

Abstract
Nature applies enzymatic assembly lines to synthesize bioactive compounds. Inspired by such capabilities, we have developed a facile method for spatially segregating attached enzymes in a continuous-flow, vortex fluidic device (VFD). Fused Hisn-tags at the protein termini allow rapid bioconjugation and consequent purification through complexation with immobilized metal affinity chromatography (IMAC) resin. Six proteins were purified from complex cell lysates to average homogeneities of 76%. The most challenging to purify, tobacco epi-aristolochene synthase, was purified in only ten minutes from cell lysate to near homogeneity (>90%). Furthermore, this “reaction-ready” system demonstrated excellent stability during five days of continuous-flow. Towards multi-step transformations in continuous flow, proteins were arrayed as ordered zones on the reactor surface allowing segregation of catalysts. Ordering enzymes into zones opens new opportunities for continuous flow biosynthesis.
Keywords: Continuous flow, Biocatalysis, Thin film, Multi-step transformations, Protein purification
Over eons of time, organisms have evolved highly efficient biosynthetic pathways to construct complex molecules through enzymatic assembly lines.[1] Polyketide biosynthesis, for example, uses assembly lines with multiple active sites performing iterative synthetic transformations. In these pathways, mobile scaffolds typically carry the intermediate compounds between each active site, termed substrate channeling.[1f, 2] Such pathways can generate an enormous range of bioactive secondary metabolites, and their use of substrate channeling can enhance reaction efficiency and yields.[3] Multi-step continuous-flow offers an analogous, in vitro approach.[4]
Numerous APIs[5] including artemisinin,[5h] rufinamide[6] and efavirenz[7] have been elegantly synthesized in multi-step continuous-flow systems. Though relatively unexplored,[8] multi-step biosynthesis could provide two major benefits to continuous-flow syntheses. First, biologically derived starting materials, reagents and enzymes can reduce the associated environmental impacts.[9] Second, all natural products are synthesized through cassettes of enzymes linked in biosynthetic pathways analogous to continuous-flow systems; such examples could inspire in vitro implementation of nature's powerful synthetic technology.
Recently, we have applied a vortex fluidic device (VFD) to drive formation of thin films for continuous flow syntheses and process improvements, including single[10] and multi-step transformations,[11] protein folding,[12] and biocatalysis.[13] The VFD confines reagents to a thin film (with ≈250 μm thickness) through rapid rotation of an angled borosilicate reactor. VFD-mediated processing has several advantages over conventional processing including micromixing and vibrational effects,[10c, 10d] which can accelerate covalent and non-covalent bond formation.
We report a simple, efficient, and rapid approach to immobilize and purify proteins in a VFD reactor. Our initial approach to immobilize proteins for continuous flow biocatalysis applied non-specific glutaraldehyde-based crosslinking to the reactor surface.[14] Although this generated a highly efficient continuous flow system for individual proteins, the method proved to be unworkable for creating the distinct enzyme zones envisioned for enzymatic assembly lines. Specifically, the relatively slow kinetics of this attachment method prevented confinement of each enzyme to a specific zone of the reactor. Furthermore, yields of the covalently attached enzymes were around 1%, which was too low for applications requiring enzymes with low catalytic rates.
Here, this enzyme attachment problem is solved using the rapid kinetics of polyhistidine attachment to IMAC resin. The approach allows formation of distinct enzymatic zones for multi-step biocatalysis. Nearly quantitative yields of applied protein were immobilized. Furthermore, all proteins fused to a polyhistidine tag should be amenable to a ten-minute purification process from cell lysates. Such immobilized proteins could be either directly used for biocatalysis in continuous flow, as shown here, or eluted as a rapid purification process.
Telescoping protein immobilization, purification, and subsequent catalysis in a single reactor removes time consuming steps associated with biocatalysis. First, this approach does not require isolation of the pure protein before immobilization. Operating in non-clogging thin films allows the cell lysate to be applied directly to the sidewall of the reactor without centrifugation or other processing, which can save hours of processing time. An identical slurry of cell lysate immediately blocked conventional protein purification apparatus (FPLC) with cell debris. Using centrifuged and non-centrifuged cell lysate provided consistently high levels of protein immobilization and protein purity of ∼96% and >76%, respectively, demonstrating that this system is not only rapid, but also efficient (Figure 1C, S7 and S8). Lastly, VFD-mediated purification can avoid lengthy dialysis into a reaction buffer. The reactor-attached enzyme allows rapid buffer exchange by simply flowing through a new buffer. Here, the same reactor is used for protein immobilization, purification, buffer exchange, and then directly for quantification or catalysis.
Figure 1. Continuous flow protein purification and immobilization.
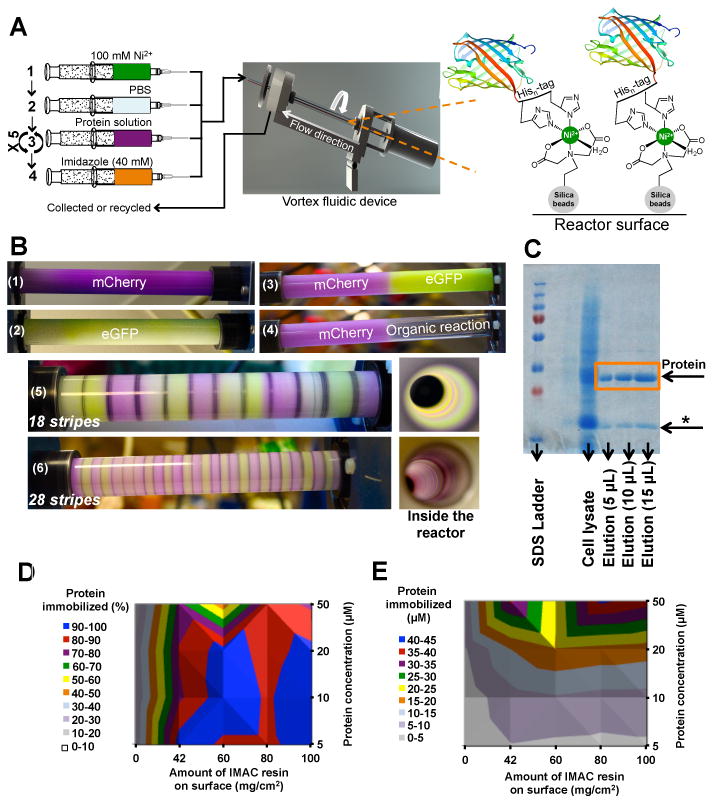
A) For thin film protein purification and immobilization, all reagents and protein solutions are fed into the reactor via peristaltic or syringe pumps (shown). The protein of interest (e.g., mCherry or eGFP) binds to the IMAC resin attached to the reactor surface as shown. B) This immobilization technique allows a range of protein zones to be created, illustrated here with mCherry (red) and eGFP (green). Possible reactor configurations include catalysis by a single enzyme (B1 and B2), half enzyme-coated plus half non-coated reactor (B3 and B4), and then finally multistep transformations with 18 or 28 stripes of enzymes (B5 and B6). C) Cell lysate can be fed directly into the IMAC resin-coated VFD reactor. The approach provides both a biosynthetic reactor for continuous flow applications, and also an effective method for protein purification. As shown by SDS-PAGE (12% acrylamide), the eluted protein (marked elution) is >90% homogeneous; in this example, an E. coli protein (indicated with a *) co-elutes with mCherry by conventional FPLC and VFD-mediated purification. D and E) These contour plots demonstrate that mCherry can reach near quantitative immobilization at various concentrations. To obtain the plotted data, solutions of mCherry (10 mL at 5, 10, 20 or 50 μM concentrations) were flowed through the reactor ten times each, and UV-Vis spectroscopy (λ = 584 nm, ε = 72000 M−1 cm−1) was used to quantify the amount of protein removed from the solution and immobilized onto the reactor surface. The protein removed from the parent solution, and immobilized onto the reactor surface is reported as either D) immobilization efficiency as a percentage of protein immobilized or E) the concentration of protein present on the reactor surface. Additional reactor optimization procedures are described in the Supplementary Information.
The model proteins mCherry and enhanced green fluorescent protein (eGFP) allowed quantification of protein immobilization during consequent purification. These fluorescent proteins allowed rapid testing of several parameters including resin and protein concentrations, purification conditions and protein stability in the reactor. Using 60 mg/cm2 of silica-based IMAC resin (100 cm2 reactor surface) resulted in high immobilization efficiencies for a wide range of protein concentrations (5-50 μM, Figure 1D and E). Furthermore, all stages of immobilization and purification are performed in continuous flow to reduce the effort required and to increase reproducibility and throughput (Figure 1A). The resultant protein coating appears uniform most likely through re-equilibration of protein concentration during the continuous flow coating process.
Optimization allows reactor construction in only ten minutes (Figure 1A and 2) with each reactor providing at least five days of stability. Painting IMAC resin onto the inner surface of the reactor is rapid and straightforward. Pipetting a homogenous solution of uncharged IMAC resin (6 mL of commercially available suspension in 20% aq. ethanol) into the rapidly rotating reactor forms a clay-like solid that adheres to the sidewall of the reactor (17.7 mm inner diameter). Removal of the buffer through thin-film room temperature evaporation leaves the IMAC resin bound to the bioreactor surface for weeks. Furthermore, hydrophobic interactions destabilize the interaction as IMAC resin fails to adhere to a hydrophobic, silanized reactor surface (treated with a solution of trichloro(octyl)silane in toluene as previously described[10d]).
Figure 2. Enzyme zones and their effects on multi-step, continuous-flow biocatalysis.
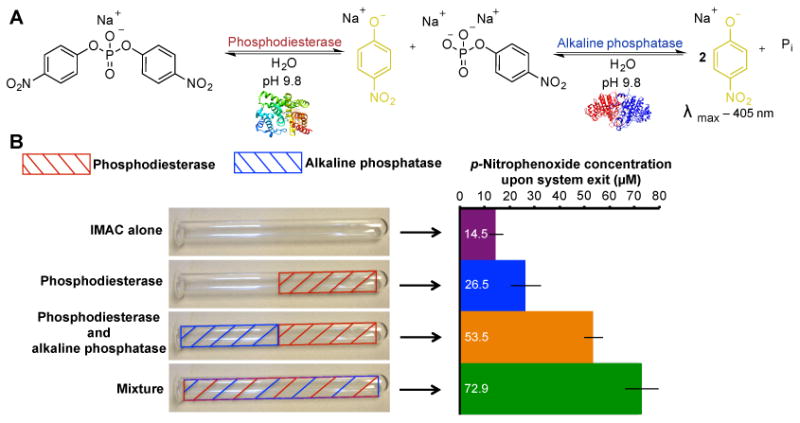
A) A two-step biosynthetic pathway was used to examine enzyme zones in multistep activity. Here, phosphodiesterase transforms the bis(p-nitrophenol)phosphate monosodium salt into p-nitrophenoxide (absorbance at 405 nm) and p-nitrophenol phosphate. The next enzyme in the pathway, alkaline phosphatase, transforms the p-nitrophenol phosphate to another molecule of p-nitrophenoxide and inorganic phosphate (Pi). Monitoring the concentration of p-nitrophenoxide liberated by the reaction determines the pathway efficiency. B) In this experiment, the reactor is divided into distinct zones, with each zone containing either phosphodiesterase (red) or alkaline phosphatase (blue). Each reactor is then tested under continuous flow conditions (0.5 mL min−1) for the quantification of p-nitrophenoxide concentration. Using a mixture of enzymes yielded 72.9 μM product (4% conversion efficiency in ≈10 min). Error is indicated as standard deviation around the mean (n=10 independent replicates).
Next, the IMAC resin is charged with Ni2+ and then the protein solution. The Ni2+solution (100 mM, 10 mL, 13.3 mL min−1, 45 s) is first flowed through the reactor using a peristaltic pump, and, to avoid interference with protein immobilization, residual Ni2+ is washed from the reactor using PBS (10 mL, 13.3 mL min−1, 45 s). Bacterial cell lysate from overexpression of polyhistidine-fused proteins is then flowed through the reactor using a peristaltic pump with the collected protein solution recycled back through the reactor four times (20 mL, 13.3 mL min−1, 450 s, Figure 1A). Finally, flowing a low concentration of imidazole (40 mM, 13.3 mL, 13.3 mL min−1, 60 s) through the reactor removes any non-specific binding. After protein purification by this continuous flow process, the protein can be eluted with a high concentration of imidazole (250 mM), and the surface-bound IMAC resin reused. Alternatively, the bioreactor with protein installed can be used directly for continuous-flow biocatalysis.
The protein bound to the reactor through the IMAC-polyhistidine complex exhibits remarkable stability. After five days of continuous flow processing with a 1.0 mL min−1 flow rate of PBS, only 0.34% of the bound protein was leached away from the reactor surface. This percentage was determined through analysis of both the flow-through and the quantities of protein retained on the reactor (Figure S12, Supplemental Information). Depending on the stability of the protein, the reactor can be stored and reused multiple times with little loss of surface-bound protein, as shown for the mCherry-modified reactor.
To demonstrate the generalizability of this approach, six proteins being used in our laboratory were purified and attached to VFD reactors. These proteins include esterase, phosphodiesterase, alkaline phosphatase, eGFP, mCherry and tobacco epi-aristolochene synthase (TEAS, a terpene cyclase).[15] Biosynthetic pathways often rely on enzymes with less than ideal properties for in vitro biochemical assays; TEAS, for example, is a poorly expressed and troublesome protein with modest stability. The rapid (10 min with a flow rate of 13.3 mL min−1) and straightforward purification of TEAS demonstrates the generalizability of this approach. The protein TEAS, which readily aggregates and precipitates out of solution, benefits from an expedient purification process to limit protein loss.
Well-defined stripes containing different immobilized proteins can be applied to the reactor surface (Figure 1 and S13). Although cell lysate can be directly added to an IMAC-treated reactor by pipette, it is more convenient to first pre-bind the protein from centrifuged cell lysates to the IMAC resin, then add this slurry to the reactor; the concentration of protein within the zone can also be controlled. Careful pipetting of the IMAC-protein slurry onto the inner surface of the rotating reactor allows fine control over enzyme zone length. Positioning the end of a Pasteur pipette at the required height for the enzymatic zone, and releasing drop-wise the pre-bound enzyme-IMAC solution (250 mg of IMAC resin in a homogeneous suspension per cm of stripe) creates a protein stripe with good positional stability. Simply moving the Pasteur pipette to a designated height in the rotating sample tube allows creation of a new stripe at this height. Thus, a range of protein stripes and patterns can be created. The approach offers exceptional versatility and potential for future applications, including for multistep biocatalysis.
Here, we report on reactors immobilized with two different proteins coating either equal or unequal zone lengths (Figure 1B1 and 1B2). This approach allowed us to examine multi-step biocatalytic activity in continuous flow, and the effects of enzyme zones on reaction performance (vide infra). Furthermore, reactors can have both dedicated enzyme catalysis zones along conventional synthetic transformation zones, aiding chemoenzymatic synthesis (Figure 1B4). For example, stripes of different IMAC resins could create arbitrary numbers of zones on the reactor surface, including the 18 or 28 stripes shown here (Figure 1B5 and 1B6). The large number of possible zones suggests multi-step transformations requiring numerous proteins could be translated directly from cell lysate to this continuous flow system.
A multi-step, biosynthetic synthesis was developed that produced colorimetric responses at each step (Figure 2). The multi-step biocatalytic pathway reported here was chosen for the following reason. First, the multi-step sequence had to be high throughput to allow rapid optimization and quantification of the system; a pathway generating a colorimetric response achieved this requirement while allowing any product localization in the reactor to be monitored (none observed). Second, the reaction had to be reliable and robust to allow the effects of enzyme stripes to be isolated. Third, alkaline phosphatase and phosphodiesterase are stable to vortexing and other potentially harsh conditions.[14] Lastly, the reaction had to take place along the length of the reactor; previous experiments had shown that alkaline phosphatase and phosphodiesterase have rapid catalytic activity.[12]
For this reaction, both alkaline phosphatase and phosphodiesterase were recombinantly expressed and purified in continuous-flow by VFD-mediated purification. Before demonstrating multi-step activity in continuous-flow, both enzymes were studied under conventional conditions to obtain Michaelis-Menten values and their preferred reaction conditions (Figure S14-23). Normalizing enzyme concentrations based upon each enzyme's kcat/Km values can equalize rates of product generation; in this example, alkaline phosphatase required a 1450-fold higher concentration to achieve roughly equivalent product generation rates.
Spatially segregating enzymes into zones could introduce new controls for multi-step biocatalysis. For example, product build-up often results in enzyme inhibition; applying additional zones with a second enzyme could transform the product from the first enzyme to enhance reaction rates. Furthermore, finely controlling the concentration of each enzyme in a zone, or the residence time of a substrate through a zone, could allow equal transformation rates in a multi-step pathway. In testing these hypotheses, alkaline phosphatase and phosphodiesterase were applied to the reactor surface in two separate 9.9 cm zones; the two enzymes operate independently on different substrates. As described above, the concentrations of the enzymes were adjusted to insure similar kcat/Km values (113 mM−1 s−1) for each enzyme in the zone. This approach was verified experimentally, as applying both enzymes in a single pathway generated roughly twice as much p-nitrophenol product at the outlet of the reactor compared to a single 9.9 cm zone of phosphodiesterase (26.5 vs. 53.5 μM, Figure 2B).
Next, we compared segregated enzyme zones to a homogeneous mixture of the enzymes. Unexpectedly, the mixture outperformed the spatially segregated enzyme zones (Figure 2B). When proteins are located in close proximity in a multi-step process, the next enzyme in the pathway could transform the product from an earlier transformation more efficiently. This rapid subsequent transformation could decrease product inhibition, and account for the experimental observation. Future experiments could involve enzymes requiring spatial segregation for a specific reaction outcome (e.g., a multi-step pathway applying the same enzyme in a specified order).
This VFD-based approach advances continuous flow multi-step biocatalysis through Hisn-tag immobilization onto the surface of a thin film reactor.[16] The VFD-mediated approach is rapid, can be easily utilized, and is the first flow system to create distinct enzymatic zones for multi-step biocatalysis. Furthermore, this method opens possibilities for rapid implementation of biosynthetic catalysis in continuous-flow. Retrosynthetic analysis of a target compound could suggest appropriate enzymes and their sequential configuration into reaction zones as illustrated here. Spatial segregation and the specific ordering of enzyme zones could facilitate such syntheses. Then, each enzyme could be synthesized in cells transformed with the appropriate DNA before coating their lysate onto specific zones of the reactor surface.
Our collaboration has now demonstrated two key fundamental concepts for in vitro, continuous flow multi-step biocatalysis. First, the VFD can accelerate enzymatic activity via an increase in kcat/Km.[10] Second, near quantitative immobilization and purification from complex cell lysate in ten minutes has been achieved. Importantly, this rapid and general approach should be amenable to any soluble protein containing a fused polyhistidine tag. In addition, given the continuous-flow nature of the processing using the VFD, this technology is highly efficient, reproducible, and scalable. Combining these advancements is now allowing us to explore biocatalytic assembly lines for generation of polyketides in continuous-flow. Such complex multi-domain enzymes are often sluggish in vitro. Combining high levels of immobilization and VFD-mediated acceleration could promote their activity. We foresee that this methodology could create efficient continuous flow systems for on-demand compound generation harnessing the power of nature. Ultimately, multi-day continuous flow could produce diverse compounds in sufficient quantities for customized therapeutics or research applications.
Supplementary Material
Acknowledgments
JB thanks the Taihi Hong Memorial award for support. GW gratefully acknowledges the National Institute of General Medical Sciences of the NIH (1RO1-GM100700-01). CR acknowledges the Australian Research Council and the Government of South Australia for their financial support during this project.
References
- 1.a) Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]; b) Wenzel SC, Muller R. Nat Prod Rep. 2007;24:1211–1224. doi: 10.1039/b706416k. [DOI] [PubMed] [Google Scholar]; c) Meier JL, Burkart MD. Chem Soc Rev. 2009;38:2012–2045. doi: 10.1039/b805115c. [DOI] [PubMed] [Google Scholar]; d) Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; e) Ladner C, Williams GJ. J Ind Microbiol Biot. 2016;43:371–387. doi: 10.1007/s10295-015-1704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Walsh CT. Accounts Chem Res. 2008;41:4–10. doi: 10.1021/ar7000414. [DOI] [PubMed] [Google Scholar]
- 2.Weissman KJ. Nat Prod Rep. 2015;32:436–453. doi: 10.1039/c4np00098f. [DOI] [PubMed] [Google Scholar]
- 3.a) Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P, Sigman M. Nat Chem. 2016;8:299–309. doi: 10.1038/nchem.2459. [DOI] [PubMed] [Google Scholar]; b) Perham RN. Annu Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]; c) Fu J, Yang YR, Johnson-Buck A, Liu M, Liu Y, Walter NG, Woodbury NW, Yan H. Nat Nano. 2014;9:531–536. doi: 10.1038/nnano.2014.100. [DOI] [PubMed] [Google Scholar]
- 4.a) Webb D, Jamison TF. Chem Sci. 2010;1:675–680. [Google Scholar]; b) McQuade DT, Seeberger PH. J Org Chem. 2013;78:6384–6389. doi: 10.1021/jo400583m. [DOI] [PubMed] [Google Scholar]
- 5.a) Adamo A, Beingessner RL, Behnam M, Chen J, Jamison TF, Jensen KF, Monbaliu JCM, Myerson AS, Revalor EM, Snead DR, Stelzer T, Weeranoppanant N, Wong SY, Zhang P. Science. 2016;352:61–67. doi: 10.1126/science.aaf1337. [DOI] [PubMed] [Google Scholar]; b) Heider PL, Born SC, Basak S, Benyahia B, Lakerveld R, Zhang H, Hogan R, Buchbinder L, Wolfe A, Mascia S, Evans JMB, Jamison TF, Jensen KF. Org Process Res Dev. 2014;18:402–409. [Google Scholar]; c) Newton S, Carter CF, Pearson CM, de L, Alves C, Lange H, Thansandote P, Ley SV. Angew Chem Int Ed. 2014;53:4915–4920. doi: 10.1002/anie.201402056. [DOI] [PubMed] [Google Scholar]; d) Bogdan AR, Poe SL, Kubis DC, Broadwater SJ, McQuade DT. Angew Chem Int Ed. 2009;48:8547–8550. doi: 10.1002/anie.200903055. [DOI] [PubMed] [Google Scholar]; e) McQuade DT, Seeberger PH. J Org Chem. 2013;78:6384–6389. doi: 10.1021/jo400583m. [DOI] [PubMed] [Google Scholar]; f) Tsubogo T, Oyamada H, Kobayashi S. Nature. 2015;520:329–332. doi: 10.1038/nature14343. [DOI] [PubMed] [Google Scholar]; g) Gutmann B, Cantillo D, Kappe CO. Angew Chem Int Ed. 2015;54:6688–6728. doi: 10.1002/anie.201409318. [DOI] [PubMed] [Google Scholar]; h) Lévesque F, Seeberger PH. Angew Chem Int Ed. 2012;51:1706–1709. doi: 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]
- 6.Zhang P, Russell MG, Jamison TF. Org Process Res Dev. 2014;18:1567–1570. [Google Scholar]
- 7.Correia CA, Gilmore K, McQuade DT, Seeberger PH. Angew Chem Int Ed. 2015;54:4945–4948. doi: 10.1002/anie.201411728. [DOI] [PubMed] [Google Scholar]
- 8.a) Kundu S, Bhangale AS, Wallace WE, Flynn KM, Guttman CM, Gross RA, Beers KL. J Am Chem Soc. 2011;133:6006–6011. doi: 10.1021/ja111346c. [DOI] [PubMed] [Google Scholar]; b) Luckarift HR, Ku BS, Dordick JS, Spain JC. Biotechnol Bioeng. 2007;98:701–705. doi: 10.1002/bit.21447. [DOI] [PubMed] [Google Scholar]; c) Mao H, Yang T, Cremer PS. Anal Chem. 2002;74:379–385. doi: 10.1021/ac010822u. [DOI] [PubMed] [Google Scholar]; d) Matosevic S, Lye GJ, Baganz F. J Biotechnol. 2011;155:320–329. doi: 10.1016/j.jbiotec.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 9.a) Clouthier CM, Pelletier JN. Chem Soc Rev. 2012;41:1585–1605. doi: 10.1039/c2cs15286j. [DOI] [PubMed] [Google Scholar]; b) Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 10.a) Yasmin L, Chen X, Stubbs KA, Raston CL. Sci Rep. 2013;3:2282. doi: 10.1038/srep02282. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yasmin L, Coyle T, Stubbs KA, Raston CL. Chem Commun. 2013;49:10932–10934. doi: 10.1039/c3cc45176c. [DOI] [PubMed] [Google Scholar]; c) Britton J, Dalziel SB, Raston CL. Green Chem. 2016;18:2193–2200. [Google Scholar]; d) Britton J, Dalziel SB, Raston CL. RSC Adv. 2015;5:1655–1660. [Google Scholar]
- 11.a) Britton J, Chalker JM, Raston CL. Chem Eur J. 2015;21:10660–10665. doi: 10.1002/chem.201501785. [DOI] [PubMed] [Google Scholar]; b) Britton J, Castle JW, Weiss GA, Raston C. Chem Eur J. 2016;22:10773–10776. doi: 10.1002/chem.201602373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan TZ, Ormonde CFG, Kudlacek ST, Kunche S, Smith JN, Brown WA, Pugliese KM, Olsen TJ, Iftikhar M, Raston CL, Weiss GA. ChemBioChem. 2015;16:393–396. doi: 10.1002/cbic.201402427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Britton J, Meneghini LM, Raston CL, Weiss GA. Angew Chem Int Ed. 2016;55:11387–11391. doi: 10.1002/anie.201604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britton J, Raston CL, Weiss GA. Chem Commun. 2016;52:10159–10162. doi: 10.1039/c6cc04210d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diaz JE, Lin CS, Kunishiro K, Feld BK, Avrantinis SK, Bronson J, Greaves J, Saven JG, Weiss GA. Protein Sci. 2011;20:1597–1606. doi: 10.1002/pro.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drager G, Kiss C, Kunz U, Kirschning A. Org Biomol Mol. 2007;5:3657–3664. doi: 10.1039/b712804e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.