

Abstract
The growing complexity of recombinant biopolymers for delivery of bioactive agents requires the ability to control the biomaterial structure with high degree of precision. Genetic engineering techniques have provided this opportunity to synthesize biomaterials in an organism such as E. coli with full control over their lengths and sequences. One class of such biopolymers is recombinant cationic biopolymers with applications in gene delivery, regenerative medicine and variety of other biomedical applications. Unfortunately, due to their highly cationic nature and complex structure, their production in E. coli expression system is marred by low expression yield which in turn complicates the possibility of obtaining pure biopolymer. SlyD and ArnA endogenous E. coli proteins are considered the major culprits that copurify with the low-expressing biopolymers during the metal affinity chromatography. Here, we compared the impact of different parameters such as the choice of expression hosts as well as metal affinity columns in order to identify the most effective approach in obtaining highly pure recombinant cationic biopolymers with acceptable yield. The results of this study showed that by using E. coli BL21(DE3) LOBSTR strain and in combination with our developed stringent expression and Ni-NTA purification protocols highly pure products in one purification step (>99% purity) can be obtained. This approach could be applied to the production of other complex and potentially toxic biopolymers with wide range of applications in biomedicine.
Keywords: Cationic recombinant biopolymer, SlyD, histidine tag, BL21(DE3) LOBSTR, low expressing protein, histone H2A
1. Introduction
The purification process is considered as one of the major contributing factors to increasing the costs associated with the production of recombinant proteins. Therefore, development of a method that could facilitate isolation and purification of target proteins in one step is highly desirable. Due to its high specificity and simplicity, the affinity chromatography is one of the most widely used single-step technique for the purification of recombinant proteins. In affinity chromatography, various affinity tags such as poly-His tag, human influenza hemagglutinin (HA) tag, and FLAG tag are utilized for the separation of target proteins [1]. Among them, poly-His tag in combination with immobilized metal ions is the most preferred one because of its high efficiency as well as ease of recycling and reusing the affinity beads. In comparison to HA- or FLAG-tag purification processes which require ligands such as monoclonal antibodies, the cost associated with the use of immobilized metal ions is also far less. In addition, the size of the poly-His tag is small, commonly around six histidine amino acids, which minimizes the possibility of interfering with protein function. Despite all these advantages, one of the major drawbacks of using poly-His tag affinity chromatography for protein purification from an E.coli expression host is non-specific binding of contaminants and co-elusion with the target protein. This problem becomes even more pronounced when the protein expression yield is low. In such cases, the major culprits are E coli’s naturally occurring histidine-rich proteins such as ArnA and SlyD [2]. SlyD is a peptidyl-prolyl cis/trans-isomerase peptide consisting of 48 amino acids with and average molecular weight of 27 kDa [3]. There is a fragment with 15 histidines at the end of the C-terminal tail of SlyD which is reported to be responsible for competing with the His-tagged target peptides for metal binding and purification [4]. ArnA is an enzyme involved in the modification of lipid A phosphates with several non-consecutive histidine residues that are exposed on the surface of the protein [5].
Our lab is specialized in the design and development of recombinant fusion cationic biopolymers for targeted gene delivery to mammalian cells [6]. Previously, we have reported the design of an efficient biopolymeric platform, namely TH4G, composed of multiple functional domains including a cancer cell targeting peptide (T), four tandem repeating units of Histone H2A (H4) and a fusogenic peptide known as GALA (G) (Figure 1). The TH4G has a C-terminal His-tag which facilitates its purification via Ni-NTA affinity chromatography. The application of this highly cationic biopolymer for in vitro and in vivo gene delivery to ovarian cancer cells has been shown before [7–9]. To develop similar biopolymers but with different molecular weights, we genetically engineered TH2G, TH6G and TH8G constructs and made an attempt to purify them from the E.coli. These biopolymers contain highly cationic histone H2A in their sequences which happen to have antimicrobial activity [10, 11]. To make matter worse, the fusogenic peptide GALA in the above mentioned biopolymers also has cell membrane disruption activity. Therefore, it is understandable that they could put an enormous amount of stress on the E.coli protein expression machinery resulting in very low expression levels. To address this challenge, the objective of this study was to develop a method that could help obtain highly pure cationic biopolymers through a single-step purification process.
Figure 1.
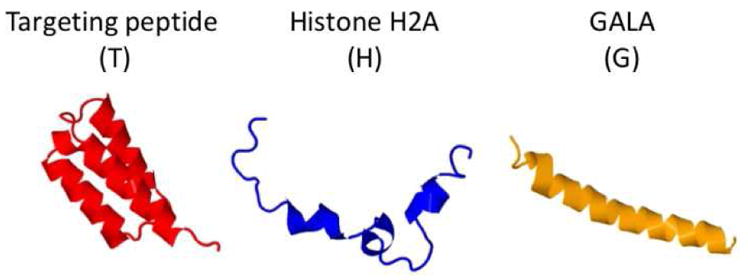
Schematic representation of each motif in the cationic histone H2A-based biopolymer structure. The structure of each motif is predicted by I-TASSER protein structure and function prediction software [12].
2. Materials and Methods
2.1. Cloning of the cationic biopolymers
The genes encoding TH2G, TH4G, TH6G and TH8G were synthesized by Integrated DNA Technologies (Coralville, IA, US) with C-terminal His-tags. The genes were digested with NdeI and XhoI restriction enzymes and cloned into the pET21b(+) vector (EMD Biosciences, Gibbstown, NJ, US) using standard cloning techniques. The detail of the cloning strategy is described previously by our group [13]. The fidelity of the genes to the original design was examined by DNA sequencing.
2.2. Expression of the biopolymers in E.coli
The plasmids encoding TH2G, TH4G, TH6G and TH8G constructs were first transformed into BL21(DE3) (Novagen, San Diego, US), BL21(DE3) pLysS (Novagen, San Diego, US) and BL21(DE3) LOBSTR (Kerafast Inc., MA, US) E. coli expression hosts.
To express biopolymers in BL21(DE3), BL21(DE3)pLysS or BL21(DE3) LOBSTR host, a single colony was picked and cultured in 5 mL Miller’s LB Broth (LB) starter culture containing 100μg/mL carbenicillin (Sigma-Aldrich Co. LLC., US). The starter culture tube was incubated overnight at 37°C under constant shaking at 350 rpm. The next morning, the whole starter culture volume was added to a flask containing 500 mL autoclaved Terrific Broth (TB) media (25.4g of TB powder, 2 mL of glycerol in 500 mL of Milli-Q water). The flask was shaken at 37°C/350 rpm and protein expression was induced at OD600 of 0.4–0.6 by 1mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). While the expressed biopolymers in BL21(DE3) and BL21(DE3) pLysS hosts were collected four hours post induction, those expressed in BL21(DE3) LOBSTR were collected 2.5 hours post induction. The E. coli pellets were collected by centrifugation and stored at −80 °C Freezer. The above mentioned protocols are an adaptation of a previously published protocol for high yield expression of recombinant peptides in E. coli [14].
2.3. Purification of biopolymers
To purify the His-tagged biopolymers, two types of Immobilized Metal Affinity Chromatography (IMAC) were used; i.e., nickel-nitrilotriacetic acid (Ni-NTA) agarose (QIAGEN Co., Maryland, US) and cobalt resin (TALON) (Takara Bio USA, Inc). In Ni-NTA purification method, the E. coli pellets were weighed and lysed with lysis buffer (5 mL of lysis buffer per gram of bacterial wet mass) composed of 8M urea, 2M NaCl,100 mM NaH2PO4,10mM Tris, 1% V/V Triton X-100, and 10 mM imidazole (pH adjusted to 8). The bacterial slurry was dispersed in the lysis buffer by vigorous stirring for one hour at room temperature. The lysate was centrifuged for one hour, at 20,000 rpm, 4°C and the supernatant was removed. The supernatant was then incubated with Ni-NTA resin for one hour on ice. The Ni-NTA resin was preconditioned with lysis buffer. Next, the mixture was diluted 3 times with the lysis buffer and gradually loaded onto a 10 mL filtered polypropylene column (Bio-Rad Inc., US) under vacuum. The column was first washed by using 100 mL of lysis buffer and then by 50 mL Wash Buffer composed of 5 M Urea, 1.5 M NaCl, 100 mM NaH2PO4, 10 mM Tris and 40 mM imidazole (pH adjusted to 8). Finally, the purified biopolymer was eluted by 5 mL of elution buffer composed of 3 M Urea, 0.5 M NaCl, 100 mM NaH2PO4, 10 mM Tris and 300 mM imidazole (pH adjusted to 8). The eluted fractions were collected in 500 μL aliquots and stored at −20°C for further analysis.
In purification method by TALON resin, we followed the supplier's “Large-Scale Batch Purification” protocol. In brief, the resin was washed and equilibrated with the equilibration buffer composed of 6M guanidine-HCl, 50mM NaH2PO4 and 300mM NaCl. Then, the bacteria pellet was lysed by equilibration buffer followed by addition of TALON resin (1 mL of resin suspension per 1.5 mg of polyhistidine-tagged biopolymer). The mixture was further incubated on ice with a gentle shaking for one hour. The TALON® resin was collected by vacuum filtration through the similar process described above for Ni-NTA method. The collected resin was then washed with ten times bed volume of equilibration buffer and the bioplymer was eluted from the column by using 5 mL of elution buffer (6M guanidine-HCl, 45mM NaH2PO4, 250mM NaCl and 150mM Imidazole). The fractions were collected in 500 μL aliquots, the biopolymer concentrations were measured by Bradford assay and then stored at −20 °C.
2.4. Assessment of the biopolymer yield and purity
The SDS-PAGE analysis was performed to determine the biopolymer purity. In brief, a 4% stacking and 12% resolving polyacrylamide gel was made from ProtoGel Stacking Buffer and ProtoGel Resolving Buffer (National Diagnostics, Atlanta, US) according to the manufacturer’s protocol. Approximately 1.5 μg of the purified biopolymer was mixed with 6x SDS Protein Loading Buffer (Thermo Fisher Scientific Inc., US) and loaded onto each well. The electrophoresis was performed by applying a constant voltage of 150 V for 45–60 min followed by gel staining with PageBlue Protein Staining Solution (Thermo Fisher Scientific Inc., US). The gel pictures were recorded by Odyssey Classic Image System (LI-COR, Inc., US) and the intensity of each band was analyzed by ImageJ image processing and analysis software (NIH, US).
3. Results and Discussion
In the past decades, various strategies have been deployed to either mitigate or completely remove the native E. coli proteins contaminants such as SlyD and ArnA from the target proteins especially in cases where the expression yield is low. These include the use of cobalt-based resin, a secondary chromatographic procedure or genetic modification of the E. coli strain [3, 15]. Since complete knock out of the ArnA and slyD in E. coli causes serious growth defects, such knockout strains are not viable options for recombinant protein expression [16]. Therefore, a practical and viable alternative would be to keep the functional sections of these proteins intact, while removing/modifying the metal-affinity segment. In one approach, Robichon and colleagues genetically modified the E. coli BL21(DE3) strain to express the endogenous proteins SlyD, Can, ArnA, and AceE fused at their C terminus to a chitin binding domain (CBD) [3]. In this approach, the CBD-tagged contaminants could be removed from the target protein through use of a chitin affinity column in tandem with IMAC. While this approach produces the desired results, but increases the complexity of purification process as well as the costs. In addition, an extra purification step could significantly reduce the yield of the purification process. Therefore, we did not examine the potential benefit of this two-step purification process for this study.
In second approach, a cobalt-based resin (TALON) instead of Ni-NTA has been utilized to remove the SlyD impurity since it is believed that it may have lower affinity towards this contaminant. Due its simplicity, we examined the use of this approach to purify the biopolymers. In third approach, the E.coli expression host is genetically modified to remove/change the histidine rich tails of the native ArnA and SlyD proteins resulting in less interaction with the immobilized nickel resin. Here, we examined the potential application of this approach as well in order to identify the most appropriate technique for complete removal of the ArnA and SlyD impurities from the cationic recombinant biopolymers.
3.1. Construction of expression plasmids
The genes encoding TH2G, TH4G, TH6G and TH8G constructs were cloned into a pET21b vector and the DNA sequencing results confirmed the fidelity of the sequences to the original design (Supplementary Table 1). Here we chose a pET21b vector as the prokaryotic expression system because of its tightly regulated T7 lac promoter. As shown in Table 1, all four biopolymers are rich in Lys, Arg and His residues; thereby, making the biopolymers highly cationic. The theoretical protein parameters calculations indicate that the estimated net charge of biopolymers increases as the molecular weight (Mw) increases.
Table 1.
The biopolymer physicochemical parameters as calculated by the ProtParam tool from the ExPASy Bioinformatics Resource Portal (http://web.expasy.org/protparam/).
| Biopolymer | Mw (Da) | Charge | No. of Cationic Residues | Theoretical pI |
|---|---|---|---|---|
| TH2G | 19,827 | +22 | 45 | 11.27 |
| TH4G | 27,625 | +46 | 71 | 11.99 |
| TH6G | 35,422 | +70 | 97 | 12.26 |
| TH8G | 43,219 | +94 | 123 | 12.42 |
3.2. Biopolymer expression in BL21(DE3) pLysS host and purification by Ni-NTA
Expression of any recombinant protein in E. coli could interfere with the normal functioning of the cell and therefore may be “toxic” to the bacteria. The level of toxicity will vary from protein to protein depending on its physicochemical characteristics. If the level of toxicity is sufficiently high to E. coli, even the basal level expression can be enough to prevent vigorous growth and protein overexpression. Based on the information shown in Table 1, it can be observed that all four constructs are highly cationic and potentially toxic to E. coli. In the past decade we have examined, optimized and reported a reliable method for the production and purification of TH4G biopolymer with minim impact on bacterial growth [9, 13]. To minimize the negative impact of biopolymer toxicity on E. coli growth and protein expression, we used BL21(DE3) pLysS strain for biopolymer production. Unlike parental BL21(DE3), the modified E. coli BL21(DE3) pLysS strain contains an additional plasmid, pLysS, which expresses the gene encoding T7 lysozyme. T7 lysozyme provides a tight control over the background expression of target genes especially before IPTG induction making it suitable for the production of toxic proteins. This is in contrast to BL21(DE3) system which is considered leaky where proteins continue to express, although at low levels, even before IPTG induction. To examine the potential use of this approach in expressing the other three constructs (i.e., TH2G, TH6G and TH8G), we first transformed them into a BL21(DE3) pLysS strain and then expressed and purified. The results of the protein expression and purification study showed that as the number of cationic residues in the biopolymer sequence increased, the amount of purified biopolymer (i.e., yield) decreased (Figure 2A). For example, from each 500mL culture we could obtain on average approximately 2.8mg of pure TH2G versus 0.8mg of TH8G. In addition, the SDS-PAGE results revealed emergence of impurity signals corresponding to the molecular weights of ~27 kDa and ~70 kDa in purified TH6G and TH8G biopolymers (Figure 2B and C). The molecular weights of these two impurity signals are very close to the theoretical molecular weights of SlyD and ArnA proteins. The western blot analysis using anti-His tag primary antibody showed that the impurities were not his-tagged indicating that they were E. coli native proteins (Supplementary Figure 1). Since we did not see these two contaminants in purified TH2G biopolymer, we hypothesized that by increasing the yield of production we may be able to eliminate the problem. It is worth noting that the molecular weight of the SlyD impurity is very close to TH4G biopolymer; therefore, we could not measure the amount of the contamination under the TH4G band.
Figure 2.

A) The amounts of purified biopolymers from each 500 mL of BL21(DE3) pLysS culture (Yield). B) The SDS-PAGE picture of the Ni-NTA purified TH2G, TH4G, TH6G and TH8G. C) The quantification of biopolymer purity using Image J software. TH4G purity is not determined since the molecular weights of SlyD and TH4G are very close.
3.2. Biopolymer expression in BL21(DE3) host and purification by Ni-NTA
To identify the most optimum method for the elimination of the impurities, we performed a study to first enhance the yield of production. To achieve this goal, we examined the potential use of parent BL21(DE3) host instead of tightly regulated BL21(DE3) pLysS system. For this, we selected the TH2G biopolymer as our negative control (high yield and low impurity) and TH8G construct as positive control (low yield and high impurity). Both constructs were transformed into BL21(DE3) host and the bacterial growth curves with and without IPTG induction were monitored over a 24 hour period (Figure 3A). The results of this study showed significant reduction in bacterial growth rate after IPTG induction which indicates the bacteria transferred the majority of its energy source to produce the biopolymers instead of growth. Based on this information, we proceeded to purify the biopolymers. Here, the BL21(DE3) was induced by IPTG when the OD600 reached ~0.4–0.6 and the pellet was collected four hours post induction. As shown in Figure 3B, the yield of production of TH8G was ~1.1 mg which is significantly less than ~4.4mg of TH2G. The SDS-PAGE results also showed the presence of an impurity around 27 kDa in the purified TH8G biopolymer, whereas the impurity band around 70kDa disappeared (Figure 3C). These results indicate that the use of BL21(DE3) instead of BL21(DE3) pLysS significantly improved the expression level of the TH8G increasing it from 0.8mg±0.07 to 1.1±0.1mg (p<0.05). While this approach resulted in production of more pure TH8G with less impurity (ArnA eliminated), but the SlyD impurity was still significant and measured to be ~35% of the total mass (Figure 3D).
Figure 3.

A) The growth curves of BL21(DE3) bacteria transformed with TH2G and TH8G constructs with and without IPTG induction. B) The amounts of purified TH2G and TH8G from 500mL of culture. C) The SDS-PAGE picture of the purified TH2G and TH8G biopolymers. D) The quantitative analysis of impurities in purified TH2G and TH8G biopolymers using Image J software. The data are presented as mean±s.d, n=3.
3.3 Biopolymer expression in BL21(DE3) host and purification by TALON resin
So far, the data shows that by changing the expression host we could increase the yield of biopolymer production and reduce the impurity, although we failed to eliminate it completely. To go one step further, in combination with BL21(DE3) host, we utilized TALON resin instead of Ni-NTA for purification which has been claimed to have less affinity towards non-specific E. coli native proteins such as SlyD. Cobalt-based (Co(II)) TALON metal affinity resin has high affinity towards histidine residues that are spatially positioned adjacent to each other such as 6xHis-tag. In a technical note, McMurry et al. (2004), reported that the cobalt-based TALON resin could remove non-specific contamination and produce the target peptide with much higher purity as compared to Ni-NTA beads [17]. Therefore, in the next step, we expressed TH8G biopolymer in BL21(DE3) host as mentioned above but purified using TALON resins. Interestingly, the results of this study revealed that TH8G biopolymer with significantly higher purity could be obtained, even though the yield of production was reduced (Figure 4A–C). Although significant improvement in purity increasing from 65% to 80% was observed, this approach also did not completely eliminate the SlyD impurity. In a study by Kaluarachchi et al. (2011), the affinity of SlyD to a series of transition metals including Mn(II), Fe(II), Co(II), Cu(I), and Zn(II) was measured. The dissociation constant of Ni(II) and Co(II) were determined to be approximately 0.1nM and 4nM, respectively [18]. Although the ion cobalt showed less affinity towards SlyD than nickel ion, but the difference was still not sufficient to completely remove the SlyD impurity. Our observations in Figure 4 also show that TALON resin was moderately helpful.
Figure 4.

Comparison of the yield and purity of TH8G biopolymer after expression in BL21(DE3) host and purification by Ni-NTA and TALON resins. A) The amount of purified TH8G obtained from 500 mL of culture. B) The SDS-PAGE picture of the purified TH8G. C) The quantification of TH8G purity using the Image J software. The data are presented as mean±s.d. (n= 3). * indicates significance, p<0.05, student t-test.
3.3 Biopolymer expression in BL21(DE3) LOBSTR host and purification by Ni-NTA
Since improving the expression yield by using BL21(DE3) host and utilization of TALON resin did not provide satisfactory results, we changed strategy and examined the use of a newly developed E. coli strain. Andersen et al. (2013), have recently reported the development of a new E. coli expression host, namely LOBSTR (low background strain) [19]. LOBSTR is derived from the E. coli BL21(DE3) strain with genetically modified copies of ArnA and SlyD. These modifications have resulted in E. coli native proteins with reduced affinities toward Ni and Co resins allowing the purification of low-expressing target proteins by reducing background contamination. To examine the potential use of this strain, the TH2G and TH8G constructs were transformed into BL21(DE3) LOBSTR, expressed and purified by Ni-NTA affinity chromatography. Here, we used Ni-NTA resins first because it is far more cost-effective than cobalt-based ones. The results of bacterial growth curves confirmed the expression of biopolymer after induction (Figure 5A), and the amount of expressed TH2G and TH8G was measured to be on average ~3.0mg and ~0.9mg, respectively (Figure 5B). While the yield of production is statistically the same as what we obtained with BL21(DE3) pLysS host (Figure 2A), but the SDS-PAGE results showed complete removal of impurities and obtaining >99% pure biopolymers (Figure 5C and D).
Figure 5.
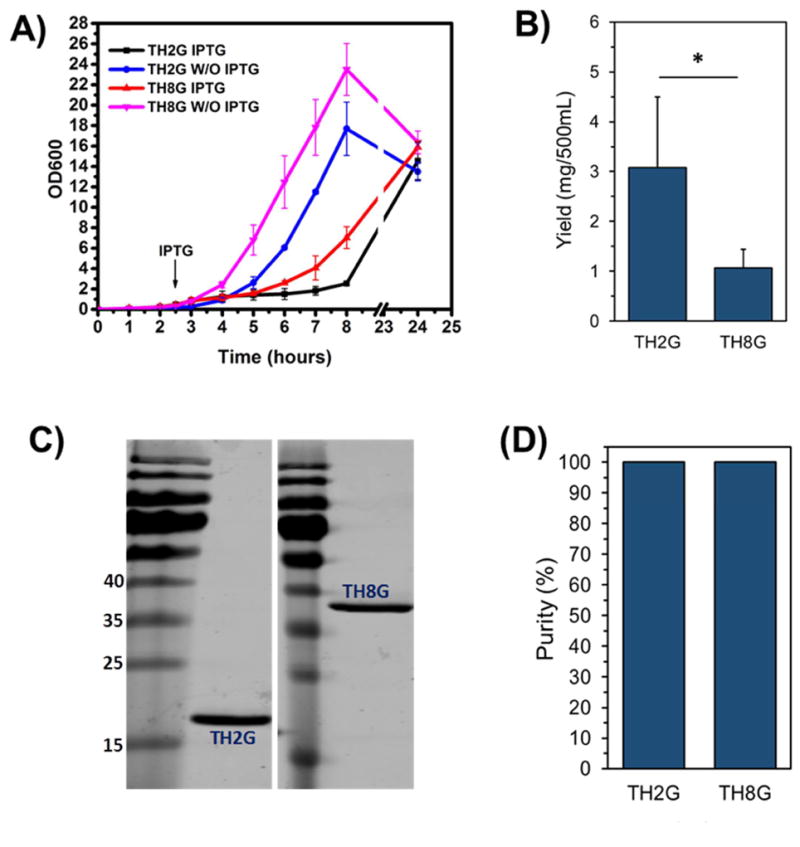
Peptide TH2G and TH8G were expressed in BL21(DE3) LOBSTR and purified by Ni-NTA. A) The growth curves of TH2G and TH8G with and without IPTG induction. B) The amounts of purified TH2G and TH8G obtained from 500 mL of culture. C) The SDS-PAGE picture of the purified TH2G and TH8G. D) The quantification of TH2G and TH8G purity using the Image J software. The data are presented as mean±s.d. (n= 3).
To validate the expression process and examine its use to purify the other two constructs (i.e., TH4G and TH6G), we used the same protocol for their expression and purification. The SDS-PAGE results confirmed that the developed protocol for the expression and purification of low-expressing cationic biopolymers in this study can produce target peptides with high purity (Figure 6). Overall, the results of our studies show that this E. coli strain facilitates the production of the cationic low expressing biopolymers with high purities.
Figure 6.
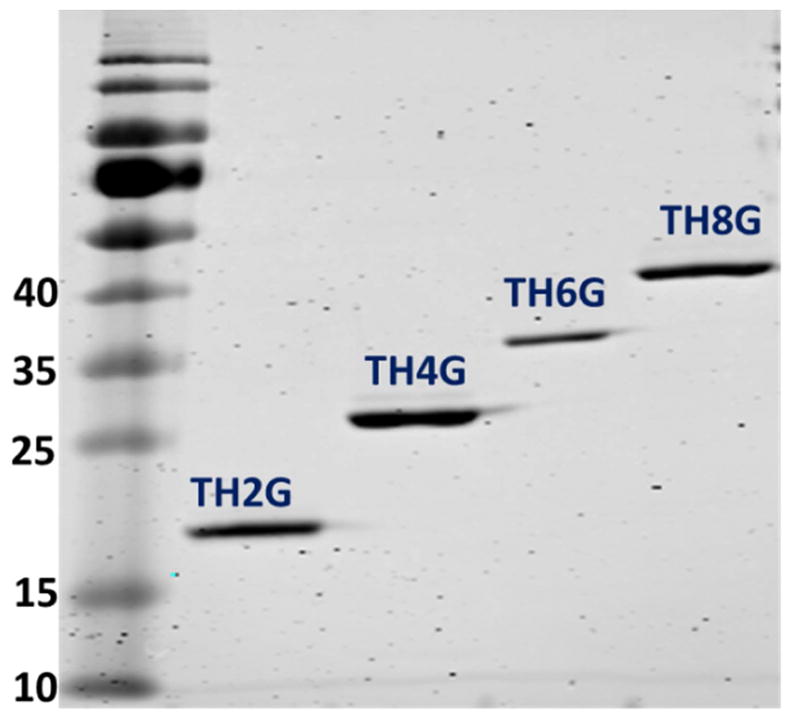
The SDS-PAGE picture of the purified TH2G, TH4G, TH6G and TH8G that were expressed in BL21(DE3) LOBSTR host. All biopolymers were purified by Ni-NTA affinity chromatography.
Conclusion
For high-expressing recombinant proteins, the endogenous E. coli proteins are a small problem because they are out-competed by the high amounts of the target protein. In contrast, when protein expression is low, endogenous host proteins such as ArnA and SlyD could have a similar abundance and compete with the target His-tagged proteins for binding onto nickel or cobalt resins. As a result, obtaining a high purity target protein becomes a challenge. The results of this study demonstrated that the developed expression method in E. coli BL21(DE3) LOBSTR in combination with our optimized one-step purification method could help completely remove endogenous E. coli contaminants from a low-expressing cationic biopolymers. Considering the complexity of the structure of the biopolymers in this study and their extreme physicochemical properties, we believe that the developed approach could be applied to express and purify the majority of other low-expressing and potentially toxic proteins.
Supplementary Material
Supplementary Table 1: The amino acid sequences of the recombinant cationic biopolymers
Supplementary Figure 1: Western blot analysis of expressed TH2G and TH8G using anti-his-tag primary antibody (abcam). This figure shows that only TH2G and TH8G are his-tagged and the primary antibody does not recognize impurities.
Highlights.
Recombinant cationic biopolymers with increasing number of cationic residues ranging from 22 to 96 were genetically engineered.
As the number of cationic residues increased the yield of production decreased.
The major contaminant during the purification process proved to be SlyD endogenous E. coli protein.
TALON cobalt based resin helped reduce the impurity during the purification process but did not eliminate completely.
Genetically modified E. coli BL21(DE3) LOBSTR facilitated production of all cationic recombinant biopolymers with above 99% purity in a single-step Ni-NTA purification process.
Acknowledgments
This work was supported by grants from the National Institutes of Health/National Cancer Institute (R01CA175318) and National Institutes of Health/National Institute of Biomedical Imaging and Bioengineering (R21EB016792) to A. Hatefi.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Saraswat M, Musante L, Ravida A, Shortt B, Byrne B, Holthofer H. Preparative purification of recombinant proteins: current status and future trends. Biomed Res Int. 2013;2013:312709. doi: 10.1155/2013/312709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolanos-Garcia VM, Davies OR. Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. Biochim Biophys Acta. 2006;1760:1304–1313. doi: 10.1016/j.bbagen.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 3.Robichon C, Luo J, Causey TB, Benner JS, Samuelson JC. Engineering Escherichia coli BL21(DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Appl Environ Microbiol. 2011;77:4634–4646. doi: 10.1128/AEM.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weininger U, Haupt C, Schweimer K, Graubner W, Kovermann M, Bruser T, Scholz C, Schaarschmidt P, Zoldak G, Schmid FX, Balbach J. NMR solution structure of SlyD from Escherichia coli: spatial separation of prolyl isomerase and chaperone function. J Mol Biol. 2009;387:295–305. doi: 10.1016/j.jmb.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 5.Gatzeva-Topalova PZ, May AP, Sousa MC. Structure and mechanism of ArnA: conformational change implies ordered dehydrogenase mechanism in key enzyme for polymyxin resistance. Structure. 2005;13:929–942. doi: 10.1016/j.str.2005.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarthy HO, Wang Y, Mangipudi SS, Hatefi A. Advances with the use of bio-inspired vectors towards creation of artificial viruses. Expert Opin Drug Deliv. 2010;7:497–512. doi: 10.1517/17425240903579989. [DOI] [PubMed] [Google Scholar]
- 7.Canine BF, Wang Y, Hatefi A. Biosynthesis and characterization of a novel genetically engineered polymer for targeted gene transfer to cancer cells. J Control Release. 2009;138:188–196. doi: 10.1016/j.jconrel.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Canine BF, Hatefi A. HSV-TK/GCV cancer suicide gene therapy by a designed recombinant multifunctional vector. Nanomedicine. 2011;7:193–200. doi: 10.1016/j.nano.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karjoo Z, McCarthy HO, Patel P, Nouri FS, Hatefi A. Systematic engineering of uniform, highly efficient, targeted and shielded viral-mimetic nanoparticles. Small. 2013;9:2774–2783. doi: 10.1002/smll.201300077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets. 2008;8:195–205. doi: 10.2174/1871526510808030195. [DOI] [PubMed] [Google Scholar]
- 11.Cho JH, Sung BH, Kim SC. Buforins: histone H2A-derived antimicrobial peptides from toad stomach. Biochim Biophys Acta. 2009;1788:1564–1569. doi: 10.1016/j.bbamem.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Mangipudi SS, Canine BF, Hatefi A. A designer biomimetic vector with a chimeric architecture for targeted gene transfer. J Control Release. 2009;137:46–53. doi: 10.1016/j.jconrel.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sivashanmugam A, Murray V, Cui C, Zhang Y, Wang J, Li Q. Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci. 2009;18:936–948. doi: 10.1002/pro.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsy CB, Chapman CJ, Barnes AC, Robertson JF, Murray A. Two-step method to isolate target recombinant protein from co-purified bacterial contaminant SlyD after immobilised metal affinity chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;853:314–319. doi: 10.1016/j.jchromb.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 16.Roof WD, Fang HQ, Young KD, Sun J, Young R. Mutational analysis of slyD, an Escherichia coli gene encoding a protein of the FKBP immunophilin family. Mol Microbiol. 1997;25:1031–1046. doi: 10.1046/j.1365-2958.1997.5201884.x. [DOI] [PubMed] [Google Scholar]
- 17.McMurry J, Macnab RM. BD Talon Resin Does Not Bind E. coli SlyD, a Common Contaminant in Ni-NTA IMAC. Clontechniques. 2004;19 http://www.biosciencetechnology.com/article/2004/2002/cobalt-based-protein-purification-resin-does-not-bind-e-coli-slyd-common-contaminant-ni-nta-imac. [Google Scholar]
- 18.Kaluarachchi H, Siebel JF, Kaluarachchi-Duffy S, Krecisz S, Sutherland DE, Stillman MJ, Zamble DB. Metal selectivity of the Escherichia coli nickel metallochaperone, SlyD. Biochemistry. 2011;50:10666–10677. doi: 10.1021/bi2014882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersen KR, Leksa NC, Schwartz TU. Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins. 2013;81:1857–1861. doi: 10.1002/prot.24364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: The amino acid sequences of the recombinant cationic biopolymers
Supplementary Figure 1: Western blot analysis of expressed TH2G and TH8G using anti-his-tag primary antibody (abcam). This figure shows that only TH2G and TH8G are his-tagged and the primary antibody does not recognize impurities.