

Abstract
Identifying sources of genetic variation and reconstructing invasion routes for non-native introduced species is central to understanding the circumstances under which they may evolve increased invasiveness. In this study, we used genome-wide single nucleotide polymorphisms to study the colonization history of Centaurea solstitialis in its native range in Eurasia and invasions into the Americas. We leveraged this information to pinpoint key evolutionary shifts in plant size, a focal trait associated with invasiveness in this species. Our analyses revealed clear population genomic structure of potential source populations in Eurasia, including deep differentiation of a lineage found in the southern Apennine and Balkan Peninsulas and divergence among populations in Asia, eastern Europe, and western Europe. We found strongest support for an evolutionary scenario in which western European populations were derived from an ancient admixture event between populations from eastern Europe and Asia, and subsequently served as the main genetic ‘bridgehead’ for introductions to the Americas. Introductions to California appear to be from a single source region, and multiple, independent introductions of divergent genotypes likely occurred into the Pacific Northwest. Plant size has evolved significantly at three points during range expansion, including a large size increase in the lineage responsible for the aggressive invasion of California’s interior. These results reveal a long history of colonization, admixture, and trait evolution in C. solstitialis, and suggest routes for improving evidence-based management decisions for one of the most ecologically and economically damaging invasive species in the western United States.
Keywords: admixture, phylogeography, rapid evolution, biological invasion, invasion routes, restriction site associated sequencing
Introduction
Rapid adaptation of introduced species to a new environment appears to be common in successful biological invasions (Thompson et al. 1998; Cox 2004; Bossdorf et al. 2005; Prentis et al. 2008; Colautti & Lau 2015). By increasing fitness, adaptive evolution is likely to increase the abundance and spread of introduced species, contributing directly to their invasiveness (e.g. Colautti & Barrett 2013). We should expect adaptation to occur frequently when strong selection associated with a new environment can act on pre-existing standing genetic variation (Koskinen et al. 2002; Lee 2002; Bock et al. 2015). Many successful invasions are indeed associated with high levels of standing genetic variation resulting from multiple introductions and subsequent intraspecific genetic admixture, or from introgression of alleles from other species via interspecific hybridization (Ellstrand & Schierenbeck 2000; Kolbe et al. 2004; Dlugosch & Parker 2008; Hufbauer 2008; Bock et al. 2015). Understanding when this variation fuels evolutionary and ecological change during invasion requires reconstructing invasion routes, pinpointing sources of variation, and identifying when key transitions in ecological characters have evolved (Rosenthal et al. 2008; Colautti & Lau 2015; Cristescu 2015; Dlugosch et al. 2015a).
The highly invasive plant yellow starthistle, Centaurea solstitialis (hereafter YST), appears to have benefited from adaptive evolution during its spread in the western US (Dlugosch et al. 2015b). Native to Eurasia, YST is a diploid, obligately outcrossing annual that has spread into Argentina, Chile, the United States, South Africa, and several oceanic islands (DiTomaso et al. 2006). Previous work has demonstrated evolutionary increases in seed size, growth rate, and adult plant size in invading genotypes of YST, particularly in its aggressive invasion of California (Widmer et al. 2007; Eriksen et al. 2012; Graebner et al. 2012; Hierro et al. 2012; Dlugosch et al. 2015b). These evolutionary changes are associated with fitness benefits to YST: larger seeds improve seedling growth (Widmer et al. 2007) and/or survival (Hierro et al. 2012), and larger size triggers earlier flowering and increased reproduction (Dlugosch et al. 2015b).
A thorough analysis of the population structure and history of YST across its range is fundamental to identifying invasion routes, sources of genetic variation, and evolutionary transitions in traits of invading populations. Historical records indicate that YST was introduced accidentally as a contaminant of alfalfa seed to Chile from Spain in the mid-1600s, and then to the San Francisco bay area in California from Chile in the 1850s (Stewart 1926; Howell 1959; Gerlach 1997). Secondary introductions of YST may also have occurred when additional alfalfa seed was imported into the western US from multiple regions throughout Eurasia beginning in the early 1900s (Gerlach 1997). Previous genetic studies have identified high levels of genetic diversity and putative admixture in western US populations (Sun 1997; Dlugosch et al. 2013; Eriksen et al. 2014), but genetic structure across YST’s Eurasian range and its contribution to invading populations has remained poorly resolved. Resolving the introduction history of YST requires more comprehensive sampling of invading populations, their potential native source populations, and co-distributed congeners which may have contributed to invasions through hybridization. In general, comprehensive geographic sampling and the analysis of hundreds or thousands of unlinked loci may be necessary for resolving past population relationships in putatively complex colonization scenarios (Garrick et al. 2015).
It may be particularly important to resolve YST’s history in western Europe, a region where populations are hypothesized to represent an ancient human-mediated range expansion (Wagenitz 1975; Maddox et al. 1985). This region may have served as a ‘bridgehead’ (Lombaert et al. 2010) of successful colonizing genotypes that were pre-adapted for invasion of the Americas. A coarse-scale study based on transcriptome-derived single nucleotide polymorphisms (SNPs) of YST reported that human-mediated introductions to Spain from independent sources in eastern Europe and Asia may explain signatures of admixture found in populations there and in the subsequent introduction of these genotypes to the Americas (Dlugosch et al. 2013). In contrast, a microsatellite-DNA study of YST found significant differentiation of populations in Spain from the rest of Europe, with little evidence for admixture (Eriksen et al. 2014). They hypothesized that YST underwent a continuous stepwise expansion through Europe prior to human activity, admixing only later during introduction to the Americas (Eriksen et al. 2014). Resolving the history of western European populations is essential for understanding the context from which contemporary introductions have evolved.
In this study, we resolve the invasion routes of YST and its population history in Eurasia, and we leverage this information to identify evolutionary shifts in plant size associated with invasiveness. We use double digest restriction-site associated sequencing (ddRADseq) to generate genome-wide polymorphism information across a broad geographic sample of YST and its co-occurring congeners. Using this genomic data set, we identify population structure and admixture of YST across its range in both Eurasia and the Americas, test for evidence of hybridization with related species, and evaluate evolutionary scenarios using coalescent simulations in an approximate Bayesian computation (ABC) framework. Finally, we test for congruence between genetic and phenotypic patterns of divergence and identify instances of putatively adaptive trait shifts during range expansion, improving our understanding of evolutionary factors responsible for the invasion of one of the most ecologically and economically damaging invasive species in the western US (DiTomaso & Healy 2007).
Methods
Sampling and ddRADseq library preparation
We sampled seeds of 732 YST individuals from 61 sites in the native and introduced range (Table S1 and Fig. S1, Supporting information). Samples spanned introductions in the western US (California, Oregon, Washington, and Idaho), introductions in South America (Chile and Argentina), and native populations in southern and western Europe (Spain, France, Italy, and Greece), central-eastern Europe [Hungary, Greece, Bulgaria, Romania, Thrace (western Turkey), Ukraine, Russia], and Asia [Anatolia (eastern Turkey), Armenia, and Uzbekistan]. We also collected individuals of closely-related taxa C. melitensis (N = 3), C. nicaeensis (N = 5), and C. pallescens (N = 5) to assess the relationship of YST populations to these species and reveal any evidence for hybridization. Centaurea melitensis, the putative sister species of YST (Garcia-Jacas et al. 2006), is co-distributed with YST in southern Europe and North America, C. nicaeensis is co-distributed with YST in parts of Spain, Italy, and Sardinia, and C. pallescens may co-occur with YST in the Middle East, northern Africa, and France (Dostál 1976).
Genomic DNA was isolated from leaves of greenhouse reared seedlings with a modified CTAB/PVP (cetyl trimethylammonium bromide/polyvinylpyrrolidone) DNA extraction protocol (Webb & Knapp 1990). We used a modified version of Peterson et al.’s (2012) ddRADseq protocol to construct reduced-representation libraries of genomic DNA of each of our individuals. Following fluorometric quantification (Qubit® 2.0 high-sensitivity or broad-range assay; Invitrogen, Inc., Carlsbad, CA, USA), 500ng of each sample was digested with Pst1 and Mse1. Unique combinations of individual P1 and P2 barcoded adapters were annealed to genomic DNA of each sample [P1 adapter as in Etter et al. 2011; P2 adapter top oligo: 5′-[Phos]TAxxxxxxAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCAGAACAA-3′ (xxxxxx = barcode); bottom oligo: 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTxxxxx*x-3′ (* = phosphorothioate bond)]. Barcodes used to differentiate multiplexed individuals were six base pairs long and differed by at least two nucleotides. Barcoded DNA was pooled, purified using AmpureXP beads (Beckman Coulter, Inc., Brea, CA, USA), and size selected for 350–650 bp using the Pippin Prep automated DNA size selection system (Sage Science, Beverly, MA, USA). We enriched adapter-ligated fragments in size-selected libraries using 12 PCR cycles and purified the resulting product using AmpureXP beads. The ddRADseq library protocol used for Run 1 used 100ng of each sample and enriched adapter-ligated fragments using 26 PCR cycles prior to DNA size selection. Pooled libraries were run on a total of five lanes of the Illumina HiSeq 2000 or 2500 platforms (Illumina, Inc., San Diego, CA, USA) to generate 100 base-pair paired-end reads. In total, we obtained ~1.5 billion paired-end Illumina reads of 100 bp length each, across 745 individuals. The list of samples and sequencing run information are available in Dryad (doi:10.5061/dryad.pf550).
Data processing
We quality-filtered and de-multiplexed reads using the SNOWHITE 2.0.2 package (Dlugosch et al. 2013). In SNOWHITE, sequences composed of primer and/or adapter contaminants were removed using TAGDUST (Lassmann et al. 2009) and SEQCLEAN (Chen et al. 2007), and bases with quality scores less than 20 were removed from the 3′ ends. Due to the poor quality of R2 reads after ca. 20 bp in two of the five Illumina data sets, we conducted all downstream analyses using only R1 reads. We end-trimmed reads to a standard length of 76bp after removing barcode and enzyme recognition sequences. The number of reads varied substantially across individuals (median 395.7K reads per individual, range 3.4K–5.6M). We removed 132 individuals with ≥ 75% missing SNP data. The resulting “FULL” data set contained 591 YST individuals (1–19 samples per site) from 61 sites (Table S1, Supporting information), with an average of 16% missing SNP data per sample (range 0–72%).
We delineated two additional RAD data sets for subsequent analyses. We defined a “CORE” data set that excluded YST individuals from five sites in the southern Apennine and Balkan Peninsulas (Italy, southern Greece, and Crete; Table S1 and Fig. S1, Supporting information) that we found to be strongly differentiated from remaining individuals (see Results). Finally, we analysed a third “OUTGROUPS” data set that included individuals of YST, C. melitensis, C. pallescens, and C. nicaeensis. We excluded individuals with ≥ 25% missing data from the “OUTGROUPS” data set to increase the number of loci shared across species, which may increase the accuracy of ancestry assessments (Vähä & Primmer 2006; Bohling et al. 2013). This resulted in the inclusion of 382 YST individuals from the “FULL” data set, as well as two C. melitensis, five C. pallescens, and two C. nicaeensis individuals.
We used the denovo_map.pl pipeline program in STACKS 1.20 (Catchen et al. 2011, 2013) to merge stacks (i.e. sets of identical reads) into RAD loci within individuals, identify polymorphic sites, create a catalog of loci across individuals, determine the allelic state at each locus in each individual, and exclude reads with likely sequencing errors. We inferred RAD loci in each sample in the “FULL” and “CORE” data sets using a minimum coverage depth (-m) of five to create a stack, a maximum mismatch distance of two nucleotides between loci when processing a single individual (-M), and a maximum of two stacks at a single locus (-X), because YST is diploid. To account for the possibility of fixed differences at loci in individuals when creating the catalog of loci (Catchen et al. 2011), we allowed a mismatch distance of two nucleotides between loci (-n). A transcriptome-based population genomics study of YST observed an average of one SNP per 89.6 bp of coding sequence across individuals in the native and introduced range (Dlugosch et al. 2013), which informed our choice of -M and -n parameters, and tests of alternative settings suggested that our parameters minimized under- or over-clustering of alleles (Fig. S2, Supporting information). Settings for the “OUTGROUPS” data set were identical to the “FULL” and “CORE” data sets except that -M and -n were increased to 4 to account for potentially greater levels of sequence divergence across species. In all analyses, we enforced the lumberjack stacks (-r) to remove stacks with excessive numbers of reads (more than two standard deviations above the mean) and the deleveraging (-d) algorithm to resolve over merged loci.
Analyses of population structure and demographic history assume that loci are unlinked and selectively neutral. To limit the effect of linkage disequilibrium in these analyses, we exported from STACKS only the first SNP from each locus for each SNP data set. In addition, we searched for SNP loci that exhibited FST coefficients that were significantly different from those expected under neutral expectations with three independent runs of BAYESCAN 2.1 (Foll & Gaggiotti 2008). Pairwise FST values were calculated among YST individuals that were grouped by geographic region and their assignment to the same genetic cluster according to both BAPS and STRUCTURE for individual SNP data sets generated from the “FULL” and “CORE” data set, and among different Centaurea species and the two differentiated lineages of YST for the “OUTGROUPS” data set (see Results). Each BAYESCAN analysis employed 20 pilot runs of each 5000 iterations followed by an additional burn-in of 5 × 104 and then 5 × 104 output iterations with a thinning interval of 10. An outlier analysis with FDR-corrected P-values (q-values) < 0.05 for all three replicate runs was used to detect outlier loci. Five SNPs that differentiated divergent southern Apennine and Balkan Peninsulas individuals in analyses of population structure of the “FULL” data set, and one SNP differentiating taxa in analyses of the “OUTGROUPS” data set, were identified as outliers in BAYESCAN analyses. These loci were removed from the data sets prior to subsequent analyses.
Population structure
We quantified population structure in YST to define genetically-differentiated native regions for subsequent tests of evolutionary scenarios in an ABC framework. When exporting SNPs in the ‘populations’ module in STACKS, we required that a locus was independent (i.e. a single locus per RAD locus), was selectively neutral, had a minimum coverage depth of 10 (m = 10), and had < 25% missing data (quantified and removed manually). We also required that each locus was present across all geographic regions and was present in at least 70% of the individuals within those regions (r = 0.7). We defined geographic regions as follows: western US (California, Oregon, Washington, and Idaho), South America (Chile and Argentina), western Europe (Spain and France), eastern Europe (Hungary, northern Greece, Thrace, Bulgaria, Romania, Ukraine, and Russia), Asia (Anatolia, Armenia, and Uzbekistan), and the southern Apennine and Balkan Peninsulas (Italy, southern Greece, and Crete). We excluded individuals from the southern Apennine and Balkan Peninsulas when exporting SNPs for the “CORE” data set. In total, SNP datasets for analyses of population structure of the “FULL” and “CORE” data sets included 752 SNPs from 591 individuals and 1013 SNPs from 550 individuals, respectively.
We visualized population structure in the “FULL” and “CORE” data sets using a discriminant analysis of principal components (DAPC), a multivariate method that maximizes genetic differentiation between pre-defined groups (Jombart et al. 2010). We grouped individuals by the same geographic regions as for STACKS output, with additional separate groups for individuals from the Pacific Northwest and Anatolia, because of multiple genetic clusters in these regions (see Results). DAPC analyses were performed using the ADEGENET 1.4 and ADE4 R packages (Dray & Dufour 2007; Jombart 2008; Jombart & Ahmed 2011) in R 3.0.2 (R Development Core Team 2013). We retained 180 principal components of PCA in the data transformation step and 10 discriminant functions for displaying differences between groups.
We used two Bayesian clustering methods to further partition population structure in YST. First, we quantified population structure in the “FULL” and “CORE” data sets in STRUCTURE 2.3.4 (Pritchard et al. 2000). For each analysis, we implemented a model of correlated allele frequencies (Falush et al. 2003) and admixture, and applied the default setting for all other parameters. Ten independent runs for all values of K (number of genetic clusters) between 1 and 10 were run using an MCMC length of 106 generations following a burn-in of 105 generations. For each K value, we used CLUMPP v. 1.1.2 (Jackobsson & Rosenberg 2007) to examine consistency across replicate cluster analyses by estimating the highest value of pairwise similarity (H’ value), and averaged assignment probabilities for each individual. We applied the Greedy algorithm for K = 1–8 and the LargeKGreedy algorithm for K = 9–10, using 1000 random input orders. The best K value was chosen by examining the log probability of the data [ln Pr(X|K)] and plots of ΔK (Evanno et al. 2005) produced by STRUCTURE HARVESTER (**Earl & vonHoldt 2011). Second, we used BAPS 6 (Corander et al. 2003; Corander & Marttinen 2006) to conduct a mixture analysis, which identifies the optimal value of K from a list of the ten best visited partitions according to their log(ml) values and assigns individuals to genetic clusters. For the mixture analysis, we set the maximum value of K to 20 and executed 10 independent runs for each value of K. We calculated assignment probabilities of each individual to each cluster. The simulations consisted of a minimum population size of 40, 100 iterations to estimate assignment probabilities for genotypes, 40 reference individuals from each genetic cluster, and 20 iterations to estimate assignment probabilities for reference individuals. We created bar plots showing assignment probabilities for each individual inferred by STRUCTURE (averaged across 10 replicates) and BAPS with DISTRUCT 1.1 (Rosenberg 2004).
Testing for interspecific hybridization
We used Bayesian clustering methods in STRUCTURE and BAPS to explore the potential role of hybridization in the evolutionary history of YST. We exported SNPs from the “OUTGROUPS” data set in the ‘populations’ module in STACKS, requiring that each locus was independent, selectively neutral, present in C. melitensis, C. pallescens, C. nicaeensis, and each of the two YST lineages (p = 5), genotyped in at least 70% of the individuals in each species/lineage (r = 0.7), had a minimum coverage depth of 10, and had < 25% missing data. This resulted in 440 SNP loci. Settings applied in STRUCTURE, CLUMPP, and BAPS were identical to those used for the population structure analyses, with the following exceptions. In STRUCTURE, we implemented a model of independent allele frequencies, because allele frequencies are likely to differ strongly among species. In BAPS, we set the minimum size of populations and number of reference individuals from each cluster to two to accommodate the smallest sample size (C. melitensis, N = 2).
Testing scenarios of evolutionary history
We conducted ABC analyses of the “CORE” data set in DIYABC 2.0.4 (Cornuet et al. 2010) to make inferences about the history of YST populations in Eurasia and the Americas. First, we generated four evolutionary scenarios likely to describe the history of divergence and admixture among YST populations in Eurasia (Fig. 1a), with a focus on evaluating the support for hypotheses regarding the sources and history of populations in western Europe, a primary source for many introductions to the Americas (Gerlach 1997). Specifically, we tested whether western European populations 1) have a long history of isolation, diverging from other regions of Eurasia during the Pleistocene (e.g. in an isolated glacial refuge), 2) have a more recent origin as a result of gradual range expansion from eastern Europe post-Pleistocene (Eriksen et al. 2014), 3) have a more recent origin as a result of long-distance dispersal from Asia post-Pleistocene, or 4) are derived from recent admixture between eastern European and Asian lineages post-Pleistocene (Dlugosch et al. 2013). To reduce the number and complexity of evolutionary scenarios to be compared, we pooled populations in Eurasia that belonged to the same genetic group (c.f. Lombaert et al. 2014). Based on results of DAPC and groupings by STRUCTURE and BAPS at K = 4, we delineated western Europe, eastern Europe, and Asia as separate groups (Table S1, Supporting information).
Fig. 1.
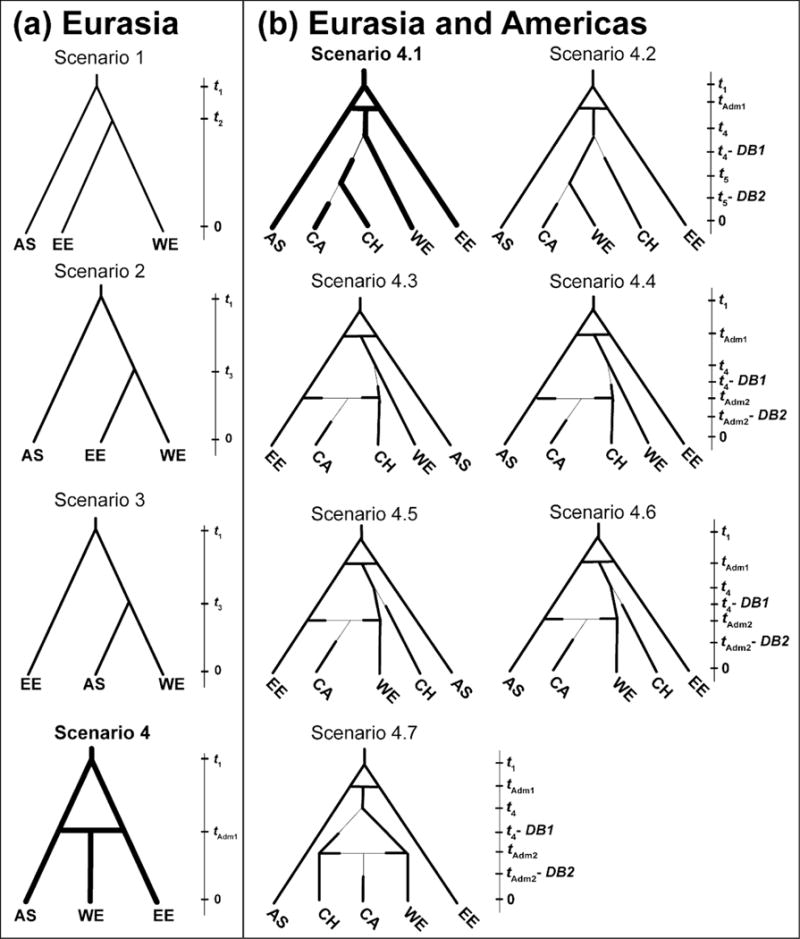
Evolutionary scenarios tested for two successive steps of ABC analyses of range expansions of the core lineage of YST. The first analysis (a) tested among four evolutionary scenarios for populations in Eurasia. The second analysis (b) incorporated the most highly supported scenario from (a) to test among seven competing evolutionary scenarios for the invasion of California. Scenarios are shown together with their respective tree topologies and relative times of divergence (t1 represents the oldest split and 0 represents the present day) and admixture (tAdm1 and tAdm2). Narrow lines in topologies of (b) indicate population bottlenecks at times DB1 and DB2. Times are not to scale. The best-fit scenario for each analysis is indicated with a bold topology. Additional details regarding evolutionary scenarios and prior distributions for their demographic parameters are described in Appendix S1 and Table S2 (Supporting information) respectively.
Next, we used the most highly supported scenario from this analysis (Scenario 4, see Results; c.f. Lombaert et al. 2014) to test seven scenarios regarding the invasion route(s) of YST to California (Fig. 1b). The first two models tested whether Californian populations have a single origin as a secondary introduction from Chile (Scenario 4.1, consistent with historical records of the first introduction to North America) or a primary introduction from western Europe (Scenario 4.2, consistent with historical records of additional introductions from western Europe to North America; Gerlach 1997). In contrast, remaining models tested whether Californian populations are derived from admixture of these introductions with those from Eurasia (Gerlach 1997), including models of admixture between Chile and eastern Europe (Scenario 4.3), Chile and Asia (Scenario 4.4), western Europe and eastern Europe (Scenario 4.5), western Europe and Asia (Scenario 4.6), or western Europe and Chile (Scenario 4.7). Each model incorporated a population bottleneck during colonization of both Chile and California, because founders there would have included only a subset of individuals from these source regions. Including populations from the Pacific Northwest in ABC analyses was computationally infeasible because multiple genetic clusters occurred in this region, and individual sampling sites varied greatly in their assignment (see Results). Inferring YST’s introduction history in the Pacific Northwest will require increased sampling and testing of multiple evolutionary scenarios for individual populations in this region. Detailed descriptions of all evolutionary scenarios and prior distributions for demographic parameters are described Appendix S1 and Table S2 (Supporting information), respectively.
For each of the two ABC analyses, we exported SNPs from the “CORE” data set in the ‘populations’ module in STACKS, requiring that each locus was independent, selectively neutral, present in all regions, genotyped in at least 70% of the individuals in each group, had a minimum coverage depth of 10, and had < 25% missing data. The analysis of Eurasian populations included 1069 SNPs, and the analysis of both Eurasian and American populations included 943 SNPs.
For both DIYABC analyses, we simulated 1.5 × 105 data sets for each scenario and generated summary statistics as in Cornuet et al. (2014), which are described in Appendix S1 (Supporting information). To select the best-fit scenario, posterior probabilities were computed via logistic regression on the 1% of simulated data sets closest to the empirical data (Cornuet et al. 2008). Summary statistics were transformed by linear discrimination analysis prior to logistic regression to reduce correlation among explanatory variables and provide conservative estimates of scenario discrimination (Estoup et al. 2012). We used a model checking computation to evaluate the goodness of fit of the most highly supported scenario and evaluated confidence in scenario choice as empirical verifications of the performance of the ABC procedure (Cornuet et al. 2010). These analyses are detailed in Appendix S1 (Supporting information).
Population genomic diversity
We quantified several aspects of genomic diversity at the 1050 SNPs used for analyses of population structure of the “CORE” data set. The proportion of variable loci (P), observed heterozygosity (HO), and nucleotide diversity (π) within each region were calculated using ARLEQUIN 3.5 (Excoffier & Lischer 2010). We inferred the number of private alleles, mean allelic richness (NAR), and mean private allelic richness (NPAR) in each region with HP-RARE version 1.0 (Kalinowski 2005), which uses rarefaction and hierarchical sampling to adjust for uneven sample sizes. Seven individuals (14 allele copies) were sampled at random from each region to match the smallest regional sample size. When calculating NPAR, we allowed a private allele to also be present in regions where YST was later introduced (e.g. a private allele in South America could also be present in California and the Pacific Northwest).
Trait differentiation
We have previously published data from a large common garden experiment, demonstrating divergence in plant size between the native range and western US invasion of YST (Dlugosch et al. 2015b). We reanalysed these data to test for trait differentiation among genetically differentiated regions revealed by our analyses here, including two sites from Chile that were part of the experiment but not previously reported. Seeds used in the common garden experiment and seeds for this study were obtained from the same field collections. In total, we included genotypes from 40 sites (Table S1, Supporting information) that span Asia (excluding Anatolia), eastern Europe, western Europe, Chile, California coast, and California interior. Briefly, single offspring from 7–20 different mothers per site were reared in a randomized glasshouse common garden, and their size at 5.5 weeks measured using a linear morphological index that strongly correlates with biomass (Dlugosch et al. 2015b): Size Index = [leaf number * (maximum leaf length*maximum leaf width)1/2]. Regional differentiation in ln-transformed size indices was compared using REML ANOVA, with fixed effects of region, site nested within region, and observer (c.f. Dlugosch et al. 2015b). We calculated pairwise FST among regions in ARLEQUIN 3.5 using 1000 permutations. Pairwise differences in least squares means for the size index were tested for a correlation with FST using a Mantel test in the ADE4 R package (with 1000 permutations).
Results
Population structure
DAPC analyses revealed clear differentiation among populations across YST’s range. Individuals from the southern Apennine and Balkan Peninsulas (Italy, southern Greece, and Crete) were clearly separated from all other native and introduced individuals (Fig. 2a). This divergence was unambiguous in all population genetic analyses (see below). Henceforth, we refer to the two divergent groups of YST as the Apennine-Balkan lineage and the core lineage. In the analysis of the “CORE” data set, eastern Europe and Californian populations were differentiated from other populations, and there was clear overlap between South America and the Pacific Northwest populations with those of western Europe and Asia (including Anatolia; Fig. 2b).
Fig. 2.
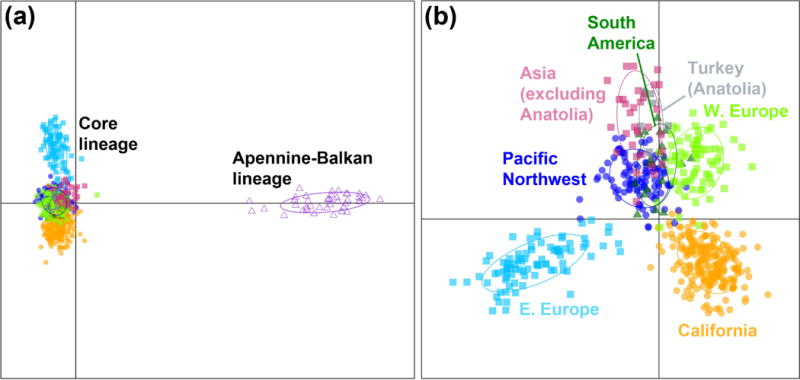
Discriminant analysis of principal components (DAPC) based on single nucleotide polymorphisms, using regions as prior clusters. Results are shown for (a) the analysis that included YST individuals from across the entire sampled range (i.e. the “FULL” data set) and (b) the analysis that included only individuals belonging to the core lineage (i.e. the “CORE” data set). Ovals are 95% inertia ellipses. Individual genotypes are depicted with closed squares (Eurasia), closed triangles (South America), dots (western US), and open triangles (southern Apennine and Balkan Peninsulas), and are colored according to the geographic region from which they were sampled.
In STRUCTURE analyses of the “FULL” data set, individuals from the southern Apennine and Balkan Peninsulas were separated from remaining individuals across all replicates at each K value (Fig. S3, Supporting information). The mean log probability of the data increased with the successive addition of clusters to K = 5, after which it plateaued, and the ΔK statistic was greatest for K = 2 (Fig. S4, Supporting information). CLUMPP revealed identical clustering solutions across replicates for K = 2–3 (H’ = 1) and lower similarity across replicates at K = 4–10 (mean H’ = 0.76; Fig. S5, Supporting information).
STRUCTURE analyses of the “CORE” data set depicted finer geographic structuring of the core lineage of YST (Figs 3a and 4). The mean log probability of the data increased with the successive addition of clusters to K = 5, after which it plateaued, and the ΔK statistic was greatest for K = 2 (Fig. S4, Supporting information). With the exception of K = 3, K = 2–5 had high similarity of clustering solutions across replicates (mean H’ = 0.95; Fig. S5, Supporting information). We focus on results for K = 4 because BAPS identified this K value as the most probable number of genetic clusters (see below), and we used this clustering solution to define genetically-differentiated native regions for testing evolutionary scenarios in DIYABC. Population structure at K = 4 clearly delineated eastern Europe, Asia, and western Europe, although individuals from Anatolia had partial assignment to the western Europe cluster (Figs 3a and 4a). For introduced populations at K = 4, a unique cluster predominated in California, the Asia cluster occurred in Washington, the eastern Europe cluster occurred in both Washington and Idaho, and the western Europe cluster was prevalent in South America, the California coast, and throughout the Pacific Northwest (Figs 3a and 4b,c). Most individuals in the western US exhibited assignment to two or more clusters (i.e. multiple assignment).
Fig. 3.
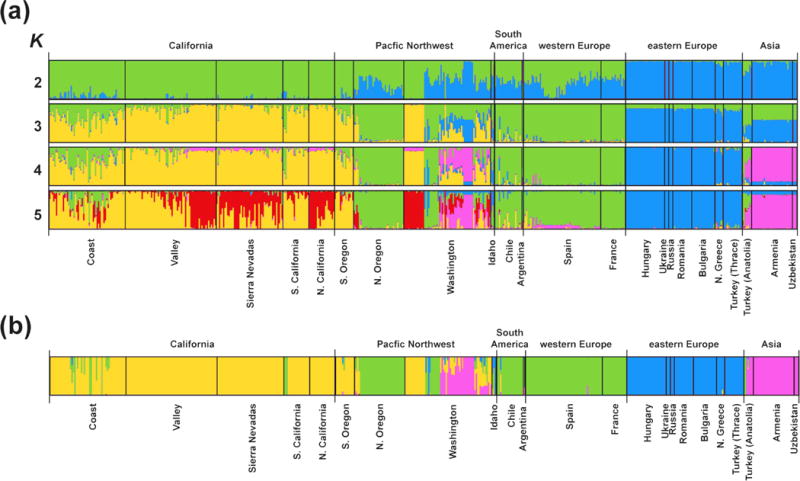
Individual assignments from STRUCTURE analyses and a BAPS admixture analysis based on 1013 SNP loci of 550 individuals belonging to the core lineage of YST, which excludes populations from the southern Apennine and Balkan Peninsulas (i.e. the “CORE” data set). (a) STRUCTURE bar plots of assignment probabilities averaged across ten runs (calculated by CLUMPP) are shown for K = 2 to K = 5, where K is the number of genetic clusters. CLUMPP H’ values indicated low heterogeneity at K = 2 (0.998), K = 4 (0.940), and K = 5 (0.923; Fig. S5, Supporting information). (b) BAPS bar plots are shown for the best estimate of K (4), where K is the number of genetic clusters. Each vertical bar shows the proportional representation of the estimated cluster membership for a single individual.
Fig. 4.
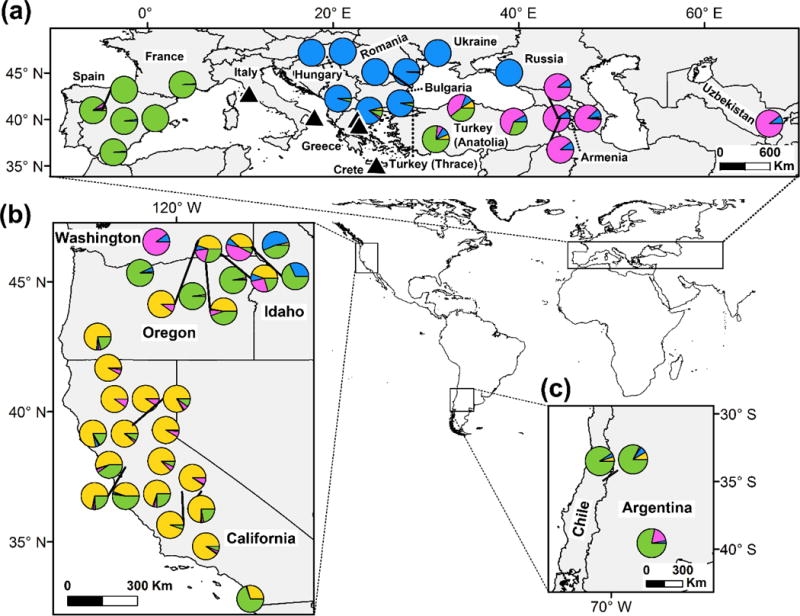
Pie charts depicting the average assignment probabilities of individuals of the core lineage of YST at each sampling site in Eurasia (a), the western US (b), and South America (c) to a genetic cluster inferred by STRUCTURE analyses at K = 4 (the average of ten replicates), where K is the number of genetic clusters. Black leader lines indicate the geographic location of nearby sampling sites. Black triangles in Eurasia indicate sampling sites for populations of the divergent Apennine-Balkan lineage of YST.
In general, results of BAPS analyses were consistent with those of STRUCTURE. BAPS identified K = 4 as the most probable number of genetic clusters for both data sets, and these clusters had similar geographic distributions as those inferred by STRUCTURE (Fig. 3b and Fig. S3, Supporting information). However, BAPS inferred lower levels of multiple assignment in Anatolia and California. Individuals in western Anatolia (site 49; Fig. S1, Supporting information) were assigned to the western Europe cluster, whereas those in central and eastern Anatolia (sites 50 and 51, respectively; Fig. S1, Supporting information) were assigned to the Asia cluster. A single individual from central Anatolia exhibited multiple assignment. In California, only individuals from the coast exhibited multiple assignment.
Interspecific hybridization
BAPS identified outgroup species as distinct and suggested no evidence of recent interspecific hybridization. BAPS identified K = 6 as the most probable number of genetic clusters, assigning individuals of each outgroup species (C. melitensis, C. pallescens, and C. nicaeensis) and the two YST lineages to separate clusters (Fig. S6, Supporting information).
STRUCTURE demonstrated less power to distinguish among species, consistent with previous work showing that clustering does not always correspond to the branching pattern of a species tree (Carstens et al. 2013), but similarly identified little evidence of recent interspecific hybridization among the core lineage of YST (responsible for invading populations) and outgroup species (Fig. S6, Supporting information). The mean log probability of the data increased with the successive addition of clusters to K = 6, after which it plateaued, and the ΔK statistic was greatest for K = 2 (Fig. S4, Supporting information). CLUMPP revealed high similarity of clustering solutions across replicates for K = 2 and K = 7–10 (mean H’ = 0.97) and lower similarity across replicates at K = 3–6 (mean H’ = 0.74; Fig. S5, Supporting information). We focus on results for K = 2 and K = 7 due to high similarity across replicates. At K = 2, all outgroup species clustered with the Apennine-Balkan lineage of YST. At K = 7, C. melitensis and C. pallescens formed a separate cluster, whereas C. nicaeensis individuals exhibited partial assignment to the Apennine-Balkan lineage of YST (mean assignment probability = 0.46) and to the western Europe cluster of the core lineage of YST (mean assignment probability = 0.30).
Scenarios of evolutionary history with approximate Bayesian computation
The ABC analysis of the history of the core lineage of YST in Eurasia identified Scenario 4 as the best-fit scenario [posterior probability (PP) = 0.95 based on logistic regression (95% confidence interval (CI): 0.93–0.96); Table S3, Supporting information]. In Scenario 4, admixture occurred between eastern European and Asian populations after their divergence from one another, creating a lineage that gave rise to populations in western Europe (Fig. 1a; Table S4, Supporting information). The observed data set was closely matched by many simulated data sets from the posterior sample of Scenario 4, indicating that the simulations produced data sets similar to the observed data (Fig. S7, Supporting information). In assessing confidence in scenario choice, the type I error was 0.12 and the mean type II error was 0.04 (minimum = 0.01, maximum = 0.06). The low type II error indicates very high (96%) statistical power to distinguish between the alternative evolutionary scenarios.
The ABC analysis of the history of populations of the core lineage of YST in both Eurasia and the Americas identified Scenario 4.1 as the best-fit scenario (PP = 0.73) and its 95% CI (0.64–0.82) did not overlap with the 95% CI of the next best scenario [Scenario 4.7; PP = 0.13 (CI: 0–0.36); Table S3, Supporting information]. Scenario 4.1 depicts a bottleneck in Chilean populations followed by their divergence from western European populations, and a second relatively recent bottleneck in Californian populations followed by their divergence from Chilean populations (Fig. 1b). Both bottlenecks were inferred to be weak, with effective population sizes similar to that of western Europe (thousands of individuals) before population expansion in each of the invasions (Table S4, Supporting information). Again, the simulations produced data sets similar to the observed one (Fig. S7, Supporting information). In assessing confidence in scenario choice, the type I error was 0.24 and the mean type II error was 0.04 (minimum = 0.00, maximum = 0.21). The low type II error indicates very high (96%) statistical power to distinguish between the alternative evolutionary scenarios.
Population genomic diversity
In general, genomic diversity of the core lineage of YST was similar across regions in the native and introduced ranges (Table 1). In the native range, populations in Asia had the lowest allelic richness and private allelic richness corrected for uneven sample size (NAR and NPAR, respectively) relative to other Eurasian populations. Introduced populations in the Americas had lower NPAR, and similar levels of NAR, nucleotide diversity (π), and observed heterozygosity (HO) as most native populations.
Table 1.
Summary statistics for 1013 single nucleotide polymorphism loci collected from populations of the core lineage of YST in the invaded range in the Americas (Pacific Northwest, California interior, California coast, and South America) and in Eurasia [western Europe, eastern Europe, Turkey (Anatolia), and Asia].
| Region | N | P | Priv. | NAR | NPAR | HO (±SE) | π (±SE) |
|---|---|---|---|---|---|---|---|
| Pacific Northwest | 104 | 0.56 | 51 | 1.19 (0.009) | 0.028 (0.002) | 0.0887 (0.0972) | 0.0566 (0.0271) |
| California interior | 168 | 0.45 | 31 | 1.18 (0.009) | 0.027 (0.002) | 0.102 (0.0995) | 0.0430 (0.0207) |
| California coast | 56 | 0.41 | 7 | 1.17 (0.009) | 0.021 (0.002) | 0.128 (0.1226) | 0.0485 (0.0234) |
| South America | 21 | 0.33 | 43 | 1.16 (0.009) | 0.024 (0.002) | 0.1570 (0.1330) | 0.0436 (0.0214) |
| Western Europe | 75 | 0.46 | 201 | 1.18 (0.009) | 0.035 (0.003) | 0.0974 (0.0981) | 0.0476 (0.0229) |
| Eastern Europe | 86 | 0.45 | 210 | 1.14 (0.008) | 0.042 (0.003) | 0.0825 (0.0979) | 0.0347 (0.0168) |
| Turkey (Anatolia) | 7 | 0.24 | 60 | 1.18 (0.010) | 0.041 (0.004) | 0.2313 (0.1535) | 0.0602 (0.0308) |
| Asia (excluding Anatolia) | 33 | 0.22 | 59 | 1.13 (0.009) | 0.025 (0.003) | 0.1684 (0.1219) | 0.0301 (0.0148) |
N = number of individuals analysed; P = proportion of variable loci; number of private alleles (Priv.); NAR = mean allelic richness corrected for uneven sample size; NPAR = mean private allelic richness corrected for uneven sample size; HO = observed heterozygosity for polymorphic loci; π = nucleotide diversity. Standard errors (SE) for NAR, NPAR, HO, and π are indicated in parentheses.
Trait differentiation
Our analyses of population structure (above) indicated clear differentiation of native populations from Asia, eastern Europe, and western Europe, and of introduced populations from California. According to STRUCTURE and BAPS, there was also differentiation between populations from California’s coast and interior. Plant size was significantly different among these regions (ANOVA: P < 0.0001; Fig. 5b), but these phenotypic differences were not correlated with FST (Mantel test: P = 0.85; Table S5, Supporting information). Instead, introduced genotypes from California and Chile differ in growth from their closest relatives in the native range (western Europe), and three specific morphological transitions appear to have occurred during YST’s colonization history (Fig. 5). The most recent native expansion into western Europe was associated with a shift to smaller plant size relative to other Eurasian regions. Assuming that this smaller size reflects western European phenotypes at the time of their introduction into the Americas, the invasion into Chile and the California coast corresponded to a single evolutionary shift (back) to larger size. Finally, the evolution of the most extreme and novel size increase in YST was associated with the range expansion from the California coast to the California interior.
Fig. 5.
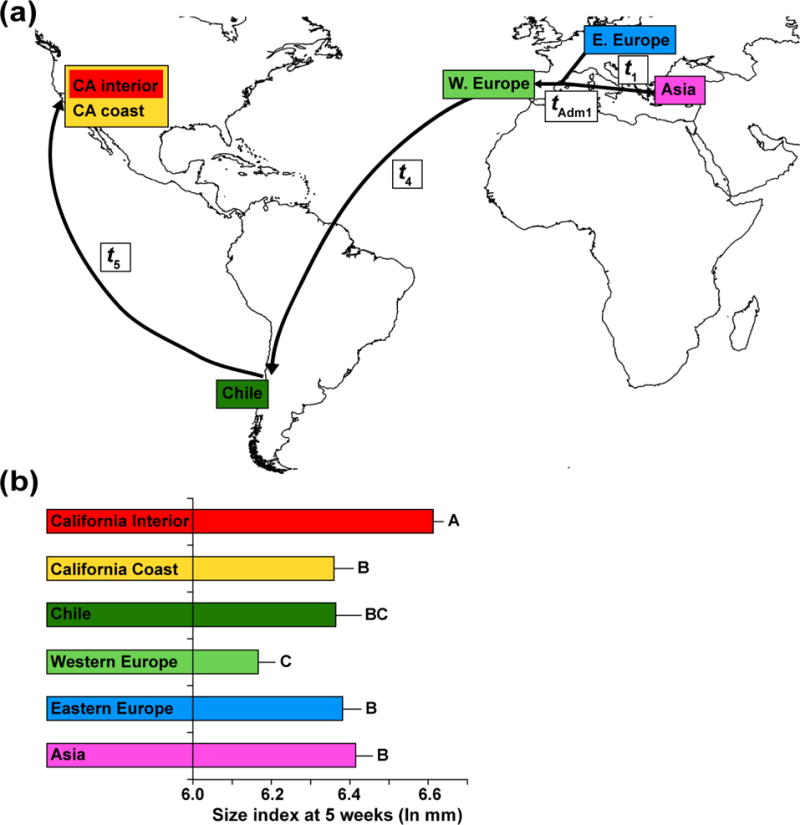
Illustration of the most highly supported evolutionary scenario (Scenario 4.1) in the ABC analysis of YST in Eurasia and the Americas, in relation to variation in plant size among regions. (a) Within Europe, eastern European populations diverged from those in Asia, and subsequently these two lineages admixed to form western European populations. The western European lineage then served as the primary source of the YST introduction to Chile, followed by introduction of the Chilean lineage to California. (b) Least Squares Means (+/− standard error of the mean) of a linear size index of biomass. Letters indicate significantly different regions based on Tukey’s HSD post-hoc tests.
Discussion
There is mounting evidence that introduced YST populations have been evolving adaptively in the Americas, potentially contributing to invasiveness in this species (Widmer et al. 2007; Hierro et al. 2009; Eriksen et al. 2012; Graebner et al. 2012; Hierro et al. 2012; Dlugosch et al. 2015b). Using broad population genomic sampling across the range of YST, we clearly identify invasion routes and the sources of genetic variation in invading populations. We find evidence to support the hypothesis that populations in western Europe represent an ancient colonization event that has served as a ‘bridgehead’ for much of the invasions into the Americas. Our reconstruction of the population history of YST reveals that colonization events into western Europe and the Americas are associated with multiple evolutionary transitions in plant size, a focal trait associated with invasiveness in YST.
Population structure and colonization history of yellow starthistle in Eurasia
The Apennine-Balkan lineage of YST, which consisted of individuals from Italy, southern Greece, and Crete, was deeply differentiated from a core lineage that included individuals from the rest of Eurasia (Fig. 2a; Fig. S3, Supporting information). There was no evidence for gene flow between these lineages despite their close geographic proximity in Greece and Italy. Additional work to understand the taxonomic status and history of the Apennine-Balkan lineage are needed. While all characters of this unique lineage conform to morphological specifications for YST in the Flora Europaea (Dostál 1976) taxonomic key (pers. obs.), our data suggest that it has not contributed to the expansion of YST across its introduced range.
Our analyses of population genomic structure of the core lineage in Eurasia clearly defined genetic differentiation among populations in eastern Europe and Asia (Figs 2b, 3, and 4a). Portions of eastern Europe and Asia are known to have provided refugia for plant and animal species during cool and dry periods of the Pleistocene (Médail & Diadema 2009; Bilgin 2011; Stöck et al. 2012; Dufresnes et al. 2016), and may have contributed to the isolation of YST populations. The narrow strip of land connecting eastern Europe to Anatolia (i.e. Thrace) is associated with a genetic discontinuity in YST, a finding consistent with previous work showing that the Sea of Marmara, the Black Sea, and a waterway connecting these seas have acted as dispersal barriers to certain insect and mammal species (Bilgin 2011). Several geologic features in Anatolia, including the Central Anatolian Plateau, the Anatolian Diagonal, and the Central Anatolian Lake system acted as barriers to dispersal to species of plants, fish, amphibians, and reptiles during the Pliocene and/or Pleistocene (Bilgin 2011; Kapli et al. 2013; Dufresnes et al. 2016).
STRUCTURE analyses suggested the possibility of admixture in YST populations in Anatolia. A previous microsatellite survey of YST (Eriksen et al. 2014) did not detect admixture in this region, nor did our BAPS analysis; however, BAPS is less likely than STRUCTURE to detect admixed genotypes (Bohling et al. 2013; Neophytou 2014). Evidence for admixture might indicate that previously isolated populations have reconnected in Anatolia, a phenomenon documented in other species in this region (Bilgin 2011; Dufresnes et al. 2016). Assignment to multiple clusters may also be derived from the contribution of populations in Anatolia to admixture elsewhere (e.g. in western Europe) and further exploration of population genetics of YST there is likely to be informative about its history of range expansion in Europe.
The origin(s) of western European populations and their possible history of admixture have been a subject of past debate (Dlugosch et al. 2013; Eriksen et al. 2014). Prior to the availability of molecular genetic data, it was hypothesized that western European populations represented an ancient human-mediated introduction of YST (Wagenitz 1975; Maddox et al. 1985). This hypothesis predicts that western European populations diverged relatively recently from other Eurasian populations, and it has been supported by a previous analysis of transcriptome data which further suggested that they formed through admixture of eastern European and Asian populations (Dlugosch et al. 2013). In contrast, a survey of microsatellite markers suggested deep divergence of western European populations, without admixture (Eriksen et al. 2014). Our ABC inference provided the strongest support for a scenario in which the western European lineage arose most recently from an admixture event between eastern European and Asian populations (Fig. 1a; Table S3, Supporting information). This scenario is consistent with high allelic richness in this region (Table 1). A rapid range expansion facilitated by human-mediated dispersal could explain these results. YST is strongly associated with human disturbed habitats (Bottema & Woldring 1990; DiTomaso et al. 2006) and it occurs in areas that have a long history of human occupation (Dennell 2008). Palynological data indicate a sudden appearance and increase in C. solstitialis-type pollen in Anatolia and south-eastern Greece ca. 5500 years ago as agropastoral activities by Neolithic farmers increased (Bottema & Woldring 1990). Neolithic plant cultivation advanced rapidly along the north coast of the Mediterranean, reaching southern Spain by ca. 6650 years ago (Waterbolk 1982; Behre 1988). Additional opportunities for human-mediated dispersal of YST likely occurred as cultivated plants were extensively introduced across southern, central and western Europe beginning in the Roman period ca. 2000 years ago (Behre 1988).
Population structure and colonization history of yellow starthistle in the Americas
Our ABC analysis provided strongest support for a scenario in which introductions from a single source genepool established YST in Chile and California (Fig. 1b; Table S3, Supporting information). This finding implies that unique allele frequencies in California (Figs 2b, 3, and 4b) did not arise via additional introductions of divergent source material but instead evolved post-introduction. The presence of the western Europe cluster in both Chile and the California coast corroborates introduction records indicating that YST was introduced to Chile from Spain in the mid-1600s, and then to the San Francisco bay area in California from Chile in the 1850s (Stewart 1926; Howell 1959; Gerlach 1997). ABC models inferred weak genetic bottlenecks (effective population sizes of thousands of individuals) during each of these introduction events, followed by effective size expansion (Table S4, Supporting information). Increasing connectivity and population sizes during a range expansion can increase population genetic diversity (Dlugosch & Parker 2008; Excoffier et al. 2009; Ramakrishnan et al. 2010), and may explain high allelic richness in Californian populations. Following its initial introduction to the San Francisco bay area, YST spread into the Sacramento and San Joaquin Valleys and began rapidly expanding into rangelands in the Sierra Nevada foothills beginning in the 1930s or 1940s (Maddox & Mayfield 1985; Gerlach 1997). Gene flow among repeated introduction events from the same source (i.e. Chile) could also have contributed to high diversity of Californian populations.
In contrast, our analyses of population structure and genomic diversity of YST in the Pacific Northwest imply multiple, independent introduction sources there. Allelic richness and nucleotide diversity were higher in the Pacific Northwest than in any other region in the Americas (Table 1), multiple genetic clusters occurred there (Figs 3 and 4b), and the 95% inertia ellipses of Pacific Northwest populations overlapped substantially with those of Asia (including Anatolia) and South America (Fig 2b). The western Europe cluster was widespread in the Pacific Northwest, a result that supports records indicating that initial introductions to the Pacific Northwest in the 1870s and 1880s likely originated from the same Spanish stock as Californian populations (Roché 1965; Gerlach 1997). Subsequently, a large proportion of alfalfa seed imported into Washington during the early 1900s originated from ‘Turkestan’ (Roché & Talbott 1986), consistent with the presence of the Asia cluster in this state. The presence of the eastern Europe cluster in Idaho supports the hypothesis of another independent introduction of YST to this area (Sun 1997). Multiple clusters were found at most sites in the Pacific Northwest, which may imply gene flow among sites and/or multiple introductions to the same site. The presence of the unique California cluster in the Pacific Northwest implies long-distance dispersal across the western US, which is consistent with evidence that YST seed is readily dispersed across long distances in the western US by vehicles, contaminated crop seed, hay or soil, road maintenance, and movement of livestock (DiTomaso et al. 2006). However, the rareness of the Asia and eastern Europe clusters in California suggests that they were not introduced in significant numbers to this region, or that they did not establish there.
Interspecific hybridization among yellow starthistle and Centaurea species
Hybridization has played a major role in the evolution of the Centaurea genus (Garcia-Jacas et al. 2006; Suárez-Santiago et al. 2007), and has been associated with an increase in invasiveness in a variety of plant taxa (Abbott 1992; Ellstrand & Schierenbeck 2000; Hovick and Whitney 2014), including Centaurea species (Hahn et al. 2012; Mráz et al. 2012). For YST in particular, hybridization with other Centaurea species has been documented in Turkey (Wagenitz 1955), and a sterile hybrid has been formed between YST and a distantly related congener, C. moncktoni, in Oregon (Roché & Susanna 2011), suggesting that hybridization could be an important component of recent evolution in this species. However, our STRUCTURE and BAPS analyses did not provide any compelling evidence for interspecific hybridization between the core lineage of YST and either C. melitensis, C. nicaeensis, or C. pallescens (Fig. S6, Supporting information). BAPS depicted no evidence of interspecific hybridization, and STRUCTURE suggested a small amount of hybridization (mean assignment probability = 0.30) between C. nicaeensis and the western Europe cluster of YST. These contrasting results are likely explained by the different clustering algorithms used by these programs (Neophytou 2014), as well as by variation in our sample sizes across species. Simulations have shown that clusters inferred by STRUCTURE are often not consistent with the evolutionary history of populations when it is forced to place individuals into too few clusters (Kalinowski 2011). Additionally, our data set consisted of substantially more YST individuals than those of outgroup species, and such unbalanced sampling can result in spurious clustering solutions (Kalinowski 2011; Neophytou 2014). Better sampling of co-occurring congeners will be necessary to fully test for evidence of interspecific gene flow in this system, but our data suggest that hybridization has not been a major feature of YST’s invasion into the Americas.
Evolution of invasiveness
We have recently shown that YST has evolved a novel increase in size and fitness in its invasion into California, and this may be a key component of its invasiveness (Dlugosch et al. 2015b). Our analyses here suggest that plant size has evolved significantly at three points during YST’s range expansion (Fig. 5). Smaller size appears to have evolved during the colonization of western Europe. Our previous research indicated that smaller size is associated with greater drought tolerance in YST, and is favored in warm climates with consistent summer droughts, including those in Mediterranean western Europe (Dlugosch et al. 2015b). In contrast, populations in Chile and the California coast that are closely derived from western Europe populations show an evolutionary transition back to larger size, a phenotype that is typical of the rest of Eurasia. While it might be parsimonious to infer that it was western European populations that evolved smaller size after introductions to the Americas occurred, it seems more plausible that larger size re-evolved from standing variation in the invasion.
The third and largest evolutionary increase in size is associated with populations in the California interior (Table S6, Supporting information), an area where YST has rapidly spread and attained high densities (Swope & Parker 2010; Andonian et al. 2011). Our analyses of population structure identified only slight genomic divergence in this region, and little evidence of significant admixture with other parts of the range. Moreover, although YST’s most recent native expansion into western Europe served as the primary genetic ‘bridgehead’ for introductions to the Americas, its traits in terms of plant size do not appear to have been pre-adapted for increased invasiveness there. Instead, YST appears to have experienced an exceptional increase in size during the expansion of a single lineage in the Americas, and in California’s interior in particular.
Management implications
Controlling the spread of YST is critical for minimizing its negative impacts on native biodiversity, water cycles, and crop, forest and livestock productivity (DiTomaso et al. 2006). The presence of multiple distinct genetic clusters and divergent phenotypes of YST in the western US is likely to have implications for managing this invasive plant. The efficacy of biological control and chemical control, both of which are primary avenues for managing and reducing the abundance and spread of YST (DiTomaso et al. 2006), can be influenced by plant traits, the genetic diversity of an invading population, and the origin of invading genotypes (Hokkanen & Pimentel 1989; Wilson et al. 2009; Paynter et al. 2012; Kuester et al. 2015). None of the seven established biological control agents (six insect and one fungal species) for YST were chosen based on the origin of invading plant genotypes, and all have had limited success (DiTomaso et al. 2006; Pitcairn et al. 2008). YST has evolved resistance to herbicide treatments in at least one population in Dayton, Washington (Sabba et al. 2003), a site where we found multiple genetic clusters. Future work that addresses the extent to which the origins and genetic diversity in invading YST shapes the success of biological and chemical control programs offers clear opportunities for improving management plans for this highly invasive species.
Supplementary Material
Appendix S1 Details regarding evolutionary scenarios, demographic parameters, and confidence in scenario choice in DIYABC.
Table S1 Sampling sites for seeds of yellow starthistle (Centaurea solstitialis) and related taxa, including sampling site information (nearest city, country/state, continent, latitude, and longitude), regional groups as defined for the ABC and trait differentiation analyses, the number of individuals sequenced for this study (N), the number of individuals included in analyses (n), species (CESO = C. solstitialis, CENE = C. nicaeensis, CEPA = C. pallescens, CEME = C. melitensis), and year of collection. Site numbers correspond to those depicted in Fig. S1, Supporting information.
Table S2 Prior distributions for effective population size (Ne) and divergence time parameters used for two successive steps of ABC analyses of yellow starthistle populations (performed in DIYABC 2.0.4). The first analysis (Eurasia) tested among four evolutionary scenarios for populations in Eurasia, whereas the second analysis (Eurasia and Americas) incorporated the most highly supported scenario from the first analysis to test among seven competing evolutionary scenarios regarding the introduction history of yellow starthistle to California. We used uniform prior distributions for effective population size and divergence time parameters. Divergence time is in generations, with a generation length of one year. We used the ‘set condition’ option to impose the rule that t1 > t2, t1 ≥ t3, and t1 > tAdm1 for the first analysis, and the rule that t1 > tAdm1, t1 > t4, t4 ≥ t5, tAdm1 > t4, tAdm1 > t5, t4 ≥ tAdm2, and tAdm1 ≥ tAdm2 for the second analysis. WE = western European; EE = eastern European; AS= Asian; CH = Chilean; CA = Californian.
Table S3 Posterior probability estimates (95% confidence interval) for evolutionary scenarios tested for yellow starthistle populations in Eurasia, and for populations in both Eurasia and the Americas, based on logistic regression on the 1% of simulated data sets closest to the empirical data (performed in DIYABC 2.0.4).
Table S4 Median estimates with 5% and 95% quantiles for posterior distributions of effective population size (Ne) and divergence time parameters for the best-fit scenario of ABC analyses of the analysis of Eurasian populations (Scenario 4), and of the best-fit scenario of ABC analyses of the analysis of Eurasian and American populations (Scenario 4.1). WE = western European; EE = eastern European; AS= Asian; CH = Chilean; CA = Californian.
Table S5 Pairwise divergence matrices used in the Mantel test. a) Pairwise FST values and b) pairwise differences in Least Squares Means in a ln-transformed size index were calculated for populations in California (CA), Chile (CH), western Europe (WE), eastern Europe (EE), and Asia (AS). All FST values have P-values of < 0.01.
Table S6 Least Squares Means (LSM) and standard errors (SE) in a ln-transformed size index were calculated for yellow starthistle populations in California (CA) coast, CA interior, Chile (CH), western Europe (WE), eastern Europe (EE), and Asia (AS), from data in Dlugosch et al. 2015b.
Fig. S1 Sampling sites of the genotyped yellow starthistle in Eurasia, the western United States, and South America are indicated by black circles. Sampling sites of the genotyped individuals of Centaurea nicaeensis, C. pallescens, and C. melitensis are indicated by black triangles, black squares, and black stars, respectively. Known introduction dates of yellow starthistle to regions of South America and the western US are indicated. site numbers correspond to those presented in Table S1, Supporting information.
Fig. S2 Boxplots of the inbreeding coefficient (FIS) based on RAD loci in individuals in the “FULL” data set (white) and “CORE” data set (gray) at different -M and -n values in STACKS, where -M is the number of nucleotide differences allowed between loci when processing a single individual and -n is the number of nucleotide differences allowed between loci when creating the catalog of loci. The RAD loci were present in Centaurea melitensis, C. pallescens, and C. nicaeensis, and each of the two yellow starthistle lineages (p = 5), genotyped in at least 60% of the individuals in each species/lineage (r = 0.6), and had a minimum coverage depth of 10. Box plots show median (horizontal line), interquartile range (box), and minimum and maximum values (whiskers). Positive values for FIS may indicate excess homozygosity due to failure to cluster loci under restrictive mismatch values, whereas negative values for FIS may indicate excess heterozygosity due to clustering of paralogous loci.
Fig. S3 Results from STRUCTURE analyses and a BAPS admixture analysis based on 747 single nucleotide polymorphism (SNP) loci of 591 yellow starthistle individuals from across the entire sampled range (Eurasia, South America, and the western US; i.e. the “FULL” data set). (a) STRUCTURE bar plots of assignment probabilities averaged across ten runs (calculated by CLUMPP) are shown for K = 2 to K = 5, where K is the number of genetic clusters. (b) BAPS bar plots are shown for the best estimate of K (K = 4). Each vertical bar shows the proportional representation of the estimated cluster membership for a single individual.
Fig. S4 The mean log probability of the data (ln Pr(X | K)) plus or minus the standard deviation for each value of K from 1 to 10 (black squares), and the ΔK statistic (gray squares), where K is the number of genetic clusters, are depicted for ten replicate STRUCTURE analyses of the (a) “FULL” and (b) “CORE” data set for yellow starthistle, and of (c) the “OUTGROUPS” data set that includes yellow starthistle and three other Centaurea species.
Fig. S5 The highest value of average pairwise similarity (H’) calculated by CLUMPP for each value of K from 1 to 10, where K is the number of genetic clusters, are depicted for ten replicate STRUCTURE analyses of the (a) “FULL” and (b) “CORE” data set for yellow starthistle, and of (c) the “OUTGROUPS” data set that includes yellow starthistle and three other Centaurea species.
Fig. S6 Results from STRUCTURE and BAPS analyses of 440 SNPs of the two lineages of yellow starthistle and three outgroup species (Centaurea nicaeensis, C. melitensis, and C. pallescens). (a) Bar plots of assignment probabilities averaged across ten STRUCTURE runs (calculated by CLUMPP) are shown for K = 2–6, where K is the number of genetic clusters. (b) BAPS bar plots are shown for the best estimate of K (K = 6). Each vertical bar shows the proportional representation of the estimated cluster membership for a single individual.
Fig. S7 Results of a principal component analysis conducted for model-checking of the best-fit scenario of ABC analyses of (a) Eurasian populations (Scenario 4) and of (b) both Eurasian and American populations (Scenario 4.1), performed in DIYABC 2.0.4. The observed data, data sets simulated with the prior distributions of parameters, and data sets simulated from the posterior sample are represented on each plot.
Acknowledgments
We thank M.S. Barker, M. Cristofaro, A. Guggisberg, J. Hierro, and B.M. McTeague for seed collection and S. Anderson, C. Carpenter, J. Cocio, K. Gibson, M. Rivera, and S. Tran for plant propagation and DNA extraction. We acknowledge technical support from the Genetics Core at the University of Arizona (UA) and the DNASU Next Generation Sequencing Core at the Biodesign Institute at Arizona State University (particularly J. Steel). Allocation of computer time from the Research Computing High Performance Computing (HPC) and High Throughput Computing (HTC) at UA is gratefully acknowledged. This study was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award #K12GM000708 through the Center for Insect Science at UA to B.S.B, and USDA grant #2015-67013-23000 to K.M.D. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author contributions
K.M.D. conceived of and outlined the project with input from B.S.B., and B.S.B., K.A., S.M.S., D.G.L., and K.M.D. collected samples. B.S.B performed the molecular laboratory work. B.S.B. and K.M.D. analysed data and drafted the manuscript. All authors contributed to and approved the final manuscript.
Data accessibility
The ddRADseq data are deposited to the NCBI sequence read archive (BioProject for C. solstitialis: PRJNA275986, C. nicaeensis: PRJNA275992, C. melitensis: PRJNA275988, C. pallescens: PRJNA317005). The following data are accessible at Dryad (http://dx.doi.org/10.5061/dryad.pf550): list of sample and sequencing run information, STACKS denovo commands, and SNP matrices used for FST outlier tests, ABC analyses, and analyses of population structure and population genomic diversity.
Supporting information
Additional supporting information may be found in the online version of this article.
References
- Abbott RJ. Plant invasions, interspecific hybridization and the evolution of new plant taxa. Trends in Ecology & Evolution. 1992;7:401–405. doi: 10.1016/0169-5347(92)90020-C. [DOI] [PubMed] [Google Scholar]
- Andonian K, Hierro JL, Khetsuriani L, et al. Range-expanding populations of a globally introduced weed experience negative plant-soil feedbacks. PLoS ONE. 2011;6:e20117. doi: 10.1371/journal.pone.0020117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behre K-E. The role of man in European vegetation history. In: Huntley B, Webb T III, editors. Handbook of Vegetation Science. Klewer Academic Publishers; Dordrecht, The Netherlands: 1988. pp. 633–672. [Google Scholar]
- Bilgin R. Back to the suture: the distribution of intraspecific genetic diversity in and around Anatolia. International Journal of Molecular Sciences. 2011;12:4080–4103. doi: 10.3390/ijms12064080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock DG, Caseys C, Cousens RD, et al. What we still don’t know about invasion genetics. Molecular Ecology. 2015;24:2277–2297. doi: 10.1111/mec.13032. [DOI] [PubMed] [Google Scholar]
- Bohling JH, Adams JR, Waits LP. Evaluating the ability of Bayesian clustering methods to detect hybridization and introgression using an empirical red wolf data set. Molecular Ecology. 2013;22:74–86. doi: 10.1111/mec.12109. [DOI] [PubMed] [Google Scholar]
- Bossdorf O, Auge H, Lafuma L, et al. Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia. 2005;144:1–11. doi: 10.1007/s00442-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Bottema S, Woldring H. Anthropogenic indicators in the pollen record of the Eastern Mediterranean. In: Bottema S, Entjes-Nieborg G, van Zeist W, editors. Man’s Role in the Shaping of the Eastern Mediterranean Landscape. CRC Press, Balkema; Rotterdam, The Netherlands: 1990. pp. 231–264. [Google Scholar]
- Carstens BC, Pelletier TA, Reid NM, Satler JD. How to fail at species delimitation. Molecular Ecology. 2013;22:4369–4383. doi: 10.1111/mec.12413. [DOI] [PubMed] [Google Scholar]
- Catchen JM, Amores A, Hohenlohe P, et al. Stacks: building and genotyping loci de novo from short-read sequences. G3: Genes, Genomes, Genetics. 2011;1:171–182. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Hohenlohe PA, Bassham S, et al. Stacks: an analysis tool set for population genomics. Molecular Ecology. 2013;22:3124–3140. doi: 10.1111/mec.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-A, Lin C-C, Wang C-D, et al. An optimized procedure greatly improves EST vector contamination removal. BMC Genomics. 2007;8:416. doi: 10.1186/1471-2164-8-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colautti RI, Barrett SCH. Rapid adaptation to climate facilitates range expansion of an invasive plant. Science. 2013;342:364–366. doi: 10.1126/science.1242121. [DOI] [PubMed] [Google Scholar]
- Colautti RI, Lau JA. Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Molecular Ecology. 2015;24:1999–2017. doi: 10.1111/mec.13162. [DOI] [PubMed] [Google Scholar]
- Corander J, Waldmann P, Sillanpää MJ. Bayesian analysis of genetic differentiation between populations. Genetics. 2003;163:367–374. doi: 10.1093/genetics/163.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Marttinen P. Bayesian identification of admixture events using multilocus molecular markers. Molecular Ecology. 2006;15:2833–2843. doi: 10.1111/j.1365-294X.2006.02994.x. [DOI] [PubMed] [Google Scholar]
- Cornuet J-M, Santos F, Beaumont MA, et al. Inferring population history with DIYABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics. 2008;24:2713–2719. doi: 10.1093/bioinformatics/btn514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet J-M, Ravigné V, Estoup A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v. 1.0) BMC Bioinformatics. 2010;11:401. doi: 10.1186/1471-2105-11-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet J-M, Pudlo P, Veyssier J, et al. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics. 2014;30:1187–1189. doi: 10.1093/bioinformatics/btt763. [DOI] [PubMed] [Google Scholar]
- Cox GW. Alien Species and Evolution: The Evolutionary Ecology of Exotic Plants, Animals, Microbes, and Interacting Native Species. Island Press; Washington, DC: 2004. [Google Scholar]
- Cristescu M. Genetic reconstructions of invasion history. Molecular Ecology. 2015;24:2212–2225. doi: 10.1111/mec.13117. [DOI] [PubMed] [Google Scholar]
- Dennell RW. Human migration and occupation of Eurasia. Episodes. 2008;31:207–210. [Google Scholar]
- DiTomaso J, Kyser GB, Pitcairn MJ. Cal-IPC Publication 2006-03. California Invasive Species Council; Berkeley, California: 2006. Yellow starthistle management guide; p. 78. [Google Scholar]
- DiTomaso JM, Healy EA. Weeds of California and Other Western States. University of California Agriculture and Natural Resources; Oakland, California, USA: 2007. [Google Scholar]
- Dlugosch KM, Parker IM. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Molecular Ecology. 2008;17:431–449. doi: 10.1111/j.1365-294X.2007.03538.x. [DOI] [PubMed] [Google Scholar]
- Dlugosch KM, Lai Z, Bonin A, et al. Allele identification for transcriptome-based population genomics in the invasive plant Centaurea solstitialis. G3: Genes, Genomes, Genetics. 2013;3:359–367. doi: 10.1534/g3.112.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosch KM, Anderson SR, Braasch J, et al. The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Molecular Ecology. 2015a;24:2095–2111. doi: 10.1111/mec.13183. [DOI] [PubMed] [Google Scholar]
- Dlugosch KM, Cang FA, Barker BS, et al. Evolution of invasiveness through increased resource use in a vacant niche. Nature Plants. 2015b;1:15066. doi: 10.1038/nplants.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostál J. Plantaginaceae to Compositae (and Rubiaceae) In: Tutin TG, Heywood VH, Burges NA, et al., editors. Flora Europaea. Cambridge University Press; Cambridge, London, UK: 1976. pp. 254–301. [Google Scholar]
- Dray S, Dufour A-B. The ade4 package: implementing the duality diagram for ecologists. Journal of Statistical Software. 2007;22:1–20. [Google Scholar]
- Dufresnes C, Litvinchuk SN, Leuenberger J, et al. Evolutionary melting pots: a biodiversity hotspot shaped by ring diversifications around the Black Sea in the Eastern tree frog (Hyla orientalis) Molecular Ecology. 2016;25:4285–4300. doi: 10.1111/mec.13706. [DOI] [PubMed] [Google Scholar]
- Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources. 2012;4:359–361. [Google Scholar]
- Ellstrand NC, Schierenbeck KA. Hybridization as a stimulus for the evolution of invasiveness in plants? Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7043–7050. doi: 10.1073/pnas.97.13.7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen RL, Desronvil T, Hierro JL, Kesseli R. Morphological differentiation in a common garden experiment among native and non-native specimens of the invasive weed yellow starthistle (Centaurea solstitialis) Biological Invasions. 2012;14:1459–1467. [Google Scholar]
- Eriksen RL, Hierro JL, Eren Ö, et al. Dispersal pathways and genetic differentiation among worldwide populations of the invasive weed Centaurea solstitialis L. (Asteraceae) PLoS ONE. 2014;9:e114786. doi: 10.1371/journal.pone.0114786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estoup A, Lombaert E, Marin J-M, et al. Estimation of demo-genetic model probabilities with Approximate Bayesian Computation using linear discriminant analysis on summary statistics. Molecular Ecology Resources. 2012;12:846–855. doi: 10.1111/j.1755-0998.2012.03153.x. [DOI] [PubMed] [Google Scholar]
- Etter PD, Preston JL, Bassham S, et al. Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS ONE. 2011;6:e18561. doi: 10.1371/journal.pone.0018561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annual Review of Ecology, Evolution, and Systematics. 2009;40:481–501. [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics. 2008;180:977–993. doi: 10.1534/genetics.108.092221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Jacas N, Uysal T, Romashchenko K, et al. Centaurea revisited: a molecular survey of the Jacea group. Annals of Botany. 2006;98:741–753. doi: 10.1093/aob/mcl157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick RC, Bonatelli IAS, Hyseni C, et al. The evolution of phylogeographic data sets. Molecular Ecology. 2015;24:1164–1171. doi: 10.1111/mec.13108. [DOI] [PubMed] [Google Scholar]
- Gerlach JD., Jr How the west was lost: reconstructing the invasion dynamics of yellow starthistle and other plant invaders of western rangelands and natural areas. Proceedings of the California Exotic Pest Plant Council. 1997;3:67–72. [Google Scholar]
- Graebner RC, Callaway RM, Montesinos D. Invasive species grows faster, competes better, and shows greater evolution toward increased seed size and growth than exotic non-invasive congeners. Plant Ecology. 2012;213:545–553. [Google Scholar]
- Hahn MA, Buckley YM, Müller-Schärer H. Increased population growth rate in invasive polyploid Centaurea stoebe in a common garden. Ecology Letters. 2012;15:947–954. doi: 10.1111/j.1461-0248.2012.01813.x. [DOI] [PubMed] [Google Scholar]
- Hierro JL, Eren Ö, Villarreal D, Chiuffo MC. Non-native conditions favor non-native populations of invasive plant: demographic consequences of seed size variation? Oikos. 2012;122:583–590. [Google Scholar]
- Hokkanen HMT, Pimentel P. New associations in biological control: theory and practice. The Canadian Entomologist. 1989;121:829–840. [Google Scholar]
- Hovick M, Whitney KD. Hybridisation is associated with increased fecundity and size in invasive taxa: meta-analytic support for the hybridisation-invasion hypothesis. Ecology Letters. 2014;17:1464–1477. doi: 10.1111/ele.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JT. Distributional data on weedy thistles in western North America. Leaflets of Western Botany. 1959;9:17–32. [Google Scholar]
- Hufbauer RA. Biological invasions: paradox lost and paradise gained. Current Biology. 2008;18:R246–R247. doi: 10.1016/j.cub.2008.01.038. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T, Ahmed I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski ST. HP-RARE: A computer program for performing rarefaction on measures of allelic richness. Molecular Ecology Notes. 2005;5:187–189. [Google Scholar]
- Kalinowski ST. The computer program STRUCTURE does not reliably identify the main genetic clusters within species: simulations and implications for human population structure. Heredity. 2011;106:652–632. doi: 10.1038/hdy.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapli P, Botoni D, Ilgaz C, et al. Molecular phylogeny and historical biogeography of the Anatolian lizard Apathya (Squamata, Lacertidae) Molecular Phylogenetics and Evolution. 2013;66:992–1001. doi: 10.1016/j.ympev.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Koskinen MT, Haugen TO, Primmer CR. Contemporary fisherian life-history evolution in small salmonid populations. Nature. 2002;419:826–830. doi: 10.1038/nature01029. [DOI] [PubMed] [Google Scholar]
- Kolbe JJ, Glor RE, Schettino LRG, et al. Genetic variation increases during biological invasion by a Cuban lizard. Nature. 2004;431:177–181. doi: 10.1038/nature02807. [DOI] [PubMed] [Google Scholar]
- Kuester A, Chang S-M, Baucom RS. The geographic mosaic of herbicide resistance evolution in the common morning glory, Ipomoea purpurea: evidence for resistance hotspots and low genetic differentiation across the landscape. Evolutionary Applications. 2015;8:821–833. doi: 10.1111/eva.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T, Hayashizaki Y, Daub CO. TagDust–a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CE. Evolutionary genetics of invasive species. Trends in Ecology & Evolution. 2002;17:386–391. [Google Scholar]
- Lombaert E, Guillemaud T, Cornuet J-M, et al. Bridgehead effect in the worldwide invasion of the biocontrol harlequin ladybird. PLoS ONE. 2010;5:e9743. doi: 10.1371/journal.pone.0009743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombaert E, Guillemaud T, Lundgren J, et al. Complementarity of statistical treatments to reconstruct worldwide routes of invasion: the case of the Asian ladybird Harmonia axyridis. Molecular Ecology. 2014;23:5979–5997. doi: 10.1111/mec.12989. [DOI] [PubMed] [Google Scholar]
- Maddox DM, Mayfield A. Yellow starthistle infestations are on the increase. California Agriculture. 1985;40:10–13. [Google Scholar]
- Maddox DM, Mayfield A, Poritz NH. Distribution of yellow starthistle (Centaurea solstitialis) and Russian knapweed (Centaurea repens) Weed Science. 1985;33:315–327. [Google Scholar]
- Médail F, Diadema K. Glacial refugia influence plant diversity patterns in the Mediterranean Basin. Journal of Biogeography. 2009;36:1333–1345. [Google Scholar]
- Mráz P, Garcia-Jacas N, Gex-Fabry E, et al. Allopolyploid origin of highly invasive Centaurea stoebe s.l. (Asteraceae) Molecular Phylogenetics and Evolution. 2012;62:612–623. doi: 10.1016/j.ympev.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Neophytou C. Bayesian clustering analyses for genetic assignment and study of hybridization in oaks: effects of asymmetric phylogenies and asymmetric sampling schemes. Tree Genetics and Genomes. 2014;10:273–285. [Google Scholar]
- Paynter Q, Overton JM, Hill RL, et al. Plant traits predict the success of weed biocontrol. Journal of Applied Ecology. 2012;49:1140–1148. [Google Scholar]
- Peterson BK, Weber JN, Kay EH, et al. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcairn MJ, Villegas B, Woods DM, et al. Evaluating implementation success for seven seed head insects on Centaurea solstitialis in California. USA: 2008. pp. 610–616. [Google Scholar]
- Prentis PJ, Wilson JRU, Dormontt EE, et al. Adaptive evolution in invasive species. Trends in Plant Science. 2008;13:288–294. doi: 10.1016/j.tplants.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. [Google Scholar]
- Ramakrishnan AP, Musial T, Cruzan MB. Shifting dispersal modes at an expanding species’ range margin. Molecular Ecology. 2010;19:1134–1146. doi: 10.1111/j.1365-294X.2010.04543.x. [DOI] [PubMed] [Google Scholar]
- Roché BF., Jr . PhD dissertation. University of Idaho; Moscow, Idaho: 1965. Ecological studies of yellow starthistle (Centaurea solstitialis L.) [Google Scholar]
- Roché BF, Jr, Talbott CJ. Agricultural Research Center Bulletin XB 0978. Agriculture Research Center, College of Agriculture and Home Economics, Washington State University; Pullman, Washington: 1986. The collection history of Centaureas found in Washington state; p. 36. [Google Scholar]
- Roché CT, Susanna A. New habitats, new menaces: Centaurea × kleinii (C. moncktonii × C. solstitialis), a new hybrid species between two alien weeds. Collectanea Botanica. 2011;29:17–23. [Google Scholar]
- Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Molecular Ecology Notes. 2004;4:137–138. [Google Scholar]
- Rosenthal DM, Ramakrishnan AP, Cruzan MB. Evidence for multiple sources of invasion and intraspecific hybridization in Brachypodium sylvaticum (Hudson) Beauv. in North America. Molecular Ecology. 2008;17:4657–4669. doi: 10.1111/j.1365-294X.2008.03844.x. [DOI] [PubMed] [Google Scholar]
- Sabba RP, Ray IM, Lownds N, Sterling TM. Inheritance of resistance to clopyralid and picloram in yellow starthistle (Centaurea solstitialis L.) is controlled by a single nuclear recessive gene. Journal of Heredity. 2003;94:523–527. doi: 10.1093/jhered/esg101. [DOI] [PubMed] [Google Scholar]
- Stewart G. Alfalfa growing in the United States and Canada. MacMillan Co; New York: 1926. [Google Scholar]
- Suárez-Santiago VN, Salinas MJ, Garcia-Jacas N, et al. Reticulate evolution in the Acrolophus subgroup (Centaurea L., Compositae) from the western Mediterranean: origin and diversification of section Willkommia Blanca. Molecular Phylogenetics and Evolution. 2007;43:156–172. doi: 10.1016/j.ympev.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Stöck M, Dufresnes C, Litvinchuk SN, et al. Cryptic diversity among Western Palearctic tree frogs: postglacial range expansion, range limits, and secondary contacts of three European tree frog lineages (Hyla arborea group) Molecular Phylogenetics and Evolution. 2012;65:1–9. doi: 10.1016/j.ympev.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Sun M. Population genetic structure of yellow starthistle (Centaurea solstitialis), a colonizing weed in the western United States. Canadian Journal of Botany. 1997;75:1470–1478. [Google Scholar]
- Swope SM, Parker IM. Widespread seed limitation affects plant density but not population trajectory in the invasive plant Centaurea solstitialis. Oecologia. 2010;164:117–128. doi: 10.1007/s00442-010-1641-1. [DOI] [PubMed] [Google Scholar]
- Thompson JN. Rapid evolution as an ecological process. Trends in Ecology and Evolution. 1998;13:329–332. doi: 10.1016/s0169-5347(98)01378-0. [DOI] [PubMed] [Google Scholar]
- Vähä J-P, Primmer CR. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Molecular Ecology. 2006;15:63–72. doi: 10.1111/j.1365-294X.2005.02773.x. [DOI] [PubMed] [Google Scholar]
- Wagenitz G. Pollenmorphologie und systematik in der gattung Centaurea L. s.1. Flora. 1955;142:213–279. [Google Scholar]
- Wagenitz G. Centaurea L. In: Davis PH, editor. Flora of Turkey and the East Aegean Islands. Edinburgh University Press; Edinburgh, Scotland: 1975. pp. 465–585. [Google Scholar]
- Waterbolk HT. The spread of food production over the European continent. In: Sjøvold T, editor. Symp Introduksjonen av jordbruk i Norden. Universitetsforlaget; Oslo, Norway: 1982. pp. 19–37. [Google Scholar]
- Webb DM, Knapp SJ. DNA lysis from a previously recalcitrant plant genus. Plant Molecular Biology Reporter. 1990;8:180–185. [Google Scholar]
- Widmer TL, Guermache F, Dolgovskaia MY, Reznick SY. Enhanced growth and seed properties in introduced vs. native populations of yellow starthistle (Centaurea solstitialis) Weed Science. 2007;55:465–473. [Google Scholar]
- Wilson JRU, Dormontt EE, Prentis PJ, et al. Something in the way you move: dispersal pathways affect invasion success. Trends in Ecology and Evolution. 2009;24:136–144. doi: 10.1016/j.tree.2008.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Details regarding evolutionary scenarios, demographic parameters, and confidence in scenario choice in DIYABC.
Table S1 Sampling sites for seeds of yellow starthistle (Centaurea solstitialis) and related taxa, including sampling site information (nearest city, country/state, continent, latitude, and longitude), regional groups as defined for the ABC and trait differentiation analyses, the number of individuals sequenced for this study (N), the number of individuals included in analyses (n), species (CESO = C. solstitialis, CENE = C. nicaeensis, CEPA = C. pallescens, CEME = C. melitensis), and year of collection. Site numbers correspond to those depicted in Fig. S1, Supporting information.
Table S2 Prior distributions for effective population size (Ne) and divergence time parameters used for two successive steps of ABC analyses of yellow starthistle populations (performed in DIYABC 2.0.4). The first analysis (Eurasia) tested among four evolutionary scenarios for populations in Eurasia, whereas the second analysis (Eurasia and Americas) incorporated the most highly supported scenario from the first analysis to test among seven competing evolutionary scenarios regarding the introduction history of yellow starthistle to California. We used uniform prior distributions for effective population size and divergence time parameters. Divergence time is in generations, with a generation length of one year. We used the ‘set condition’ option to impose the rule that t1 > t2, t1 ≥ t3, and t1 > tAdm1 for the first analysis, and the rule that t1 > tAdm1, t1 > t4, t4 ≥ t5, tAdm1 > t4, tAdm1 > t5, t4 ≥ tAdm2, and tAdm1 ≥ tAdm2 for the second analysis. WE = western European; EE = eastern European; AS= Asian; CH = Chilean; CA = Californian.
Table S3 Posterior probability estimates (95% confidence interval) for evolutionary scenarios tested for yellow starthistle populations in Eurasia, and for populations in both Eurasia and the Americas, based on logistic regression on the 1% of simulated data sets closest to the empirical data (performed in DIYABC 2.0.4).
Table S4 Median estimates with 5% and 95% quantiles for posterior distributions of effective population size (Ne) and divergence time parameters for the best-fit scenario of ABC analyses of the analysis of Eurasian populations (Scenario 4), and of the best-fit scenario of ABC analyses of the analysis of Eurasian and American populations (Scenario 4.1). WE = western European; EE = eastern European; AS= Asian; CH = Chilean; CA = Californian.
Table S5 Pairwise divergence matrices used in the Mantel test. a) Pairwise FST values and b) pairwise differences in Least Squares Means in a ln-transformed size index were calculated for populations in California (CA), Chile (CH), western Europe (WE), eastern Europe (EE), and Asia (AS). All FST values have P-values of < 0.01.
Table S6 Least Squares Means (LSM) and standard errors (SE) in a ln-transformed size index were calculated for yellow starthistle populations in California (CA) coast, CA interior, Chile (CH), western Europe (WE), eastern Europe (EE), and Asia (AS), from data in Dlugosch et al. 2015b.
Fig. S1 Sampling sites of the genotyped yellow starthistle in Eurasia, the western United States, and South America are indicated by black circles. Sampling sites of the genotyped individuals of Centaurea nicaeensis, C. pallescens, and C. melitensis are indicated by black triangles, black squares, and black stars, respectively. Known introduction dates of yellow starthistle to regions of South America and the western US are indicated. site numbers correspond to those presented in Table S1, Supporting information.
Fig. S2 Boxplots of the inbreeding coefficient (FIS) based on RAD loci in individuals in the “FULL” data set (white) and “CORE” data set (gray) at different -M and -n values in STACKS, where -M is the number of nucleotide differences allowed between loci when processing a single individual and -n is the number of nucleotide differences allowed between loci when creating the catalog of loci. The RAD loci were present in Centaurea melitensis, C. pallescens, and C. nicaeensis, and each of the two yellow starthistle lineages (p = 5), genotyped in at least 60% of the individuals in each species/lineage (r = 0.6), and had a minimum coverage depth of 10. Box plots show median (horizontal line), interquartile range (box), and minimum and maximum values (whiskers). Positive values for FIS may indicate excess homozygosity due to failure to cluster loci under restrictive mismatch values, whereas negative values for FIS may indicate excess heterozygosity due to clustering of paralogous loci.
Fig. S3 Results from STRUCTURE analyses and a BAPS admixture analysis based on 747 single nucleotide polymorphism (SNP) loci of 591 yellow starthistle individuals from across the entire sampled range (Eurasia, South America, and the western US; i.e. the “FULL” data set). (a) STRUCTURE bar plots of assignment probabilities averaged across ten runs (calculated by CLUMPP) are shown for K = 2 to K = 5, where K is the number of genetic clusters. (b) BAPS bar plots are shown for the best estimate of K (K = 4). Each vertical bar shows the proportional representation of the estimated cluster membership for a single individual.
Fig. S4 The mean log probability of the data (ln Pr(X | K)) plus or minus the standard deviation for each value of K from 1 to 10 (black squares), and the ΔK statistic (gray squares), where K is the number of genetic clusters, are depicted for ten replicate STRUCTURE analyses of the (a) “FULL” and (b) “CORE” data set for yellow starthistle, and of (c) the “OUTGROUPS” data set that includes yellow starthistle and three other Centaurea species.
Fig. S5 The highest value of average pairwise similarity (H’) calculated by CLUMPP for each value of K from 1 to 10, where K is the number of genetic clusters, are depicted for ten replicate STRUCTURE analyses of the (a) “FULL” and (b) “CORE” data set for yellow starthistle, and of (c) the “OUTGROUPS” data set that includes yellow starthistle and three other Centaurea species.
Fig. S6 Results from STRUCTURE and BAPS analyses of 440 SNPs of the two lineages of yellow starthistle and three outgroup species (Centaurea nicaeensis, C. melitensis, and C. pallescens). (a) Bar plots of assignment probabilities averaged across ten STRUCTURE runs (calculated by CLUMPP) are shown for K = 2–6, where K is the number of genetic clusters. (b) BAPS bar plots are shown for the best estimate of K (K = 6). Each vertical bar shows the proportional representation of the estimated cluster membership for a single individual.
Fig. S7 Results of a principal component analysis conducted for model-checking of the best-fit scenario of ABC analyses of (a) Eurasian populations (Scenario 4) and of (b) both Eurasian and American populations (Scenario 4.1), performed in DIYABC 2.0.4. The observed data, data sets simulated with the prior distributions of parameters, and data sets simulated from the posterior sample are represented on each plot.