

Abstract
Infectious diseases have serious impacts on human and wildlife populations, but the effects of a disease can vary, even among individuals or populations of the same host species. Identifying the reasons for this variation is key to understanding disease dynamics and mitigating infectious disease impacts, but disentangling cause and correlation during natural outbreaks is extremely challenging. This study aims to understand associations between symbiotic bacterial communities and an infectious disease, and examines multiple host populations before or after pathogen invasion to infer likely causal links. The results show that symbiotic bacteria are linked to fundamentally different outcomes of pathogen infection: host–pathogen coexistence (endemic infection) or host population extirpation (epidemic infection). Diversity and composition of skin-associated bacteria differed between populations of the frog, Rana sierrae, that coexist with or were extirpated by the fungal pathogen, Batrachochytrium dendrobatidis (Bd). Data from multiple populations sampled before or after pathogen invasion were used to infer cause and effect in the relationship between the fungal pathogen and symbiotic bacteria. Among host populations, variation in the composition of the skin microbiome was most strongly predicted by pathogen infection severity, even in analyses where the outcome of infection did not vary. This result suggests that pathogen infection shapes variation in the skin microbiome across host populations that coexist with or are driven to extirpation by the pathogen. By contrast, microbiome richness was largely unaffected by pathogen infection intensity, but was strongly predicted by geographical region of the host population, indicating the importance of environmental or host genetic factors in shaping microbiome richness. Thus, while both richness and composition of the microbiome differed between endemic and epidemic host populations, the underlying causes are most likely different: pathogen infection appears to shape microbiome composition, while microbiome richness was less sensitive to pathogen-induced disturbance. Because higher richness was correlated with host persistence in the presence of Bd, and richness appeared relatively stable to Bd infection, microbiome richness may contribute to disease resistance, although the latter remains to be directly tested.
Keywords: microbiome, enzootic, wildlife disease, Batrachochytrium dendrobatidis, chytridiomycosis, defensive symbiosis
1. Introduction
Infectious diseases are important players in ecological systems and are of increasing concern to the conservation of biodiversity [1]. The effects of infectious disease on host populations can vary dramatically, and understanding natural variation in disease dynamics is important for predicting and managing effects of disease in wild populations. The fungal pathogen Batrachochytrium dendrobatidis (Bd) infects the skin of amphibians and causes the disease chytridiomycosis, which has led to widespread amphibian declines [2–6]. However, the effects of Bd infection vary among host species, and even among populations of the same species, with outcomes ranging from asymptomatic infection to lethal disease [7–10]. Understanding the mechanisms by which some species or populations resist lethal disease could inform management strategies for threatened amphibians, but currently these mechanisms are poorly understood.
Symbiotic microbial communities are ubiquitous inhabitants of multicellular organisms [11,12]. Resident symbiotic microbes (referred to as the microbiome) can interact with invading pathogens and may play a role in resistance to disease [13]. In the case of Bd, researchers are actively pursuing probiotics as a potential approach to mitigate the impacts of Bd on threatened amphibians. Some bacteria isolated from amphibian skin can inhibit the growth of Bd in laboratory culture (in vitro). However, laboratory experiments using bacteria as probiotics have yielded varying results: augmentation of amphibians with bacterial cultures reduced rates of Bd infection or host mortality in some cases [14,15], but failed to prevent disease in others [16–18]. In addition, extrapolating laboratory results to field dynamics without the guidance of field data is problematic because both microbiome and pathogen dynamics are affected by many interacting factors in nature, such as environment, host behaviour and host population density. Our understanding of the links between symbiotic bacteria and Bd infection in wild populations is limited, but recent studies have examined correlations between skin-associated bacterial communities and Bd infection status, intensity, prevalence or susceptibility [19–22]. In this study, we ask if the skin microbiome can explain fundamentally different outcomes of Bd infection: host–pathogen coexistence or host population extirpation.
Rana sierrae is a frog species with striking differences in Bd infection outcomes among wild populations. Rana sierrae and its close relative Rana muscosa are severely threatened by Bd, and hundreds of populations have suffered rapid decline and extirpation due to Bd infection, referred to as epizootic disease dynamics or population die-off [2,6]. However, some R. sierrae populations show long-term coexistence with Bd [8,23], and are referred to as enzootic or persistent populations. (The terms ‘enzootic’ and ‘epizootic’ refer to epidemic and endemic infection dynamics in animal populations.) Notably, most frogs in persistent populations are infected with Bd (i.e. prevalence is high) but usually maintain low Bd loads (the density of Bd cells infecting the skin), which is likely how they escape lethal disease because mortality is linked to high-Bd loads [8]. It is thought that widespread high-Bd loads result in epizootics [2], but the factors controlling Bd load are unclear. More broadly, what leads to enzootic or epizootic disease dynamics is not known. Disease models indicate that demographic traits such as population density can influence the likelihood of catastrophic outbreaks leading to host population extinction [8], although empirical tests of this hypothesis are currently lacking. Studies of population genetics and phenotypic variation among Sierra Nevada Bd strains found no consistent difference between strains isolated from enzootic and epizootic populations [23,24]. The outcomes of infection do show geographical patterns, with enzootics in northern and epizootics in southern populations (figure 1a) [2,23], but whether this geographical distinction is due to spatial environmental gradients, intrinsic host differences predating Bd invasion, host evolution after Bd invasion or time since Bd invasion is not clear [23]. Some evidence suggests that frogs from enzootic populations have reduced susceptibility to infection, suggesting that susceptibility is partially determined by traits inherent to frogs, such as innate or acquired immunity or characteristics of the skin [23]. Skin-associated bacteria have the potential to contribute to disease resistance [14], and previous work found that the proportion of frogs with at least one Bd-inhibitory bacterial isolate differed between an epizootic and an enzootic population, and did not differ between an enzootic and naive population [25,26]. These results are consistent with bacteria playing a role in disease outcomes, but because only one population of each type has been studied in this context so far, it is difficult to draw firm conclusions. In addition, it is not known whether observed differences in the microbiomes among frogs could be a result, rather than a cause, of differences in Bd infection severity. Bd infection can alter the R. sierrae skin-associated bacterial community (hereafter microbiome for brevity; [20]), and higher Bd loads during epizootics could drive shifts in the microbiome that result in differences in the microbiomes on frogs in enzootic and epizootic populations. It is therefore important to consider the possibility that differences in the skin microbiome of enzootic and epizootic R. sierrae populations could in fact be caused by differences in severity of Bd-induced disturbance of the microbiome.
Figure 1.
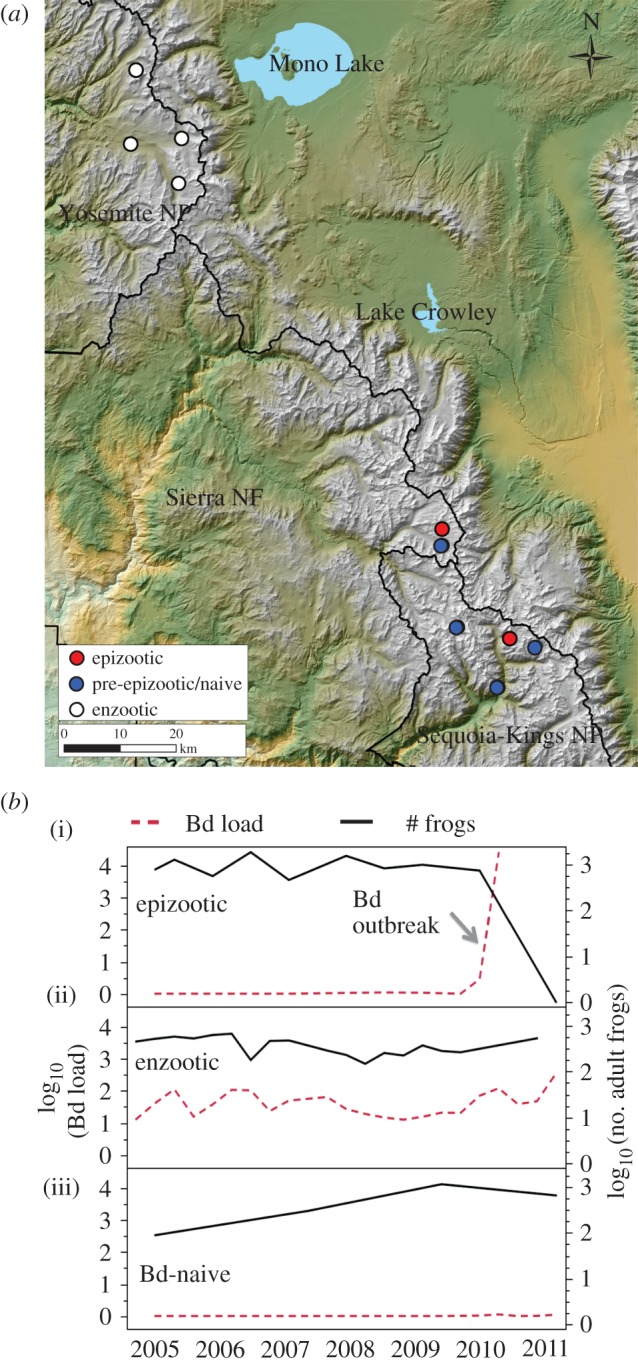
(a) Map showing division between ‘northern’ (Yosemite) and ‘southern’ (Kings Canyon National Park and Sierra National Forest) R. sierrae populations in this study. Northern populations exhibit enzootic dynamics (white markers). Southern locations include populations that were epizootic (red markers) as well as populations that were Bd-naive (pre-epizootic; blue markers). (b) Examples of long-term mean Bd load and population dynamics in three populations. (i) An uninfected population showing stable population numbers until invasion by Bd that resulted in epizootic dynamics and population decline. (ii) An enzootic population exhibiting stable Bd loads and population numbers through time. (iii) A Bd-naive population maintained stable population numbers through time.
This study tests the hypothesis that populations with fundamentally distinct infection dynamics—host–pathogen coexistence or host extirpation—harbour different skin microbiomes. We then identify factors that potentially influence landscape-level microbiome variation by testing how well environmental variables, geographical variables and Bd infection predict among-population microbiome variation. We apply these insights into what controls the microbiome to determine if differences between enzootic and epizootic populations are more consistent with Bd-induced disturbance or, alternatively, microbiome-mediated protection against disease.
2. Material and methods
(a). Population surveys and determination of disease dynamic type
This study was conducted in 10 R. sierrae populations across Yosemite and Kings Canyon National Parks and Sierra National Forest, California (figure 1a). Population sizes were estimated by visual encounter surveys as described in [27]. We used long-term population census and Bd infection data to categorize populations at each sampling time point in 2010 as enzootic, epizootic or pre-epizootic with respect to Bd dynamics (figure 1b). All populations in this study have been censused regularly for over a decade (since 2002 or earlier). Beginning in 2005, surveys included collection of standardized skin swabs for quantification of Bd loads, as described previously [8]. Populations that remained stable through time, despite Bd infection (adult frogs observed for at least 5 years after Bd invasion), were categorized as enzootic. Populations in which Bd load trajectories on adult frogs increased dramatically through time, followed by frog population collapse after which zero adult frogs were observed for at least 2 years, were categorized as epizootic or ‘die-off’ [2]. Four populations were categorized as pre-epizootic at the time of microbiome sampling in 2010: these populations were Bd-naive (no Bd-positive samples or only an occasional low-Bd load sample detected, and no Bd outbreak) prior to 2010, and remained Bd-naive through early summer of 2010. We classified these populations as pre-epizootic (as opposed to pre-enzootic) based on our observations that formerly naive populations in nearby regions consistently suffered epizootics due to Bd invasion (R.A.K. 2017, unpublished data, [2]). Indeed, several of the populations that were Bd-naive in 2010 in this study experienced epizootics in later years (electronic supplementary material, table S1). Enzootic populations are generally located in the north (Yosemite and northwards), while epizootic and remaining pre-epizootic populations are found further to the south (figure 1a; [2,8,23]).
Previous studies have shown that epizootics result in much higher Bd loads than are characteristic of enzootic populations [2,8,23]. To determine if this was also the case with our study populations, we tested if Bd loads are significantly different during epizootic and enzootic dynamics using a linear mixed model with survey as a random factor nested in disease dynamic type.
(b). Sample collection and processing
To describe frog microbiomes and Bd infection intensities at each of the 10 R. sierrae study populations, we visited each population one to five times during the 2010 summer active season (frogs hibernate in ice-covered lakes November–June). Survey dates and sample sizes are provided in electronic supplementary material, table S1. Detailed sample collection, preservation and storage methods for bacterial community samples were described previously [20] and are summarized as follows: post-metamorphic (i.e. subadult and adult) R. sierrae were captured by dip net and swabbed for skin-associated microbes (both bacteria and Bd) using sterile synthetic swabs and wearing new nitrile gloves for each frog handled. Nets were rinsed thoroughly in lake water between each frog captured. Frogs were rinsed in clear lake water prior to swabbing, but were not rinsed with sterile water. We found in a pilot study that sterile-water rinsing did not affect skin bacterial profiles (electronic supplementary material). In addition to samples collected from frogs, we sampled planktonic microbial communities present in lake water, which is the primary habitat for these aquatic frogs and may serve as a source of colonists to frog skin. Planktonic microbes were sampled by filtration of water through 0.22 µm filters.
We quantified Bd loads from all swabs using quantitative PCR following Boyle et al. [28]. Microbiome analysis was conducted using a subset of surveys that covered all 10 populations in a short time (less than four weeks) to minimize seasonal variation. In one population, our surveys captured an epizootic that increased in intensity with time. To examine changes associated with intensification of this epizootic, we also sequenced bacterial communities from a second survey from that epizootic and three reference populations. These four populations are therefore represented by two surveys each in our microbiome dataset. However, results from analyses based on all surveys are consistent with analyses using only one survey from each population (not shown). We randomly selected 8–20 samples from each of the 14 population surveys for microbiome analysis. Surveys of enzootic, epizootic and pre-epizootic populations were interspersed through time to avoid bias that could result from temporal aggregation of samples of the same disease state.
Bacterial communities were characterized by 16S amplicon pyrosequencing as detailed previously [20] and summarized in electronic supplementary materials. For each microbiome sample, we calculated four metrics of community diversity: observed number of operational taxonomic units (OTUs) (SOBS), Chao's richness estimate, Shannon diversity and Shannon evenness. We examined microbiome community composition using weighted UniFrac distance [29], which is a measure of bacterial community distance based on the phylogenetic relatedness and abundance of OTUs. This study is focused on variation among populations at the landscape scale; within-population analyses are addressed in Jani & Briggs [20].
(c). Overview of statistical analyses
Analyses are described in detail below. To summarize: we first tested if enzootic and epizootic populations harbour different skin microbiota. We then asked if environmental factors or Bd infection best predict variation in the microbiome. In the latter analysis, we included populations spanning enzootic, epizootic and pre-epizootic disease states to tease apart effects of geography, environment and Bd infection on the microbiome. We then used our findings about potential drivers of microbiome variation to infer whether differences between enzootic and epizootic populations are more consistent with Bd-induced disturbance of the microbiome, or variation in microbiome-mediated resistance to Bd.
Details about statistical software, data transformations, Q-value thresholds, definition of common OTUs and statistical models of community structure are provided in the electronic supplementary material.
(d). Testing for differences in bacterial communities of enzootic and epizootic populations
We used linear mixed models to test if bacterial diversity (bacterial richness and evenness) differed between enzootic and epizootic populations. We used permutational MANOVA (PERMANOVA) of weighted UniFrac distance matrices to test for differences in the composition of bacterial communities between enzootic and epizootic host populations. In both cases, survey was included as a random variable, nested within population type (enzootic or epizootic) to account for non-independence of samples collected in the same population survey.
To identify bacterial taxa that differed in relative abundance between enzootic and epizootic populations, we ran a linear mixed model for each common OTU, with relative abundance as the response variable and population type as the predictor. We also tested for correlations between Bd load and each common OTU using survey-level mean Bd load and OTU relative abundances as inputs (described in the electronic supplementary material). Potential for false discoveries due to large numbers of tests was accounted for using Q-value thresholds [30].
(e). Identifying potential drivers of bacterial community variation
The analyses described below allowed us to examine what factors might drive landscape-level variation in the skin microbiome. The data for these analyses span enzootic, epizootic and pre-epizootic populations. The inclusion of pre-epizootic populations is key because it enables separation of variables that would otherwise be confounded with one another: geography, Bd infection severity (Bd load) and Bd infection outcome (enzootic or epizootic). Pre-epizootic (low or no Bd) and epizootic (high-Bd load) populations were spatially mixed, which removes spatial autocorrelation between Bd load and geographical region. Similarly, confounding between Bd load at the time of this study and eventual disease outcome (enzootic or epizootic) is removed because populations that were Bd-naive at the time of the survey succumbed to epizootics in later years (electronic supplementary material, table S1).
We used a linear model (standard least squares) to test predictors of microbiome diversity, and a non-parametric distance-based linear model (DistLM) to test predictors of microbiome composition. The following predictor variables were included: geographical region (north/south), elevation (metres), lake size (square metres), richness of aquatic bacteria (number of OTUs detected in lake water) and Bd load (electronic supplementary material, table S2). For DistLM, we used stepwise model selection to add or remove one variable at each step until the best model was reached based on AICc (Akaike information criterion adjusted to reduce the risk of over-parameterizing that can otherwise occur with smaller sample sizes). In this study, we are specifically interested in among-population variation. However, Bd load varies both within and among populations. Therefore, the mean Bd load for each survey was used as a predictor variable, ensuring that the analyses detect only drivers of among-population variation, and not drivers of within-population variation. All other predictor variables (lake size, elevation, geographical region and richness of aquatic bacteria) are measured at the population or survey level, further supporting the use of population survey rather than frog as the unit of replication in these analyses. Other response variables were converted from frog-level to population survey-level data by pooling (community composition) or averaging (community diversity) across all frog samples within each population survey.
The above analyses account for aquatic bacterial richness, but do not include aquatic bacterial community composition as a predictor variable of the frog skin microbiome because of the difficulty of simultaneously assessing linear variables and pairwise distance matrices as predictors in a single statistical model. Therefore, we assessed the potential for planktonic aquatic bacterial community composition to shape the R. sierrae microbiome as follows. We first tested if bacterial communities on R. sierrae skin are different from bacterial communities present in the surrounding lake water, using ANOSIM. Next, we tested for a correlation across R. sierrae populations in the planktonic bacterial communities and frog microbiomes, using a Mantel test. Third, we compared the degree to which aquatic bacteria and Bd load explain variation in frog-associated bacterial communities, as follows: we conducted a Mantel test for the correlation between Bd load (after conversion to a distance matrix) and frog-associated bacterial communities. We then compared the strengths of the two Mantel correlations; i.e. we compared the planktonic bacteria–microbiome correlation with the Bd load–microbiome correlation.
3. Results
(a). Disease dynamics
Enzootic, epizootic and pre-epizootic populations had distinct Bd load trajectories (figure 2). Bd loads were lower during enzootics than epizootics that occurred in the same year, 2010, and the effect was more pronounced for adults than subadults (adults p < 0.001, least-squares mean: enzootic 1.32, epizootic 4.02; subadults p = 0.040, least-squares mean enzootic 2.73, epizootic 4.36; data on a log10 scale), most likely because subadults can have high loads even in enzootic populations [8,20]. We had predicted that populations that were Bd-naive or nearly so would exhibit epizootic dynamics if and when Bd invaded. Consistent with our prediction, two of the four populations that we classified as pre-epizootic showed signs of Bd invasion in late 2010 (initially low but increasing loads and prevalence) and experienced epizootics by the following year. A third population experienced an outbreak in 2015 (electronic supplementary material, table S1). Thus, our surveys include populations that differed in Bd infection status (no/low Bd versus high-Bd) at the time of this study, but share the same response to infection (epizootic) when they do become infected with Bd.
Figure 2.
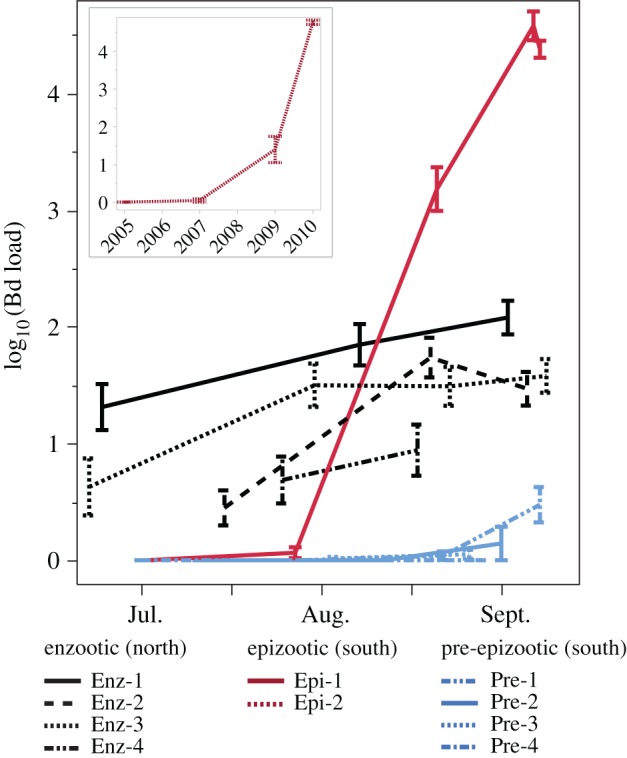
Distinct Bd load trajectories in enzootic and epizootic populations. Lines are mean Bd loads of adult frogs in each of 10 populations during the 2010 field season. (Inset) Bd load trajectory shown separately for one population (Epi-2) where the epizootic developed over several years; Epi-2 includes subadults because by 2010 no adults remained in this population. Each population is colour-coded according to its disease status. If a population changed status during the 2010 season (e.g. Epi-1 changed from naive to epizootic), the corresponding line is coloured according to status at the end of the season. Error bars are standard error of the mean.
(b). Bacterial communities in enzootic versus epizootic frog populations
Bacterial richness was higher in enzootic populations compared with epizootic populations (pSOBS = 0.023, pChao = 0.011), although diversity indices (Shannon diversity and evenness) did not differ (pShannon = 0.187, peven = 0.713). Microbiome composition also differed significantly between enzootic and epizootic frog populations (PERMANOVA p = 0.033), as visualized using NMDS ordination (figure 3a). When Bd load data are superimposed on the NMDS ordination, a pattern of correlation between Bd load and bacterial communities is apparent (figure 3b, formally tested using among-population DistLM analysis, below).
Figure 3.
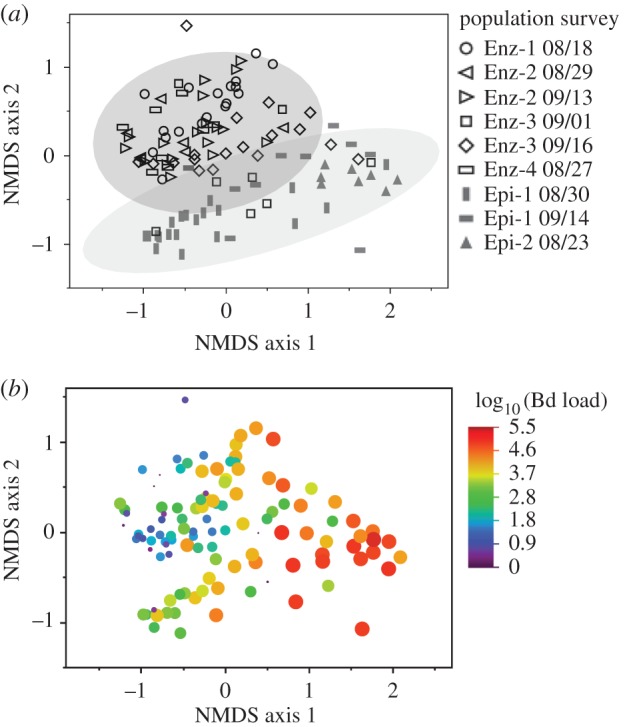
Frog skin microbiome composition differs between enzootic and epizootic populations, and is associated with Bd load. (a) NMDS ordination of skin bacterial communities from frogs in enzootic and epizootic populations. Each data point represents the microbiome from one frog. Different marker shapes represent different population surveys (see legend). Shaded areas are bivariate normal 90% density ellipses for enzootic and epizootic populations. (b) The same ordination as in (a), but here marker size and colour indicate Bd load. NMDS stress = 0.16.
(c). Bacterial taxa that differ between enzootic and epizootic populations
Only two OTUs differed in relative abundance between enzootic and epizootic populations, and these differences were marginal (p < 0.05 but 0.05 < Q < 0.1) after accounting for false discovery rate, suggesting that the difference between enzootic and epizootic bacterial populations is mostly due to small changes in abundances of many taxa. It is also possible that studying a larger number of host populations would lead to detection of more OTUs that differ between enzootic and epizootic populations. Our analyses account for non-independence among frogs within a population. This correctly represents the structure of the data (because we are interested in differences among types of populations), but also reduces power, and when combined with significance threshold correction for multiple tests (use of Q-value threshold) may limit detection of individual OTU differences. OTU F-96, classified to the genus Undibacterium (family Oxalobacteraceae), was marginally more abundant in epizootic populations (p = 0.003, Q = 0.083). OTU F-23, classified as Ideonella (family Comamonadaceae), was marginally more abundant in enzootic populations (p = 0.004, Q = 0.083). Both of these OTUs were also correlated with Bd load across populations: F-96 (Undibacterium) was significantly positively correlated with Bd load (p < 0.001, Q = 0.025, correlation coefficient = 0.90), while F-23 (Ideonella) was marginally negatively correlated with Bd load (p = 0.016, Q = 0.078, correlation coefficient = −0.77). The relative abundances of OTUs in nine genera were correlated with Bd load across infected frog populations (electronic supplementary material, table S3).
(d). Pathogen load is correlated with skin microbiome composition across populations
Bd load was a robust predictor of microbiome composition, regardless of whether analyses included all populations (DistLM, p < 0.001; figure 4a,b), only infected populations (DistLM p < 0.001), only southern (epizootic and pre-epizootic) populations (p = 0.002) or only northern (enzootic) populations (p = 0.013).
Figure 4.

Across populations, Bd load predicts microbiome composition (a,b) while geography predicts richness (c,d). Each data point represents the pooled microbiome of all frogs within a population survey. (a) NMDS ordination of bacterial communities. Shaded areas are bivariate normal 90% density ellipses. NMDS stress = 0.10. (b) Regression of NMDS axis 1 against mean Bd load. Shaded area is 95% confidence of fit. NMDS is for visualization only; DistLM is used for statistical test. (c) Microbiome richness of northern and southern populations. Each marker shows mean richness for one population survey. (d) Richness is not predicted by Bd load.
(e). Drivers of microbiome community composition
Bacterial communities present on R. sierrae skin were distinct from bacterial communities present in the water column (PERMANOVA, p< 0.001; electronic supplementary material, figures S2 and S3). However, despite the clear differences between them, frog microbiomes and aquatic bacterial communities covaried: a Mantel test showed a significant correlation between the UniFrac distance matrices of frog-associated and aquatic bacterial communities (p = 0.023, ρ = 0.342). In other words, although there was clearly a difference between the microbial communities from lake water and frog skin, there was still a tendency across sites for microbial community in water to predict the microbial community on frogs. Skin-associated bacterial communities were also correlated with Bd load (Mantel p < 0.001, ρ = 0.532). Frog-associated bacterial communities were more strongly correlated with Bd load than with aquatic bacteria. Aquatic bacterial communities were not correlated with frog Bd loads (Mantel p = 0.125).
Multi-variate linear models (DistLM) showed that microbiome composition was primarily explained by Bd load as opposed to spatial variables (elevation, lake area, geographical region) or aquatic bacterial richness. When explanatory variables were tested individually, only Bd load explained a significant proportion of variation in microbiome composition (DistLM, p < 0.001). One landscape variable (geographical region: north or south) had a significant effect (p = 0.015) only when added to the model sequentially after Bd load, indicating that latitude explains a portion of the variation not already accounted for by Bd load. The model that explained the most variation included two variables: Bd load (p < 0.001, explaining 30.5% of variation) and geographical region (p = 0.013, explaining 15.1% of variation), and explained a total of 45.6% of the variation in bacterial communities. However, based on AICc scores, the model including both Bd load and geographical region (AICc = −38.626) was not significantly better than the model including only Bd load (AICc = −38.507).
Unlike microbiome composition, microbiome richness was strongly predicted by geographical region (north or south, figure 4c; electronic supplementary material, table S2). Elevation also predicted richness, although not as strongly or consistently as geographical region. Elevation also covaries with geographical region. Bd load did not significantly predict richness or diversity (figure 4d), although it was marginally significant for one richness metric: the observed number of OTUs (PSOBS = 0.056). Furthermore, there was no difference in bacterial diversity between epizootic and pre-epizootic populations (pSOBS = 0.386, pChao = 0.428, pShannon = 0.472, peven = 0.671; electronic supplementary material, figure S4), indicating that the shift from Bd-naive to Bd outbreak was not accompanied by significant changes in microbiome diversity.
(f). Rana sierrae skin microbiome shows high variability among populations and with time
We conducted ANOSIM tests between every possible pair of the 14 population surveys. In 72 out of 91 possible comparisons, the two population surveys in question harboured different bacterial communities (electronic supplementary material, table S4). This was often the case even when two surveys were from the same type of population (e.g. both enzootic or both epizootic) or from the same population at different time points. That any two populations are likely to differ illustrates why comparing a single pair of populations provides limited insight into differences among types of populations such as enzootic and epizootic. Understanding population-level variation requires analysis of multiple populations.
4. Discussion
(a). Overview
This study aims to understand the role of symbiotic bacteria in shaping a fundamental difference in host response to infection: host–pathogen coexistence versus host population extirpation. We found that populations that coexist with Bd or suffer disease-induced die-off have distinct skin-associated bacteria. To our knowledge, this is the first demonstration that microbiomes are more similar among amphibian populations characterized by the same disease outcome (host persistence or extirpation) than between populations with different disease outcomes. This result is striking, but by itself it is difficult to interpret in terms of cause and effect because the pattern could be driven by bacteria affecting Bd infection, Bd infection altering bacterial communities and/or unmeasured factors. To aid in interpreting differences between enzootic and epizootic populations, we used separate analyses to identify factors that likely shape the microbiome across a broad set of populations, including multiple populations that were either enzootic, epizootic or pre-epizootic at the time of sampling. Including pre-epizootic populations (in which frogs had low loads at the time of this study but have since suffered epizootic decline) alleviates confounding between Bd load at the time of this study and eventual response to Bd infection. This enables us to begin to tease apart aspects of the microbiome that differ because of Bd disturbance from those that are robust to Bd infection and therefore more likely to contribute to disease resistance. We found that bacterial community composition is best predicted by Bd load, such that much of the difference in microbiome composition between enzootic and epizootic populations may be in part a result, rather than a cause, of differences in Bd infection severity. By contrast, bacterial taxonomic richness differed between enzootic and epizootic populations and appeared relatively robust to Bd-induced perturbation.
(b). Batrachochytrium dendrobatidis infection severity is the best predictor of variation in microbiome composition
We examined potential drivers of landscape-level variation in the amphibian microbiome, including geographical region, elevation, lake size, bacterioplankton taxonomic richness and Bd infection severity (Bd load). We treated each population survey (not each frog sampled) as one data point, such that the result clearly indicates landscape-level patterns among populations. In addition, Bd load is not confounded with geographical region because both the high-Bd (epizootic) and low-Bd (pre-epizootic) populations are located in the southern region. Bd load was by far the strongest predictor of microbiome composition, explaining 30.5% of among-population microbiome variation.
We compared the composition of the R. sierrae skin microbiome with the bacterial communities found in pelagic lake habitats. Because R. sierrae spend most of their time in or immediately adjacent to lakes [31], we expected the bacterial species pool of lake water to provide an important source of bacterial colonists to amphibian skin. However, we found that the bacterial communities present on R. sierrae skin and in lake water are markedly distinct (electronic supplementary material, figures S2 and S3). Consistent with a previous study [32], our results suggest that bacterial communities on amphibian skin are selected rather than randomly assembled from the aquatic environment. However, we also found that composition of frog-associated bacteria covaried with composition of aquatic bacteria. Thus, although frog-associated and aquatic bacterial communities are distinct, they have parallel patterns in landscape scale variation, suggesting that there are factors that affect both the aquatic and amphibian-associated bacterial communities. Currently, we do not know what those causal factors are, but possibilities include water chemistry or temperature [33,34], benthic bacterial, invertebrate or plant communities [35], or regional dispersal of bacteria. It is also possible that host genetics influence the microbiome (e.g. [36]), which could lead to covariation between microbiome and aquatic bacteria if the relevant (unknown) R. sierrae genes were to follow geographical patterns that coincide with environmental gradients affecting aquatic bacteria.
Together, these analyses indicate that the R. sierrae skin microbiome is distinct from, but covaries with, the aquatic bacterial community, and is also affected by host genetic or environmental factors linked to latitude. However, Bd load was more strongly correlated with microbiome composition than any spatial or environmental factor measured.
(c). Microbiome composition differs between enzootic and epizootic populations
We sought to understand if differences in the microbiome between enzootic and epizootic populations are more consistent with a protective effect by bacteria or with Bd-induced disturbance. Previous work found that Bd infection alters the R. sierrae microbiome in an experiment and is correlated with microbiome variation among frogs within wild populations [20], suggesting that differences in the microbiome of enzootic and epizootic populations could be a response to Bd infection intensity. Yet, culture-based studies found that bacteria isolated from R. sierrae can inhibit Bd growth in vitro, suggesting the potential for bacteria to play a role in disease resistance [25,26]. However, when comparing only enzootic and epizootic populations, key variables are confounded. Most notably, Bd load is confounded with outcome of infection (enzootic or epizootic dynamics) and geographical region.
To alleviate the problem of spatial autocorrelation, we first assessed the potential for Bd and environmental factors to shape the microbiome while controlling for spatial autocorrelation (described in the previous section). Once we had identified the strongest predictors of microbiome composition while controlling for spatial autocorrelation, we used the results to interpret differences between enzootic and epizootic populations. When identifying predictors of microbiome composition or diversity, we alleviated spatial autocorrelation by including pre-epizootic populations: these were virtually free of Bd at the time of this study but later experienced Bd-induced die-offs. This alleviates both the confounding between Bd load and outcome of infection, and the confounding between Bd load and regional geography, because the pre-epizootic populations (Bd-naive or nearly so at the time of the study) were located in the southern region and are spatially interspersed with the epizootic populations (with very high Bd loads). Microbiome composition was predicted by Bd load across enzootic, epizootic and pre-epizootic populations, indicating that the Bd–microbiome link is not an artefact of spatial autocorrelation. Furthermore, there was only a weak effect of geographical region, which in this dataset is a proxy for disease outcome (enzootics in the north, epizootics in the south). These results indicate that microbiome composition is more strongly tied to Bd infection severity than to disease outcome, and suggests that differences in microbiome composition of enzootic and epizootic populations likely represent Bd-induced alteration of the microbiome.
Additional lines of evidence further support the hypothesis that differences in microbiome composition between enzootic and epizootic populations are largely due to Bd-induced disturbance. First, Bd load predicted bacterial composition even when considering only populations that eventually suffered epizootics. In this case, the microbiome did not protect against population die-off, meaning that correlations between bacteria and Bd load were not linked to variation in disease resistance. Second, specific differences between the microbiomes of enzootic and epizootic populations in this study could be explained by differences in Bd load. For example, OTU-96 (Undibacterium) was enriched in epizootic populations in this study, was positively correlated with Bd load among frogs within populations in previous work, and OTUs of Undibacterium have been shown to increase in abundance in response to Bd infection in a controlled experiment [20]. In addition, many OTUs showed consistent correlations with Bd in this study of across-population variation and in a previous study that indicated disturbance of the microbiome among frogs within populations (electronic supplementary material, table S3; [20]). Together these results point to Bd-induced disturbance as an explanation for the difference in microbiome composition between enzootic and epizootic populations.
Our study does not specifically test or rule out evolution of the microbiome in enzootic populations. Similarly, we do not test if the initial persistence of northern populations was facilitated by microbiome traits that pre-dated Bd invasion; the biological samples needed to test that hypothesis are not available, as Bd is nearly ubiquitous in northern regions [23]. These hypotheses are not mutually exclusive with one another or with the hypothesis that Bd disturbs the microbiome: any combination may contribute to present-day disease dynamics. However, our results best support the hypothesis that the difference in composition between enzootic and epizootic populations is largely due to Bd-induced disturbance. Both types of populations may experience Bd-induced disturbance, but that disturbance is more severe in epizootic populations due to extremely high-Bd infection severity.
(d). Bacterial community richness differs between enzootic and epizootic populations and appears robust to pathogen disturbance
We found that bacterial richness is higher in enzootic than epizootic populations. Bd loads are consistently lower in enzootic populations, so we initially hypothesized that bacterial richness could be affected by Bd infection severity, as was found for Proteobacteria on coquí frogs [19]. But enzootic and epizootic populations occurred in different geographical regions, so the difference in bacterial richness could also be explained by other factors that vary geographically. If we consider only enzootic and epizootic populations, we confound Bd load with Bd infection outcome and geography. Inclusion of the pre-epizootic populations in this analysis is key because it disrupts the spatial autocorrelation, as detailed above. We found that variation in microbiome richness was best explained by geographical location (northern versus southern region). Unlike microbiome composition, microbiome richness was not significantly predicted by Bd load, and richness was similar among populations sampled during epizootics and populations sampled prior to epizootics, indicating no effect or negligible effect of Bd on richness. Consistent with this result, a previous experiment found no effect of Bd on Shannon diversity, evenness or Chao's richness, and only a weak effect on observed number of OTUs, while a field survey found no correlation between Bd load and richness in within-population analyses [20]. The previous study used individual frogs as the statistical unit of replication, while the current analysis uses population as the unit of replication to understand landscape-level variation. We conclude that, at the landscape scale, any effect of Bd on R. sierrae microbiome richness is small relative to the effect of geographical region. This finding suggests that at this scale, host genetics or environmental variables are more important than Bd in driving among-population variation in microbiome richness. Latitude covaries with transitions between genetic clades within R. sierrae [37], such that the north/south variation in the microbiome may be due to environmental effects, host genetic effects or both. The result that microbiome richness differs by geographical region (and disease outcome) and does not correlate with Bd infection severity across populations suggests that higher microbiome richness has the potential to contribute to R. sierrae population coexistence with Bd. This hypothesis is consistent with a recent finding that bacterial species richness affected the ability of in vitro experimental communities to suppress Bd growth [38], although additional studies are needed to directly demonstrate cause in vivo. Community resistance to invasion can be affected by richness per se (e.g. through more complete use of resources or space), or simply because richness is correlated with the probability that key species are present. Therefore, if richness is causally linked to host response to Bd infection, further work will also be required to understand the mechanism.
5. Conclusion
This study shows that symbiotic bacterial communities differ between enzootic and epizootic host populations. Cause and correlation are notoriously difficult to disentangle in observational field studies. We inferred likely causes for our field patterns by using a separate analysis of factors shaping the microbiome among host populations, which showed that Bd load is the dominant predictor of microbiome composition after alleviating spatial autocorrelation between Bd load and population response to Bd (enzootic versus epizootic). Interestingly, bacterial richness also differed between enzootic and epizootic populations, and appeared robust to Bd infection severity. These results suggest that microbiome richness is more likely than microbiome composition to influence infection outcome for R. sierrae populations, and merit further studies to experimentally test if symbiotic bacterial richness is protective. Other amphibian species have shown variable population-level responses to Bd infection [9] and future research to understand the role of the microbiome in disease dynamics in other species will be important to understanding the generality of our findings.
Supplementary Material
Acknowledgements
We thank C. M. Dorcey for dedicated assistance in the field. E. Hegeman created the map of field sites. C. E. Nelson created the Krona plots in Figure S3. We are grateful to two anonymous reviewers for their constructive critiques. Research permits were provided by the National Park Service, U.S. Forest Service, and UCSB Institutional Animal Care and Use Committee (IACUC). The Sierra Nevada Aquatic Research Laboratory (SNARL) provided field logistical support. DNA sequencing was performed by L. Tomsho and the laboratory of Stephan Schuster (Pennsylvania State University). This project was supported by the National Science Foundation (NSF IOS-1455873 and DGE-1144085 to A.J.J.; DEB-0723563 to C.J.B. and R.A.K.). This article is UH-SOEST publication 10025.
Ethics
All work with R. sierrae was approved by the UCSB Institutional Animal Care and Use Committee (IACUC). No animals were harmed in conducting this study. Surveys and sample collections were conducted in accordance with National Park Service and US Forest Service permits. To prevent researcher-mediated spread of Bd between R. sierrae populations, all field research equipment that contact lake water or frogs was disinfected with 0.1% quaternary ammonium compound 128 solution or 70% ethanol.
Data accessibility
DNA sequence data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA, accession numbers SRR1598941 and SRR1598942). All other data have been deposited in the Dryad Digital Repository at http://dx.doi.org/10.5061/dryad.6p35f [39].
Authors' contributions
A.J.J., C.J.B. and R.A.K. designed the research; A.J.J. and R.A.K. conducted the field surveys; A.J.J. conducted the laboratory analyses and analysed the data; A.J.J. wrote the manuscript with contributions from C.J.B. and R.A.K. All authors gave final approval for publication.
Competing interests
We declare we have no competing interests.
Funding
This project was supported by the National Science Foundation (NSF IOS-1455873 and DGE-1144085 to A.J.J.; DEB-0723563 to C.J.B. and R.A.K.).
References
- 1.Daszak P, Cunningham A, Hyatt A. 2000. Wildlife ecology—emerging infectious diseases of wildlife—threats to biodiversity and human health. Science 287, 443–449. ( 10.1126/science.287.5452.443) [DOI] [PubMed] [Google Scholar]
- 2.Vredenburg VT, Knapp RA, Tunstall TS, Briggs CJ. 2010. Dynamics of an emerging disease drive large-scale amphibian population extinctions. Proc. Natl Acad. Sci. USA 107, 9689–9694. ( 10.1073/pnas.0914111107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lips KR, et al. 2006. Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc. Natl Acad. Sci. USA 103, 3165–3170. ( 10.1073/pnas.0506889103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger L, et al. 1998. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc. Natl Acad. Sci. USA 95, 9031–9036. ( 10.1073/pnas.95.15.9031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford AJ, Lips KR, Bermingham E. 2010. Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proc. Natl Acad. Sci. USA 107, 13 777–13 782. ( 10.1073/pnas.0914115107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rachowicz LJ, Knapp RA, Morgan JA, Stice MJ, Vredenburg VT, Parker JM, Briggs CJ. 2006. Emerging infectious disease as a proximate cause of amphibian mass mortality. Ecology 87, 1671–1683. ( 10.1890/0012-9658(2006)87%5B1671:EIDAAP%5D2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 7.Kilpatrick AM, Briggs CJ, Daszak P. 2010. The ecology and impact of chytridiomycosis: an emerging disease of amphibians. Trends Ecol. Evol. 25, 109–118. ( 10.1016/j.tree.2009.07.011) [DOI] [PubMed] [Google Scholar]
- 8.Briggs CJ, Knapp RA, Vredenburg VT. 2010. Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc. Natl Acad. Sci. USA 107, 9695–9700. ( 10.1073/pnas.0912886107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piovia-Scott J, et al. 2015. Correlates of virulence in a frog-killing fungal pathogen: evidence from a California amphibian decline. ISME J. 9, 1570–1578. ( 10.1038/ismej.2014.241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.James TY, et al. 2015. Disentangling host, pathogen, and environmental determinants of a recently emerged wildlife disease: lessons from the first 15 years of amphibian chytridiomycosis research. Ecol. Evol. 5, 4079–4097. ( 10.1002/ece3.1672) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McFall-Ngai M, et al. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl Acad. Sci. USA 110, 3229–3236. ( 10.1073/pnas.1218525110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH. 2013. Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. ( 10.1038/nrmicro3109) [DOI] [PubMed] [Google Scholar]
- 13.Honda K, Littman DR. 2012. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 30, 759–795. ( 10.1146/annurev-immunol-020711-074937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris RN, et al. 2009. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. ( 10.1038/ismej.2009.27) [DOI] [PubMed] [Google Scholar]
- 15.Kueneman JG, Woodhams DC, Harris R, Archer HM, Knight R, McKenzie VJ. 2016. Probiotic treatment restores protection against lethal fungal infection lost during amphibian captivity. Proc. R. Soc. B 283, 20161553 ( 10.1098/rspb.2016.1553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker MH, et al. 2015. Composition of symbiotic bacteria predicts survival in Panamanian golden frogs infected with a lethal fungus. Proc. R. Soc. B 282, 20142881 ( 10.1098/rspb.2014.2881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker MH, Harris RN, Minbiole KPC, Schwantes CR, Rollins-Smith LA, Reinert LK, Brucker RM, Domangue RJ, Gratwicke B. 2011. Towards a better understanding of the use of probiotics for preventing chytridiomycosis in Panamanian golden frogs. Ecohealth 8, 501–506. ( 10.1007/s10393-012-0743-0) [DOI] [PubMed] [Google Scholar]
- 18.Woodhams DC, Geiger CC, Reinert LK, Rollins-Smith LA, Lam B, Harris RN, Briggs CJ, Vredenburg VT, Voyles J. 2012. Treatment of amphibians infected with chytrid fungus: learning from failed trials with itraconazole, antimicrobial peptides, bacteria, and heat therapy. Dis. Aquat. Organ. 98, 11–25. ( 10.3354/Dao02429) [DOI] [PubMed] [Google Scholar]
- 19.Longo AV, Savage AE, Hewson I, Zamudio KR. 2015. Seasonal and ontogenetic variation of skin microbial communities and relationships to natural disease dynamics in declining amphibians. R. Soc. Open Sci. 2, 140377 ( 10.1098/rsos.140377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jani AJ, Briggs CJ. 2014. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl Acad. Sci. USA 111, E5049–E5058. ( 10.1073/pnas.1412752111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebollar EA, Hughey MC, Medina D, Harris RN, Ibáñez R, Belden LK. 2016. Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J. 10, 1682–1695. ( 10.1038/ismej.2015.234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belden LK, et al. 2015. Panamanian frog species host unique skin bacterial communities. Front. Microbiol. 6, Article 1171 ( 10.3389/fmicb.2015.01171) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knapp RA, Fellers GM, Kleeman PM, Miller DA, Vredenburg VT, Rosenblum EB, Briggs CJ. 2016. Large-scale recovery of an endangered amphibian despite ongoing exposure to multiple stressors. Proc. Natl Acad. Sci. USA 113, 11 889–11 894. ( 10.1073/pnas.1600983113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan JAT, et al. 2007. Population genetics of the frog-killing fungus Batrachochytrium dendrobatidis. Proc. Natl Acad. Sci. USA 104, 13 845–13 850. ( 10.1073/pnas.0701838104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodhams DC, Vredenburg VT, Simon M-A, Billheimer D, Shakhtour B, Shyr Y, Briggs CJ, Rollins-Smith LA, Harris RN. 2007. Symbiotic bacteria contribute to innate immune defenses of the threatened mountain yellow-legged frog, Rana muscosa. Biol. Conserv. 138, 390–398. ( 10.1016/j.biocon.2007.05.004) [DOI] [Google Scholar]
- 26.Lam BA, Walke JB, Vredenburg VT, Harris RN. 2010. Proportion of individuals with anti-Batrachochytrium dendrobatidis skin bacteria is associated with population persistence in the frog Rana muscosa. Biol. Conserv. 143, 529–531. ( 10.1016/j.biocon.2009.11.015) [DOI] [Google Scholar]
- 27.Knapp RA, Briggs CJ, Smith TC, Maurer JR. 2011. Nowhere to hide: impact of a temperature-sensitive amphibian pathogen along an elevation gradient in the temperate zone. Ecosphere 2, Article 93 ( 10.1890/ES11-00028.1) [DOI] [Google Scholar]
- 28.Boyle DG, Boyle DB, Olsen V, Morgan JAT, Hyatt AD. 2004. Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Organ. 60, 141–148. ( 10.3354/dao060141) [DOI] [PubMed] [Google Scholar]
- 29.Lozupone C, Knight R. 2005. Unifrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. ( 10.1128/AEM.71.12.8228-8235.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445. ( 10.1073/pnas.1530509100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradford DF. 1984. Temperature modulation in a high-elevation amphibian, Rana muscosa. Copeia 1984, 966 ( 10.2307/1445341) [DOI] [Google Scholar]
- 32.Walke JB, Becker MH, Loftus SC, House LL, Cormier G, Jensen RV, Belden LK. 2014. Amphibian skin may select for rare environmental microbes. ISME J. 8, 2207–2217. ( 10.1038/ismej.2014.77) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ. 2014. The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250. ( 10.1111/mec.12510) [DOI] [PubMed] [Google Scholar]
- 34.Kohl KD, Yahn J. 2016. Effects of environmental temperature on the gut microbial communities of tadpoles: effects of temperature on tadpole gut microbiome. Environ. Microbiol. 18, 1561–1565. ( 10.1111/1462-2920.13255) [DOI] [PubMed] [Google Scholar]
- 35.Kelly LW, et al. 2014. Local genomic adaptation of coral reef-associated microbiomes to gradients of natural variability and anthropogenic stressors. Proc. Natl Acad. Sci. USA 111, 10 227–10 232. ( 10.1073/pnas.1403319111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL. 2012. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J. 6, 588–596. ( 10.1038/ismej.2011.129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vredenburg VT, Bingham R., Knapp R, Morgan JAT, Moritz C, Wake D. 2007. Concordant molecular and phenotypic data delineate new taxonomy and conservation priorities for the endangered mountain yellow-legged frog. J. Zool. 271, 361–374. ( 10.1111/j.1469-7998.2006.00258.x) [DOI] [Google Scholar]
- 38.Piovia-Scott J, Rejmanek D, Woodhams DC, Worth SJ, Kenny H, McKenzie V, Lawler SP, Foley JE. 2017. Greater species richness of bacterial skin symbionts better suppresses the amphibian fungal pathogen Batrachochytrium dendrobatidis. Microb. Ecol. 74, 217 ( 10.1007/s00248-016-0916-4) [DOI] [PubMed] [Google Scholar]
- 39.Jani AJ, Knapp RA, Briggs CJ. 2017. Data from: Epidemic and endemic pathogen dynamics correspond to distinct host population microbiomes at a landscape scale. Dryad Digital Repository. ( 10.5061/dryad.6p35f) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Jani AJ, Knapp RA, Briggs CJ. 2017. Data from: Epidemic and endemic pathogen dynamics correspond to distinct host population microbiomes at a landscape scale. Dryad Digital Repository. ( 10.5061/dryad.6p35f) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
DNA sequence data have been deposited in the National Center for Biotechnology Information Sequence Read Archive (NCBI SRA, accession numbers SRR1598941 and SRR1598942). All other data have been deposited in the Dryad Digital Repository at http://dx.doi.org/10.5061/dryad.6p35f [39].