13,14]. Patients need to be monitored closely in the newborn period for hypoglycemia, and require multi-disciplinary follow-up depending on specific anomalies seen in each patient. Furthermore, frequent physical examinations, imaging and laboratory testing for monitoring for tumor risk are indicated. Neonatal death is seen in a small percentage of males, and has been largely attributed to heart defects [1].
Case Report
We present a case of an infant who was born to a 27-year-old G4P2 mother via vaginal delivery at 36 weeks and 4 days gestational age (as estimated by first trimester ultrasound). The pregnancy was complicated by polyhydramnios (treated with amnion reduction), and congenital diaphragmatic hernia was diagnosed by ultrasound. The ultrasound was performed at 16 weeks gestation, and revealed a thick nuchal fold, diaphragmatic hernia, echogenic bowel, and absent nasal bone. This prompted amniocentesis, which showed elevated amniotic fluid alpha-fetoprotein (AFP, 2.26 MoM) with normal acetylcholinesterase (AChE), normal karyotype, and no numerical chromosomal abnormalities by aneuploidy FISH screening (which included probes for chromosomes 13, 18, 21, X and Y). In order to investigate the observed anatomic findings and elevated AFP, microarray analysis of cultured amniocytes was performed using the Agilent GGXChip+SNP v1.0. This analysis showed two variants: a likely pathogenic variant on chromosome Xq26.2 with loss of portion of one gene (GPC3) and a variant of unknown significance on chromosome 3p11.2 with gain of two genes and one microRNA. No regions of homozygosity were seen. Analysis of the maternal sample revealed the identical variant on one of the X chromosomes, indicating inheritance of this allele.
The infant was delivered with Apgar scores 1, 3, 5 at 1, 5, and 10 minutes respectively, admitted to the neonatal intensive care unit (NICU), and found to have severe pulmonary hypoplasia with severely impaired gas exchange after birth. Following discussion with the family, the decision was made to proceed with comfort care measures only, and the infant died approximately six hours after birth.
At autopsy, the infant showed macrosomia (3,300 gm, expected for >42 weeks gestational age [GA]; crown-heel length, 51 cm, >42 weeks GA; head circumference, 34 cm, 39 weeks GA) and organomegaly, with enlarged liver (275 gm, >42 weeks GA), spleen (12.3 gm, >42 weeks GA), thymus (14.2 gm, >42 weeks GA), adrenals (13.9 gm combined, >42 weeks GA) and kidneys (59.4 gm combined, >42 weeks GA). The heart (13.6 gm, 35 weeks GA) and brain (337 gm, 37 weeks GA) were not enlarged, and the lungs were significantly hypoplastic (8 gm combined, 19 weeks GA, Figure 1). The finding of small lungs as opposed to enlargement of most organs was due to the congenital diaphragmatic hernia with consequent presence of multiple abdominal organs in the thoracic cavity. In addition, several congenital anomalies were noted, including midline submucosal cleft palate, large tongue with a midline groove, coarse facial features (including large, bulging head, saddle-like and flat-bridged nose with broad, fleshy tip, and large mouth with protruding tongue), a supernumerary nipple, and Meckel’s diverticulum (Figure 1). Furthermore, abnormal phalanges (short middle phalanges of the bilateral small fingers with borderline short distal phalanges of the bilateral hands) were seen by x-ray. No other skeletal abnormalities were seen. There were no congenital heart defects or CNS abnormalities seen grossly or by microscopic examination. Microscopic examination of the pancreas revealed increased number and size of pancreatic islets, some of which were coalescing (Figure 2A, 2B). Together, these findings are consistent with Simpson-Golabi-Behmel syndrome, type I. Additional findings at autopsy included testes with hyperplastic seminiferous tubules and Mullerian remnants. One of the testes was in the abdominal cavity, while the other was in the inguinal canal. Sections of both testes (Figure 2C–2E) showed crowded, back-to-back seminiferous tubules, sometimes confluent, with increased numbers of germ cells (positive for CD117) and increased density of inhibin-positive Leydig cells in the interstitium. The Mullerian remnants were tubular structures, lined by CD10-strongly positive Fallopian tube-like epithelium (ciliated epithelium with interspersed peg cells), and associated with CD10-positive stroma (Figure 2F–2I). Section of the pituitary gland showed evidence of pseudoglandular transformation (Figure 2J). The umbilical cord consisted of a proximal thin portion (1 cm in diameter) with normal coiling (2.8 coils/10 cm), and distant hypercoiled portion (3.2 coils/10 cm) (Figure 3). The placenta was mature and showed incipient fetal thrombotic vasculopathy. A few scattered, partially fibrotic and avascular villi were seen on H&E. However, the E-cadherin/CD34 immunostain [15] highlighted a cluster of relative hypovascularity but with normal density of villous cytotrophoblasts (Figure 3).
Figure 1.

Gross findings during autopsy. Autopsy revealed diaphragmatic hernia (A) with liver in thoracic cavity bilaterally (B), leading to pulmonary hypoplasia (C). Additional findings at autopsy included supernumerary nipple on the right (D) and Meckel’s diverticulum (E).
Figure 2.

Microscopic findings. Sections of the pancreas stained by H&E (A) and chromogranin immunohistochemistry (B) highlight the pancreatic islet cells, which are increased in number, and somewhat diffuse and coalescing islets. Section of testis stained with H&E (C), inhibin (staining Sertoli cells) (D) and CD117 (staining germ cells) (E) highlights the hyperplastic seminiferous tubules. Bilaterally, Mullerian remnants are seen with H&E (F), with tubules strongly positive for CD10 (G). There is also spindle cell ovarian-type stroma (H), also positive for CD10 (I). The pituitary gland shows focal pseudoglandular transformation (J).
Figure 3.
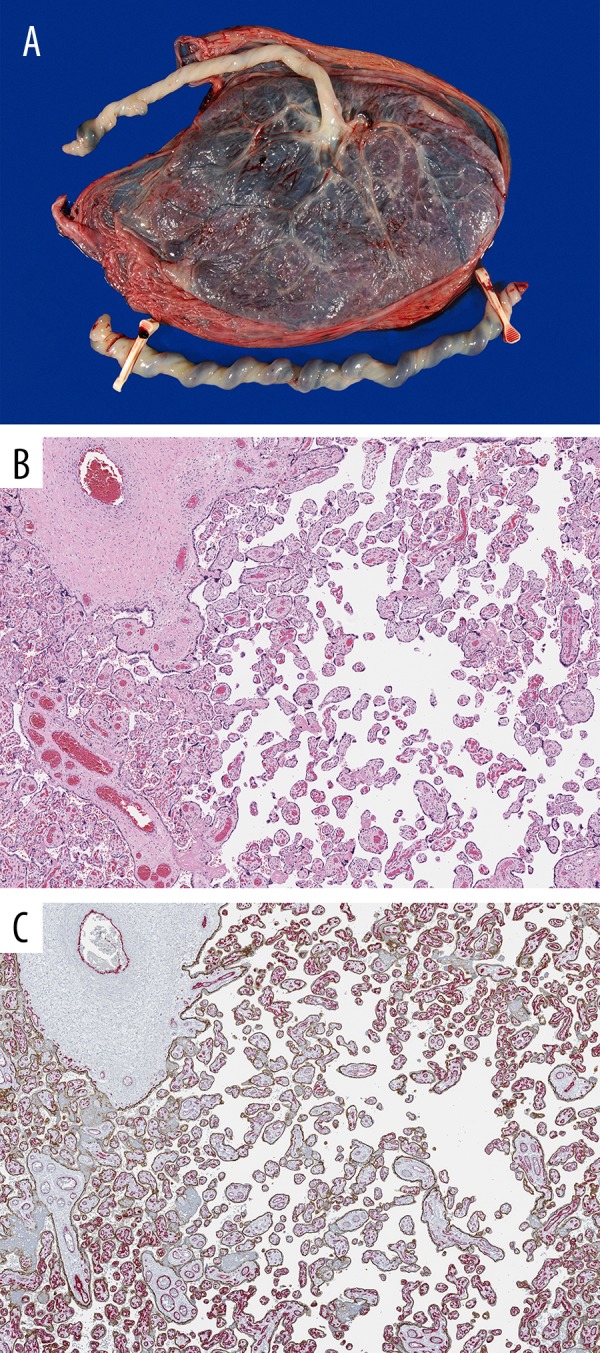
Gross and microscopic findings of the placenta. Grossly, the distal portion of the cord was hypercoiled (A), and microscopically avascular villi are seen on H&E (B) and E-cadherin (brown)/CD34 (red) double-immunohistochemistry (C).
Discussion
Many of the features seen in our patient were characteristic of the SGBS syndrome, including macrosomia, organomegaly, macroglossia with a grooved tongue, cleft palate, diaphragmatic hernia, coarse facial features, supernumerary nipples, and islet cell hyperplasia. However, our patient had some unique features that had previously not been described in the context of SGBS.
Microscopic analysis of testes revealed hyperplasia of the seminiferous tubules and Mullerian remnants. Since most patients with SGBS live into adulthood, histology of neonatal testis has not been previously described to the best of our knowledge. However, previous case descriptions have documented cryptorchidism [1] thus suggesting that growth and development of this organ is under the control of regulatory factors which are dysregulated in SGBS. Hyperplasia of the seminiferous tubules seen in our case was consistent with overall overgrowth features of the syndrome, presumably due to unchecked cell growth and division in the absence of regulator glypican-3. We were unable to detect expression of glypican-3 in gestational age-matched, non-SGBS testes (data not shown); however, this does not exclude the possibility that glypican-3 is expressed earlier in the development of the testis leading to overgrowth effects seen beyond the time frame of glypican-3 expression and effect. This is further supported by the fact that glypican-3 is expressed in testicular germ cell tumors [16]. The persistence of the paramesonephric duct could be due to disturbances in anti-Mullerian hormone, a feature previously not reported in SGBS.
Another unique feature described in our case was the fetal thrombotic vasculopathy seen on microscopic examination of the placenta. This finding was most likely due to impaired fetal blood perfusion and stasis-induced thrombotic coagulopathy, as seen previously in other severe congenital malformations [17], such as diaphragmatic hernia observed in this case. Alternatively or additionally, the fetal thrombotic vasculopathy (FTV) could be related to the stasis due to umbilical cord hypercoiling [18]. This highlights the importance of considering coagulopathy in the management of fetuses and neonates with SGBS.
While most patients with SGBS type I survive into childhood or even adulthood, some die in the neonatal period and most of those deaths have been ascribed to heart malformations and/or arrhythmias [1,19]. Our case highlights another possible cause of early mortality, as the severe course in our patient was ascribed to pulmonary hypoplasia secondary to the bilateral diaphragmatic hernia. This highlights the importance of careful monitoring of patients who have been prenatally diagnosed with SGBS, as diaphragmatic hernia and pulmonary hypoplasia may need to be managed prenatally for optimal outcomes. Furthermore, delivery should be scheduled at highly experienced medical centers that can appropriately manage possible neonatal hypoglycemia, thrombotic vasculopathy, and consequences of pulmonary hypoplasia, ideally centers with extracorporeal membrane oxygenation (ECMO) capability, and neonatal cardiothoracic surgery.
Conclusions
Our case presents several features not previously described in SGBS, including hyperplasia of the seminiferous tubules and fetal thrombotic vasculopathy of the placenta. Furthermore, our case highlights the potential lethality from diaphragmatic hernia and need for careful prenatal monitoring of patients diagnosed with SGBS.
Acknowledgments
The authors wish to thank Chris Woods for assistance with photomicrographs.
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
References:
- 1.Tenorio J, Arias P, Martinez-Glez V, et al. Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis. 2014;9:138. doi: 10.1186/s13023-014-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simpson JL, Landey S, New M, German J. A previously unrecognized X-linked syndrome of dysmorphia. Birth Defects Original Article Series. 1975;11(2):18–24. [PubMed] [Google Scholar]
- 3.Behmel A, Plochl E, Rosenkranz W. A new X-linked dysplasia gigantism syndrome: Identical with the Simpson dysplasia syndrome? Hum Genet. 1984;67(4):409–13. doi: 10.1007/BF00291401. [DOI] [PubMed] [Google Scholar]
- 4.Golabi M, Rosen L. A new X-linked mental retardation-overgrowth syndrome. Am J Med Genet. 1984;17(1):345–58. doi: 10.1002/ajmg.1320170128. [DOI] [PubMed] [Google Scholar]
- 5.Neri G, Marini R, Cappa M, et al. Simpson-Golabi-Behmel syndrome: An X-linked encephalo-tropho-schisis syndrome. Am J Med Genet. 1988;30(1–2):287–99. doi: 10.1002/ajmg.1320300130. [DOI] [PubMed] [Google Scholar]
- 6.Vaisfeld A, Pomponi MG, Pietrobono R, et al. Simpson-Golabi-Behmel syndrome in a female: A case report and an unsolved issue. Am J Med Genet Part A. 2017;173(1):285–88. doi: 10.1002/ajmg.a.38003. [DOI] [PubMed] [Google Scholar]
- 7.Brzustowicz LM, Farrell S, Khan MB, Weksberg R. Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999;65(3):779–83. doi: 10.1086/302527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet. 1995;59(3):329–33. doi: 10.1002/ajmg.1320590310. [DOI] [PubMed] [Google Scholar]
- 9.Magini P, Palombo F, Boito S, et al. Prenatal diagnosis of Simpson-Golabi-Behmel syndrome. Am J Med Genet A. 2016;170(12):3258–64. doi: 10.1002/ajmg.a.37873. [DOI] [PubMed] [Google Scholar]
- 10.Kehrer C, Hoischen A, Menkhaus R, et al. Whole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome. Prenat Diagn. 2016;36(10):961–65. doi: 10.1002/pd.4920. [DOI] [PubMed] [Google Scholar]
- 11.Slavotinek AM. Single gene disorders associated with congenital diaphragmatic hernia. Am J Med Genet C Semin Med Genet. 2007;145C(2):172–83. doi: 10.1002/ajmg.c.30125. [DOI] [PubMed] [Google Scholar]
- 12.Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: A comprehensive review. Am J Med Genet C Semin Med Genet. 2005;137C(1):53–71. doi: 10.1002/ajmg.c.30064. [DOI] [PubMed] [Google Scholar]
- 13.Mariani S, Iughetti L, Bertorelli R, et al. Genotype/phenotype correlations of males affected by Simpson-Golabi-Behmel syndrome with GPC3 gene mutations: Patient report and review of the literature. J Ped Endocrin Metabol. 2003;16(2):225–32. doi: 10.1515/jpem.2003.16.2.225. [DOI] [PubMed] [Google Scholar]
- 14.Hughes-Benzie RM, Pilia G, Xuan JY, et al. Simpson-Golabi-Behmel syndrome: Genotype/phenotype analysis of 18 affected males from 7 unrelated families. Am J Med Genet. 1996;66(2):227–34. doi: 10.1002/(SICI)1096-8628(19961211)66:2<227::AID-AJMG20>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 15.Johnson SL, Stanek J. E-cadherin/CD34 dual immunohistochemical stain in search for placental focal fetal vascular malperfusion. United States & Canadian Academy of Pathology Annual Meeting; March 4–10, 2017; San Antonio, Texas. 2017. [Google Scholar]
- 16.Ota S, Hishinuma M, Yamauchi N, et al. Oncofetal protein glypican-3 in testicular germ-cell tumor. Virchows Arch. 2006;449(3):308–14. doi: 10.1007/s00428-006-0238-x. [DOI] [PubMed] [Google Scholar]
- 17.Stanek J, Sheridan RM, Le LD, Crombleholme TM. Placental fetal thrombotic vasculopathy in severe congenital anomalies prompting EXIT procedure. Placenta. 2011;32(5):373–79. doi: 10.1016/j.placenta.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Ernst LM, Minturn L, Huang MH, et al. Gross patterns of umbilical cord coiling: Correlations with placental histology and stillbirth. Placenta. 2013;34(7):583–88. doi: 10.1016/j.placenta.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Lin AE, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999;83(5):378–81. [PubMed] [Google Scholar]