

Abstract
Human cytomegalovirus DNA polymerase comprises a catalytic subunit, UL54, and an accessory subunit, UL44, the interaction of which may serve as a target for the development of new antiviral drugs. Using a high-throughput screen, we identified a small molecule, (5-((dimethylamino)-methylene-3-(methylthio)-6,7-dihydrobenzo[c]thiophen-4(5H)-one), that selectively inhibits the interaction of UL44 with a UL54-derived peptide in a time-dependent manner, full-length UL54, and UL44-dependent long-chain DNA synthesis. A crystal structure of the compound bound to UL44 revealed a covalent reaction with lysine residue 60 and additional noncovalent interactions that cause steric conflicts that would prevent the UL44 connector loop from interacting with UL54. Analyses of the reaction of the compound with model substrates supported a resonance-stabilized conjugation mechanism, and substitution of the lysine reduced the ability of the compound to inhibit UL44–UL54 peptide interactions. This novel covalent inhibitor of polymerase subunit interactions may serve as a starting point for new, needed drugs to treat human cytomegalovirus infections.
Keywords: human cytomegalovirus, DNA polymerase, protein—protein interaction, covalent inhibitor, SGM8
Graphical abstract
Protein—protein interactions (PPIs) are central to all biological processes and present a vast class of potential therapeutic targets for pharmacological intervention. Although targeting PPIs with small molecules can be challenging, considerable progress has been made in identifying PPIs that are amenable to such inhibition.1,2
We have been studying the feasibility of pharmacologically disrupting PPIs of human cytomegalovirus (HCMV). HCMV rarely causes disease in immunocompetent individuals, but in neonates, who are immune-naïve, and immunocompromised patients, HCMV can cause serious diseases.3 Although currently approved drugs can successfully treat certain HCMV diseases, limited efficacy, toxicity, and the emergence of drug resistance limit their use.4 Thus, better anti-HCMV agents active against mutants resistant to current drugs are needed.
An important HCMV PPI is that between the viral DNA polymerase catalytic subunit, UL54,5 and an accessory subunit, UL44, which stimulates long-chain DNA synthesis, presumably by increasing processivity.6,7 There has been considerable interest in compounds that inhibit PPIs between polymerase catalytic subunits and processivity factors (e.g., refs 8–11), highlighted by the recent discovery that griselimycin inhibits Mycobacterium tuberculosis by this mechanism.10 Both UL54 and UL44 are essential for HCMV DNA replication, and their interaction is well-defined.12–18 The crystal structure of UL44 bound to a peptide corresponding to the C-terminal 22 amino acids of UL54 (P54) reveals intersubunit contacts including a crevice on UL44, into which hydrophobic residues of UL54 insert, and the UL44 “connector loop” (defined below), with which UL54 extensively hydrogen bonds.15 Importantly, single substitutions of residues on the interaction interface can dramatically impair the PPI and long-chain DNA synthesis,16,19 supporting this PPI as a potential drug target. Inhibitors of this PPI should be active against viruses resistant to current anti-HCMV drugs.
Previously, a screen of ~50,000 compounds in five libraries identified small molecules that specifically disrupt the UL54− UL44 PPI in vitro and inhibit HCMV replication in cell culture.9 However, the compounds identified had less than ideal properties for further development, with structures that include three or more fused aromatic rings (the structure provided by the supplier of one of these compounds (AL21) did not have three such rings, but further analysis showed that the compound did). Medicinal chemistry efforts to improve the properties of these compounds did not succeed.
To try to identify starting points for more “drug-like” compounds that target the UL54−UL44 PPI, we screened 45 other libraries with a total of 213,457 small molecules or extracts at the Harvard Institute of Chemistry and Cell Biology—Longwood (ICCB-L), utilizing our previously reported fluorescence polarization (FP) assay.8,9 The initial screen assayed the interaction of fluorescently labeled P54 and glutathione S-transferase (GST) fused UL44ΔC290 (herein referred to as UL44). Hits from this initial screen (414 compounds) were rescreened and subjected to two counter-screens to assess selectivity. As UL44 displays a fold strikingly similar to other processivity factors,20 such as UL42, the herpes simplex virus-1 (HSV-l) processivity subunit,21 and proliferating cell nuclear antigen (PCNA), the eukaryotic processivity factor,22 we tested the hits for inhibition of the interaction of HSV-l UL42and fluorescently labeled P30, a peptide corresponding to 36 residues of HSV-1 polymerase UL30,23 and the interaction of human PCNA and fluorescently labeled P21, a 22 residue peptide derived from the cell-cycle checkpoint protein p21,22 in FP assays similar to the UL44−P54 assay. These assays, which entailed prolonged incubation of the components prior to assay, identified 23 compounds including a small molecule, (5-((dimethylamino)methylene-3-(methyl-thio)-6,7-dihydrobenzo[c]thiophen-4(5H)-one), that we called SGM8 (Figure 1a; structure, purity, and stability verified by NMR, HRMS, and LC-MS (Figure S1 and Supporting Information (SI) text). Despite its expected reactivity, we were intrigued by its apparent selectivity and relatively small size.
Figure 1.
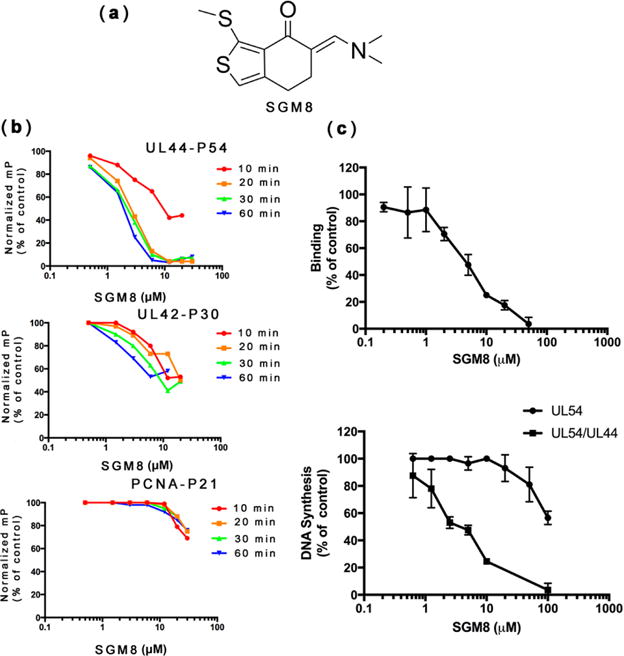
(a) Chemical structure of SGM8. (b) Activity of SGM8 against the interactions of UL44−P54, UL42−P30, and PCNA−P21 in FP assays. The normalized mP at each concentration of SGM8 relative to the control without inhibitor was plotted versus the concentration of SGM8. FP was assayed following pre-incubation for the times indicated in each panel. (c) (Top) SGM8 inhibition of binding of UL44 to full-length UL54, as analyzed by biolayer interferometry; (bottom) SGM8 inhibition of long-chain DNA synthesis mediated by UL54 in the presence of UL44 (UL54/UL44, squares; see Figure S2) or DNA synthesis by UL54 alone in a filter-binding assay (circles), relative to the signal in the absence of compound. Error bars show standard errors of the mean.
While performing dose—response studies, we found that when we incubated samples at room temperature for 10 min prior to measurement, SGM8 only weakly inhibited the P54−UL44 interaction, with an IC50 ~ 20 μM, similar to that for the P30−UL42 interaction (Figure 1b, top and middle). However, when we extended the incubation time to 20 min or beyond, SGM8 inhibited the P54−UL44 interaction with an IC50 of 2 μM(Figure 1b, top). Extending the incubation time to 60 min only slightly increased SGM8 inhibition of the P30−UL42 interaction (Figure 1b, middle) and did not meaningfully change the very weak inhibition by SGM8 of the P21−PCNA interaction (Figure 1b, bottom). Thus, SGM8 selectively inhibits the P54−UL44 interaction in a time-dependent manner.
We also found that SGM8 inhibits the interaction of UL44 with full-length HCMV GST-UL54 (herein referred to as UL54) with an IC50 of 4.5 μM in a biolayer interferometry assay (Figure 1c, top). We then tested whether SGM8 inhibits long-chain DNA synthesis by UL54−UL44,16,19 using a poly(dA)-oligo(dT) primer-template. As previously described, we observed only short-chain DNA synthesis with UL54 alone (Figure S2, lane 1), but formation of long DNA products with UL54 and UL44 (Figure S2, lane 2) that could be inhibited by P54 (Figure S2, lanes 3 and 4). SGM8 inhibited long-chain DNA synthesis in a dose-dependent manner (Figure S2, lanes 5–10; Figure 1c, bottom), with an IC50 of 3.2 μM, similar to that observed in the FP and biolayer interferometry assays. However, SGM8 exhibited much less potent inhibition (IC50 > 100 μM; Figure 1c, bottom) of DNA synthesis by UL54 alone, using the same primer-template in a filter binding assay.18 Thus, SGM8 inhibition was selective for long-chain DNA synthesis mediated by UL44.
We prepared crystals of UL44 at neutral pH in the C2221 space group (the space group of the crystals used in the structure of the UL44−P54 complex15), soaked them with SGM8, and determined the crystal structure of the complex (PDB code: 5IWD) to 2.56 Å resolution (Rwork/Rfree, 0.2125/0.2670) by molecular replacement (Table S1), initially based on the low-pH crystal form of the UL44−P54 complex structure.15
UL44 is a head-to-head dimer.20 Each monomer contains two topologically similar domains (N-terminal and C-terminal) linked by a connector loop (Figure S3A), with a “front” face (including the connector loop) that interacts with UL54.20 The structure of the complex revealed a strong covalent SGM8 binding site that includes residue K60 on the N-terminal domain below the connector loop and two weaker sites at each end of the monomer, one that includes K192 and one that includes K4 (Figure S3A). Interestingly, there are 12 other lysines visible in the structure, of which at least 10 are solvent-exposed and completely accessible. Five of these 10 lie close to hydrophobic residues. The three SGM8 substitution sites are each close to hydrophobic crevices. The weaker sites share a hydrophobic crevice created by crystal packing contacts and the hydrophobic portions of the two SGM8 molecules; thus, this hydrophobic crevice would not be expected to form in solution. Density corresponding to SGM8 at the strong site was readily visualized (Figure S3B). Notably, the density corresponding to SGM8 no longer fit the original compound, but instead fit a rearranged form of SGM8 in which the dimethylamino group is lost, and the compound forms a covalent adduct on the side chain of K60 (Figure S3B). The SGM8 adduct extends from K60 into a hydrophobic slot between the connector loop and a β sheet, making numerous noncovalent interactions (Figure 2a, stereoview in Figure S3C). Interestingly, there are no contacts between SGM8 and residues in or near the cleft of UL44 (e.g., V136 and F266 (Figure 2a and Figure S3C)) into which certain hydrophobic residues of UL54 insert.15 Instead, the secondary amino group of the covalent adduct hydrogen bonds with residue Q51, while the 3-(methylthio)-6,7-dihydrobenzo[c]-thiophen-4-one moiety makes numerous hydrophobic contacts with UL44 residues on both sides of the slot, including contacts with the main chain and side chain of I135 of the connector loop and weak but extensive contacts with the side chains of T41, L43, I49, and T79 on the β-sheet (Figure 2a and Figure S3C). These noncovalent contacts likely contribute to binding affinity and help position SGM8 for reaction with the K60 side chain.
Figure 2.
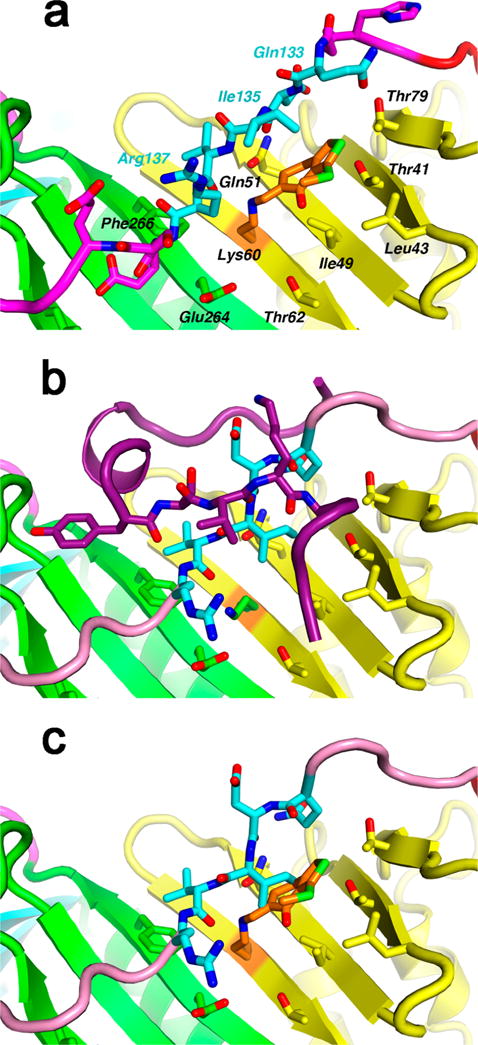
How SGM8 interacts with UL44 and interferes with P54 binding. (a) SGM8 covalently bonds with UL44 residue K60 (orange) and noncovalently interacts with UL44 in a hydrophobic slot, making contacts with residues 133–137 of the connector loop (cyan) and with side chains from the central β sheet (yellow and green). In this ribbon representation, important contacts are shown as stick models and are labeled. The Ile135 side chain extends toward the viewer. (Stereoview in Figure S3C.) (b) Binding of P54 (dark purple) to UL44 (PDB entry: 1YYP),15 includes β-sheet hydrogen bonding with the connector loop (cyan and pink). Residues 133–137 (cyan) are oriented differently from panel a, with the Ile135 side chain extending laterally. (c) In this modeling exercise, the peptide-binding connector loop (from 1YYP) replaces residues 128–143 in the SGM8−UL44 complex. A steric clash between SGM8 and Ile135 prevents the connector loop from adopting its peptide-bound conformation.
The contacts of SGM8 with I135 were particularly interesting because substitution of that residue with alanine (I135A) ablates interaction with HCMV UL54.19 In the structure of UL44 bound to P54 (Figure 2b), I135 makes extensive interactions with the β-sheet below the connector loop, particularly with residues that interact with SGM8 in the present structure, to anchor the connector loop so that it can form a hydrogen-bonding network between the connector loop and the Pol peptide P54.15 That connector loop conformation differs substantially from that when SGM8 is bound (compare Figure 2a,b). Indeed, as illustrated in Figure 2c, which superimposes the position of SGM8 from Figure 2a with the position of the connector loop bound to P54 in Figure 2b, the connector loop would clash with the covalently bound compound. This explains how SGM8 inhibits P54 binding to UL44.
We also solved the structure of unliganded UL44 at neutral pH (PDB code: 5IXA) from crystals in space group P212121 (obtained under slightly different conditions) to 2.7 Å resolution, and, in that structure, the connector loop is not prohibited from interacting with P54 (Figure S4 and accompanying SI text).
To investigate how SGM8 can form covalent bonds with lysines, we performed reactions using model substrates. In the first reaction, we incubated SGM8 either by itself or with pentylamine, which mimics the side chain of lysine, at the same pH (pH 7.5) used for crystallography. We used LC-MS to monitor this reaction (Figure 3a). When the reaction contained only SGM8, we detected a main peak at 10.2 min with MW 227, lower than what we expected for SGM8 (MW 253). This peak corresponds to a derivative of SGM8 that we term SGM8A, the identity of which we confirmed using NMR (Figure S5). SGM8A would be generated by reaction of SGM8 with water under acidic conditions during elution with 0.1% trifluoroacetic acid in acetonitrile by LC-MS (Figure 3a). Addition of pentylamine to the reaction generated a new peak at 11.2 min with MW 296 (Figure 3a), consistent with the MW expected if SGM8 reacted as an enamine with pentylamine (structure confirmed by NMR (Figure S6)), generating dimethylamine as the other product. We then investigated whether SGM8 can react with a lysine within a peptide, again at pH 7.5. MALDI-TOF/TOF MS revealed a new species with a mass 208 Da greater than that of the peptide (Figure 3b, top), consistent with covalent addition of SGM8 to the peptide, and loss of the dimethylamino group. MALDI-MS/MS analysis localized the covalent adduct to the lysine residue in the peptide (Figure 3b, bottom). These results support a mechanism in which SGM8 acts as an enamine to form a covalent bond with K60 on UL44 via a resonance-stabilized mechanism.
Figure 3.
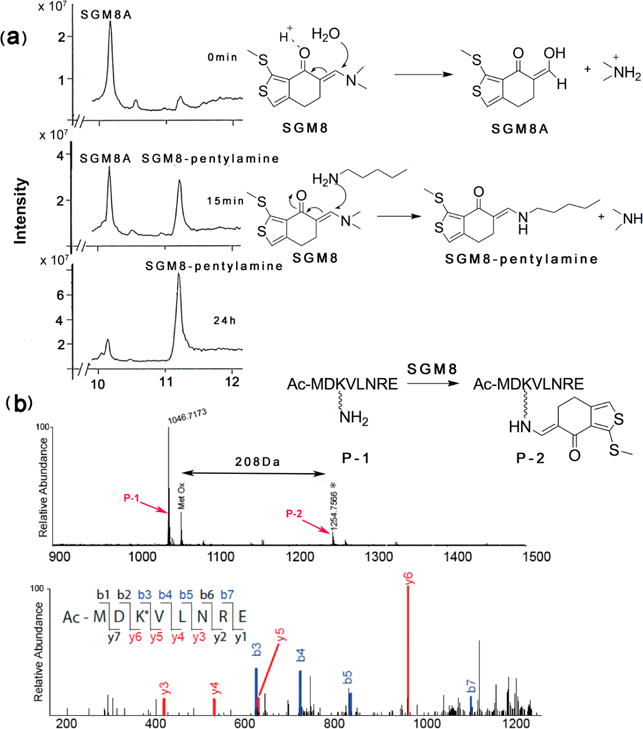
Reactivity of SGM8 with model substrates. (a) (Left) LC-MS analysis of the reaction of SGM8 with pentylamine following incubation for 0 min, 15 min, and 24 h; (right) proposed reaction mechanism for the reaction converting SGM8 to the species eluting at 10.2 min (SGM8A) and below it the proposed reaction mechanism for the reaction between SGM8 and pentylamine to produce the species eluting at 11.2 min (SGM8-pentylamine). (b) MALDI-TOF analysis of the reaction of peptide P-1 with SGM8. (Top) Peaks at 1047 and 1255 Da (*) were assigned to unreacted P-1 and SGM8-conjugated peptide P-2 (upper panel). Proposed reaction is shown in the inset. The peak labeled Met Ox corresponds to unreacted P-1 with oxidized methionine. (Bottom) MALDI-MS/MS spectrum of labeled peptide P-2 (lower panel). Ions of types b and y are shown in blue and red, respectively. The difference between y5 and y6 is 336 Da, corresponding to the mass of a lysine residue conjugated to SGM8 with loss of the dimethylamino group.
A prediction of this proposed mechanism is that substitution of K60 with alanine would greatly reduce SGM8 activity. This substitution had little or no effect on the interaction of UL44 with P54 (Figure S7). However, as predicted, the interaction of the mutant protein with P54 was substantially less sensitive to SGM8 than was the wild type UL44−P54 interaction, with <50% inhibition observed even at 20 μM SGM8 following 60 min of incubation (Figure 4; compare to Figure 1b, top). Thus, K60 is important for SGM8 inhibition of the interaction, most likely because SGM8 covalently bonds to this residue.
Figure 4.
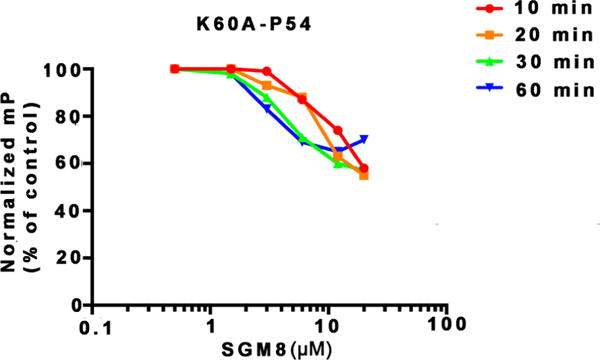
Activity of SGM8 against the interaction of mutant UL44 K60A with P54 in FP assays. FP was assayed following the different pre-incubation times indicated.
In summary, we identified a novel small compound, SGM8, which, despite its reactivity, selectively inhibits the interaction of subunits of HCMV DNA polymerase via covalent modification of K60 of the UL44 subunit. In particular, SGM8 selectively inhibits the UL44−P54 interaction relative to other processivity subunit—peptide interactions, selectively inhibits long-chain DNA synthesis mediated by UL44 relative to short-chain DNA synthesis by UL54 alone, and selectively modifies 1 lysine out of at least 13 surface-exposed lysines. That lysine is normally adjacent to a hydrophobic slot into which SGM8 can bind with reasonable affinity. As noted, SGM8 binding does not directly block UL54 from interacting with the UL44 hydrophobic crevice and connector loop. Rather, compound binding acts allosterically, clashing with the UL44 connector loop to prevent its anchoring, thereby disrupting hydrogen bonding of the connector loop to UL54. Covalent bond formation with K60 likely accounts for the time-dependent inhibition of the interaction, almost certainly increases binding affinity substantially, and, as there are no lysines in similar positions in UL42 and PCNA, may impart selectivity.
To our knowledge, there have been very few examples of allosteric inhibitors that inhibit PPIs via covalent bond formation24 and no covalent inhibitors of polymerase subunit interactions. Aside from the substantial interest in inhibitors of polymerase subunit interactions as antibacterial and antiviral drugs,8–11 there is renewed interest in covalent modifiers as drugs or as starting points for drug design.25 In this regard, it is interesting that SGM8 targets a lysine rather than a cysteine.26 We screened SGM8 for anti-HCMV activity and cytotoxicity and found similar potencies in both assays (Table S2), likely due to reactivity with cellular targets. Development of more potent and selective inhibitors may permit studies of covalent modification of UL44 in HCMV-infected cells. Whether or not advantages of covalent modification in terms of high affinity and prolonged inhibition could outweigh disadvantages in terms of time dependence of inhibition and potential off-target effects including haptenization, given its relatively small size and the opportunities to use structure-based methods to design additional contacts (possibly resulting in high affinity non-covalent inhibitors), SGM8 may provide an interesting starting point for the discovery of new drugs to treat HCMV infections.
METHODS
FP Assays and High-Throughput Screening
FP assays of the interaction of UL44 with P5418 and of UL42 with P3023 were used for high-throughput screening as described previously9 of small molecules of the libraries listed in the SI. Selected compounds were also tested in a similar FP assay of the interaction of PCNA with p21 and in dose—response assays. Details regarding preparation of the proteins and peptides, and the FP assays are provided in the SI.
UL54−UL44 Interaction Assay
The effect of SGM8 on the interaction of UL44 and full-length UL54 was assessed using biolayer interferometry with a BLItz system (fortéBIO) following the manufacturer’s instructions, with modifications and details provided in the SI.
DNA Polymerase Assays
The effects of SGM8 on long-chain DNA synthesis and basal polymerase activity of UL54 in the absence of UL44 were performed as described previously16,18 with the modifications detailed in the SI.
Crystallography
One microliter of 10 mg/mL UL44 in storage buffer (20 mM Tris (pH 7.5), 500 mM NaCl, 0.1 mM EDTA, 2 mM DTT, and 5% glycerol) and 1 μL of crystallization buffer (75 mM ammonium sulfate, 50 mM HEPES (pH 7.5), and 10% PEG4K) were mixed and crystallized in hanging drops over 1.8 M NaCl. To introduce SGM8 into the crystals, crystals were soaked in crystallization buffer supplemented with 10 mM compound for 3 days at room temperature. Soaked crystals were flash-frozen in liquid nitrogen.
Diffraction data were collected at the Argonne National Laboratory APS ID24C beamline at 100 K. Data were processed with HKL200027 and merged to yield an essentially complete data set in space group C2221, extending to 2.56 Å. Initial molecular replacement phases were based on portions of the UL44ΔC290 complex with peptide P54 (PDB code: 1YYP),15 which are nearly isomorphous. Model reconstruction and refinement were performed using Phenix,28 Refmac5,29 and Coot.30 SGM8, the connector loop, and weaker density features were added during subsequent refinement cycles. Topology and parameter files for the inhibitors were generated using PRODRUG.31 Figures depicting crystal structures were prepared using PyMol (The Pymol Molecular Graphics System, version 1.3, Schrödinger, LLC).
Analyses of SGM8 Reaction with Model Substrates
SGM8 (2.5 mM) was reacted with excess pentylamine, or 20 nmol of SGM8 was reacted with 2 nmol of the lysine containing peptide shown in Figure 3. Reaction products were analyzed by LC-MS and NMR or MALDI MS, respectively, as detailed in the SI.
Antiviral and Cytotoxicity Assays
Screening for anti-HCMV activity was conducted using a 3-day assay in HFF cells that employed a virus (HCMV-pp28-LUC) which expresses luciferase under the control of a promoter that requires viral DNA synthesis for activity. Details regarding construction of the virus and the assay are provided in the SI. Cytotoxicity was assessed in HFF cells following a 3-day incubation using a WST-1 assay (Roche), according to the manufacturer’s instructions. Details of the assay are provided in the SI.
Supplementary Material
Acknowledgments
We thank Walt Massefski of the Dana-Farber Cancer Institute NMR core, Tom Wyche of the Harvard Medical School analytical chemistry core, and Jinhua Wang of Nathanael Gray’s laboratory for help in analysis of SGM8; Michael J. Eck for kindly providing a robotic system for high-throughput screening of initial crystallization conditions; Cai-Hong Yun for assistance in crystallization and refinement; Nathanael Gray for advice, assistance, and a critical reading of the manuscript; and James Marvel-Coen for editorial help. We are especially grateful to personnel at ICCB-L for assistance with high-throughput screening, Robin Ross of the Biomolecule Production Core of NERCE (U54 AI057158) for production of UL44 and UL42, and Kelly Arnett at the Harvard Medical School Center for Molecular Interactions for help with BLItz. This work was supported by NIH Grant AI019838 to D.M.C. and J.M.H., a sponsored research agreement from Biota Holdings Limited to D.M.C., and the Dana-Farber Strategic Resources Initiative to J.A.M.
Footnotes
ORCIDDonald M. Coen: 0000-0002-2148-5671
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.6b00079.
Detailed experimental procedures, results regarding the structure of unliganded UL44; Tables S1 and S3 (crystallographic data and screened libraries, respectively); Table S2 (antiviral and cytoxicity data); and Figures S1–S7 (NMR spectra (S1, S5, S6), LC-MS data (S1), long-chain DNA synthesis inhibition data (S2), structural data (S3, S4), and peptide binding data (S7)) (PDF)
Author Contributions
D.M.C., J.M.H., M.S.M., G. K.-M., and S.B.F. conceived and designed the studies; H.C., M.C., S.B.F., M.S.M., G.K.-M., and S.B. acquired data; all authors interpreted data; H.C., D.J.F., and D.M.C. drafted the manuscript; and all authors critically revised the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Arkin MR, Tang YY, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin LY, Wang WR, Fang GW. Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- 3.Mocarski ES, Shenk T, Griffiths PD, Pass RF. Cytomegaloviruses. In: Knipe DM, Howley PM, editors. Fields Virology. 6th. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2013. pp. 1960–2014. [Google Scholar]
- 4.Coen DM, Richman DD. Antiviral agents. In: Knipe DM, Howley PM, editors. Fields Virology. 6th. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2013. pp. 338–373. [Google Scholar]
- 5.Nishiyama Y, Maeno K, Yoshida S. Characterization of human cytomegalovirus-induced DNA polymerase and the associated 3′-to-5′ exonuclease. Virology. 1983;124:221–231. doi: 10.1016/0042-6822(83)90339-2. [DOI] [PubMed] [Google Scholar]
- 6.Ertl PF, Powell KL. Physical and functional interaction of human cytomegalovirus DNA polymerase and its accessory protein (ICP36) expressed in insect cells. J Virol. 1992;66:4126–4133. doi: 10.1128/jvi.66.7.4126-4133.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiland KL, Oien NL, Homa F, Wathen MW. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 1994;34:191–206. doi: 10.1016/0168-1702(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 8.Pilger BD, Cui C, Coen DM. Identification of a small molecule that inhibits herpes simplex virus DNA polymerase subunit interactions and viral replication. Chem Biol. 2004;11:647–654. doi: 10.1016/j.chembiol.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Loregian A, Coen DM. Selective anti-cytomegalovirus compounds discovered by screening for inhibitors of subunit interactions of the viral polymerase. Chem Biol. 2006;13:191–200. doi: 10.1016/j.chembiol.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, Konig C, Ammerman NC, Barrio MB, Borchers K, Bordon-Pallier F, Brönstrup M, Courtemanche G, Gerkits M, Geslin M, Hammann P, Hernz DW, Hoffmann H, Kiieber S, Kohlmann M, Kurz M, Lair C, Matter H, Nuermberger E, Tyagi S, Fraisse L, Grosset JH, Lagrange S, Muller R. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science. 2015;348:1106–1112. doi: 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- 11.Georgescu RE, Yurieva O, Kim SS, Kuriyan J, Kong XP, O’Donnell M. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc Natl Acad Sci U S A. 2008;105:11116–11121. doi: 10.1073/pnas.0804754105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ripalti A, Boccuni MC, Campanini F, Landini MP. Cytomegalovirus-mediated induction of antisense mRNA expression to UL44 inhibits virus replication in an astrocytoma cell line: identification of an essential gene. J Virol. 1995;69:2047–2057. doi: 10.1128/jvi.69.4.2047-2057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D, Silva MC, Shenk T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A. 2003;100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A. 2003;100:14223–14228. doi: 10.1073/pnas.2334032100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Appleton BA, Brooks J, Loregian A, Filman DJ, Coen DM, Hogle JM. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. J Biol Chem. 2006;281:5224–5232. doi: 10.1074/jbc.M506900200. [DOI] [PubMed] [Google Scholar]
- 16.Loregian A, Appleton BA, Hogle JM, Coen DM. Residues of human cytomegalovirus DNA polymerase catalytic subunit UL54 that are necessary and sufficient for interaction with the accessory protein UL44. J Virol. 2004;78:158–167. doi: 10.1128/JVI.78.1.158-167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pari GS, Anders DG. Eleven loci encoding transacting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J Virol. 1993;67:6979–6988. doi: 10.1128/jvi.67.12.6979-6988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loregian A, Rigatti R, Murphy M, Schievano E, Palu G, Marsden HS. Inhibition of human cytomegalovirus DNA polymerase by C-terminal peptides from the UL54 subunit. J Virol. 2003;77:8336–8344. doi: 10.1128/JVI.77.15.8336-8344.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loregian A, Appleton BA, Hogle JM, Coen DM. Specific residues in the connector loop of the human cytomegalovirus DNA polymerase accessary protein UL44 are crucial for interaction with the UL54 catalytic subunit. J Virol. 2004;78:9084–9092. doi: 10.1128/JVI.78.17.9084-9092.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appleton BA, Loregian A, Filman DJ, Coen DM, Hogle JM. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol Cell. 2004;15:233–244. doi: 10.1016/j.molcel.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Zuccola HJ, Filman DJ, Coen DM, Hogle JM. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol Cell. 2000;5:267–278. doi: 10.1016/s1097-2765(00)80422-0. [DOI] [PubMed] [Google Scholar]
- 22.Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 23.Digard P, Williams KP, Hensley P, Brooks IS, Dahl CE, Coen DM. Specific inhibition of herpes simplex virus DNA polymerase by helical peptides corresponding to the subunit interface. Proc Natl Acad Sci U S A. 1995;92:1456–1460. doi: 10.1073/pnas.92.5.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorczynski MJ, Grembecka J, Zhou YP, Kong Y, Roudaia L, Douvas MG, Newman M, Bielnicka I, Baber G, Corpora T, Shi JX, Sridharan M, Lilien R, Donald BR, Speck NA, Brown ML, Bushweller JH. Allosteric inhibition of protein-protein interaction between the leukemia-associated proteins Runx1 and CBFß. Chem Biol. 2007;14:1186–1197. doi: 10.1016/j.chembiol.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 26.Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016;12:931–936. doi: 10.1038/nchembio.2174. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology: Macromolecular Crystallography, Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building in tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complex. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.