

Abstract
Background
Cognitive dysfunction has been increasingly recognized in chronic kidney disease (CKD) patients. Senile plaques are important pathophysiological characteristic of cognitive dysfunction. The major component of plaques is the amyloid β (Aβ) peptide released from proteolytic cleavage of amyloid precursor protein (APP). Plasma Aβ has been a focus of the growing literature on blood based biomarkers for cognitive dysfunction. Oxidative stress is prevalent in CKD and it plays an important role in cognitive dysfunction. Increased oxidative stress leads to cause cleavage of APP and Aβ production. The aim of this study is to assess the antioxidant status and Aβ42 levels in plasma of CKD patients with cognitive dysfunction compared to CKD without cognitive dysfunction.
Methods
A total of 60 subjects divided into 30 CKD without cognitive dysfunction and 30 CKD with cognitive dysfunction based on neuropsychological assessment tests. To compare antioxidant status and Aβ42 levels in plasma, the following groups such as healthy subjects (n = 30), normocytic normochromic anemia (n = 30) and Alzheimer's disease (AD, n = 10) patients were also maintained. Plasma Superoxide dismutase (SOD), Catalase (CAT), Glutathione peroxidase (GPx), Reduced glutathione (GSH) and lipid peroxidation (LPO) were determined by spectrophotometrically. Aβ level was determined by immunoblotting method. The parameters were statistically compared with healthy, normocytic normochromic anemia and AD subjects.
Results
Like AD subjects, significantly increased Aβ and LPO level while decreased SOD, CAT, GPx and GSH levels were observed in plasma of CKD patients with cognitive dysfunction when compared to healthy, CKD without cognitive dysfunction and normocytic normochromic anemic subjects.
Conclusion
Results suggest that elevated plasma oxidative stress and Aβ were seen in CKD patients with cognitive dysfunction may be attributed to pathological changes within the brain.
Keywords: Chronic kidney disease, Cognitive dysfunction, Oxidative stress, APP processing, Amyloid β
Graphical abstract
Graphical representation of the densitometric analysis of Western blot results.
Each bar represents the Mean ± SEM of three independent observations. Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups.
Highlights
-
•
Cognitive dysfunction has been increasingly recognized in chronic kidney disease (CKD) patients.
-
•
The major component of plaques is the amyloid β peptide released from proteolytic cleavage of amyloid precursor protein.
-
•
Plasma Aβ has been a focus of the growing literature on blood based biomarkers for cognitive dysfunction.
-
•
Oxidative stress is prevalent in CKD and it plays an important role in cognitive dysfunction.
-
•
Increased oxidative stress leads to cause cleavage of APP and Aβ production.
1. Introduction
CKD is a rapidly growing global health problem, with a prevalence of 15% in developed nations. It causes many complications such as anemia, pericarditis, cardiovascular disease, etc. [1] Anemia is a common feature of CKD and it is typically normocytic, normochromic and hypoproliferative [2]. Many studies demonstrated that CKD is associated with uremic encephalopathy and cognitive impairment. Cognitive impairment is reported to be as high as 20–40% in CKD [3]. Increased risk of dementia [4], and poorer performance on tests of global cognitive function, executive function, defects in language and memory [5] have been reported among CKD patients and compared to general population, CKD patients are at double the risk of developing dementia and/or cognitive impairment and studies show it has evolved as a possible new determinant of cognitive decline and dementia [6]. Dementia is a primary neurodegenerative disorder and it leads to a complete psychological and physical dependency and finally to death within one to two decades. It involves aberrant protein processing and is characterized by the presence of both intraneuronal protein clusters composed of paired helical filaments of hyperphosphorylated tau protein (Neurofibrillary tangles, NFT) and extracellular amyloid β (Aβ) protein aggregates (Senile plaques) [7]. Aβ is a 39–43 residue protein with a molecular weight of ~ 4 KDa. It is derived by proteolytic cleavage of an integral membrane protein known as amyloid precursor protein (APP) by the action of β- and γ-secretases [7].
Proteomics is a rapidly expanding field of research, which aims to characterize the protein expression in biological fluids and tissues under certain conditions. Protein expression is a very dynamic process that, in contrast to the genome, may change profoundly during disease conditions. A biomarker is an abnormal signal from a bodily fluid or tissue that can provide distinguishable pathological information for a patient. Studies show blood Aβ is an attractive biomarker of dementia and Alzheimer's disease (AD) for the well-characterized efflux mechanism to pass the blood brain barrier (BBB); the low-density lipoprotein receptor-related protein-1 in BBB allows brain Aβ to be actively transported to blood. The increased Aβ levels in blood may in turn result in a stronger Aβ accumulation in cerebral region, which reinforces neuronal degeneration [8]. Plasma is the liquid portion of blood that suspends cells such as red blood cells, white blood cells, and platelets and studies demonstrated that quantification of plasma Aβ is considered as an emerging diagnostic tool for cognitive dysfunction and dementia [9], [10] and it has received strong interest as a reliable biomarker of neurodegenerative diseases, since blood sampling is much less invasive as lumbar puncturing that is currently required for CSF Aβ measurement [8], [11]. Study shown increased plasma Aβ42 peptide levels are associated with earlier onset of AD and increased threat of death [12]. Plasma Aβ levels are elevated and mainly incorporated into lipo-proteins and different plasma proteins [13] and also, high plasma levels of Aβ fragments associated with AD when they accumulate in the brain [14]. Increased levels of plasma Aβ have been reported in AD mouse models also studied [15].
Oxidative stress is a state of imbalance between free radicals production and its degradation by antioxidant systems with increased accumulation of the radicals. Cellular stress response requires the activation of pro-survival pathways that, under control of defensive genes called vitagenes result in the formation of molecules such as heat shock proteins (HSPs), glutathione and bilirubin capable with antioxidant and antiapoptotic actions [16], [17]. Antioxidants are known to dispose, scavenge, suppress the formation of ROS or oppose their action. An important endogenous antioxidant in human is reduced glutathione (GSH), a sulphydryl containing tripeptide (gamma glutamyl cysteinyl glycine) which plays a central role in the defense against oxidative damage and toxins. It serves as a co-factor for GPx and glutathione S-transferase (GST) and can react directly with hydrogen peroxide, super oxide anion, hydroxyl and alkoxyl radicals by its free sulphydryl groups. When present extracellularly, GSH is able to react directly with cytotoxic aldehydes produced during lipid peroxidation. A decrease in the GSH content below the normal level may indicate the disturbance of cellular redox status and change in the redox dependent regulation of genes. A decrease in GSH subjects the cell to the risk of oxidative damage. It has been shown that disturbance in the regulation of GSH level is observed in a wide range of pathologies such as cancer, neurodegenerative diseases, mucoviscidosis, and HIV infection.
Oxidative stress is prevalent in CKD patients and is considered to be an important pathogenic mechanism. Study suggests that the neurotoxic properties of Aβ are mediated by oxidative stress. Oxidative stress is thought to be a key factor in the pathogenesis of AD and mild cognitive impairment [18]. Studies also shown oxidative stress and Aβ production are proportionally linked to each other because Aβ induces oxidative stress in vivo and in vitro [19] and oxidative stress increases the production of Aβ [20]. Based on literatures, till now there is no study on the level of plasma Aβ expression and its association with enzymatic (SOD, CAT, GPx) and non-enzymatic antioxidant (GSH) system in CKD patients with cognitive decline when compared with CKD without cognitive decline. The aim of this study is to determine whether the plasma Aβ expression was also altered along with antioxidant status in CKD patients with cognitive dysfunction and dementia.
2. Materials and methods
2.1. Chemicals
The chemicals and reagents to estimate enzymatic and nonenzymatic antioxidant system were purchased from Himedia and Sisco Research Laboratory (SRL), India. Primary antibodies such as Rabbit Anti Beta Amyloid (1–42) polyclonal (bs-0107R, BIOSS, USA) and Rabbit Anti beta actin polyclonal (bs – 0061R, BIOSS, USA) were purchased from BIOSS, USA, used to detect Aβ and β actin expressions in plasma by Western blot technique. Suitable secondary antibody, Goat Anti Rabbit IgG Antibody (H + L), HRP conjugated was also purchased from BIOSS, USA.
2.2. Participants
This study was carried out in Department of Nephrology, SRM Medical college Hospital, SRM University, Kattankulathur, Tamilnadu, India and approved by Institutional ethical committee (Clearance No. 58/IEC/2010, SRM Medical college Hospital and Research Centre, SRM University, India).
2.2.1. Inclusion criteria
Patients who have been diagnosed to have CKD by the nephrologists in SRM Medical college hospital and either the patient or his relatives who had given informed consent for the study. A total of 60 CKD (Stage V, Age 20 to 50) patients comprising of 30 CKD without cognitive dysfunction and 30 CKD with cognitive dysfunction based on standard neuropsychological assessment tests i.e. Mini Mental scale examination (MMSE), Wechsler memory scale (WMS I) and Tower of London test were included for this study. Separate healthy subjects were also maintained (n = 30) (Table 1).
Table 1.
Demographic and clinical characteristics of healthy subjects, Normocytic Normochromic anemic, Alzheimer's disease, CKD with cognitive dysfunction and CKD without cognitive dysfunction (n = 130).
| Variable | Control (n = 30) | Normocytic normochromic (n = 30) | Alzheimer's disease (n = 10) | CKD with cognitive dysfunction (n = 30) | CKD without cognitive dysfunction (n = 30) |
|---|---|---|---|---|---|
| Age (years) | |||||
| 20–29 | 16 | 13 | 0 | 7 | 8 |
| 30–39 | 13 | 12 | 0 | 6 | 3 |
| 40–49 | 1 | 5 | 0 | 17 | 19 |
| 50–59 | 0 | 0 | 0 | 0 | 0 |
| 60–69 | 0 | 0 | 0 | 0 | 0 |
| 70–79 | 0 | 0 | 7 | 0 | 0 |
| 80–89 | 0 | 0 | 3 | 0 | 0 |
| Gender | |||||
| No. of males | 16 | 12 | 6 | 15 | 20 |
| No. of female | 14 | 18 | 4 | 15 | 10 |
| Education level | |||||
| Middle school (6–8) | 0 | 5 | 5 | 17 | 16 |
| Secondary school (8–9) | 0 | 4 | 2 | 2 | 4 |
| High school (10 − 12) | 21 | 10 | 3 | 8 | 9 |
| Graduate | 5 | 11 | 0 | 2 | 1 |
| Any diploma | 4 | 0 | 0 | 1 | 0 |
| Place of living | |||||
| Rural | 22 | 24 | 1 | 27 | 26 |
| Urban | 8 | 6 | 9 | 3 | 4 |
| Marital status of patients | |||||
| Single | 12 | 17 | 0 | 5 | 5 |
| Married | 18 | 13 | 10 | 25 | 25 |
| Urea (mg/dl) | 22.3 ± 5.6 | 19.5 ± 2.7 | 12.5 ± 2.1 | 79.7 ± 26 | 73.3 ± 25 |
| Creatinine (mg/dl) | 0.53 ± 0.2 | 0.42 ± 0.3 | 0.3 ± 0.2 | 6.8 ± 3.6 | 6.9 ± 3.6 |
| Hemoglobin (g/dl) | |||||
| Male | 14 ± 1.7 | 6.5 ± 1.2 | 10.8 ± 1.5 | 6.8 ± 1.6 | 7.7 ± 2 |
| Female | 13 ± 1.4 | 5.8 ± 1.9 | 9.6 ± 1.4 | 6.5 ± 1.4 | 6.5 ± 3 |
Urea (mg/dl), creatinine (mg/dl) and hemoglobin (g/dl) values are expressed as Mean ± SD.
In this study, to compare the plasma Aβ expression and antioxidant status with CKD patients with and without cognitive dysfunction, we also collected blood samples from Alzheimer's disease (n = 10) and Normocytic normochromic anemic patients (n = 30). Studies shown anemia in most of the CKD patients is normocytic and normochromic type [2].
2.2.2. Exclusion criteria
Patient previously diagnosed with Uraemic encephalopathy, Type 1 diabetes mellitus, Malnutrition, Thrombocytopenia, Cerebro Vascular Accident (CVA), patient or relatives who refused to give consent to the study and patient who were dangerously ill and who had very poor medical condition were also excluded.
2.3. Neuropsychological assessment tests
CKD patients were clinically evaluated by the clinical psychologist. The patients were subjected to a battery of neuropsychological test to assess memory and functional integrity. The standard questionnaire obtained would be translated into regional language (Tamil) and were subjected to validation.
For screening of cognitive status, CKD patients were administered with standardized MMSE according to the method of Cockrell and Folstein [21]. MMSE is a test of global cognitive function with components for concentration, orientation, language, praxis and memory. After conducting MMSE, WMS I was administered to measure different memory functions according to the method of Wechsler [22]. WMS I is made up of seven subtests such as spatial addition, symbol span, design memory, general cognitive screener, logical memory, verbal paired associates and visual reproduction. Person's performance were reported as five index scores such as auditory memory, visual memory, visual working memory, immediate memory and delayed memory. Then to assess executive functioning, Tower of London test were administered to all the groups according to the method of Riccio et al. [23].
2.4. Blood collection and plasma preparation for determination of antioxidant parameters
After obtaining informed consent from subjects, blood samples was collected from all the experimental groups in after overnight fasting into standard vacuum tubes with lithium heparin, and plasma were removed after centrifuging at 3000 rpm for 10 min. The plasma aliquots were immediately used to determine antioxidant parameters.
2.4.1. Determination of plasma antioxidant enzymes
Both the enzymatic and nonenzymatic antioxidant parameters in plasma were determined by standard methods using spectrophotometrically. Superoxide dismutase (SOD, EC 1.15.1.1) was estimated by the method of Marklund and Marklund [24]. The result was expressed as Units/ml. Catalase (CAT, EC 1.11.1.6) was assayed by the method of Sinha [25]. The result was expressed as Units/L (one unit is the amount of enzyme that utilizes 1 μmol of hydrogen peroxide/min/mg protein). Glutathione peroxidase (GPx, EC 1.11.1.9) was determined by the method of Rotruck et al. [26]. The result was expressed as Units/L (one unit is the amount of enzyme that converts 1 μg GSH to GSSG in the presence of hydrogen peroxide/min/mg protein).
2.4.2. Determination of plasma GSH and LPO
Reduced glutathione (GSH) level in the plasma of all the groups was determined by the method of Moron et al. [27]. The amount of reduced glutathione was expressed as mg/dl. Lipid peroxidation (LPO) was determined using the method of Devasagayam and Tarachand [28]. The result was expressed as nmol of MDA formed/mg protein.
2.5. Determination of plasma Aβ (1–42) expression by Western blot method
Protein concentrations were determined in each sample according to Lowry method [29], before the Western blot assay.
2.5.1. Western blotting
Determination Aβ (1–42) level in plasma was carried out as per the standard protocol. For each sample, 24 μg of protein was separated by electrophoresis in 12% polyacrylamide gels and transferred to nitrocellulose membranes. After block unspecific bindings with 5% non fat dry milk, membranes were incubated for overnight with the primary antibody Rabbit Anti Beta Amyloid (1–42) polyclonal (bs-0107R, BIOSS, USA). After an overnight incubation, membranes were then incubated for 2 h with goat Anti Rabbit IgG Antibody (H + L) labeled with horse radish peroxidase (bs-0295G- HRP, BIOSS, USA), after which the enhanced chemiluminescence (ECL) reagent was poured. Imaging was performed in GELSTAN 4 × Chemi imaging system (Medicare scientific, India), which captures the chemiluminescence light emission from the reaction of the peroxidase conjugated antibody with ECL reagent. Light emission was continuously captured for 15 Min, and the densitometry of distinct bands was performed and quantified with specific software tools. As a control for detection of beta actin expression in plasma, Rabbit Anti beta actin polyclonal antibody (bs – 0061R, BIOSS, USA) was used. Then the densities of bands from CKD with and without cognitive dysfunction groups were compared with healthy, normocytic normochromic and AD subjects.
2.6. Statistical analysis
The statistical analysis of the results were conducted using SPSS software version 21.0, the independent t-test and Analysis of variance (ANOVA) followed by Turkey's multiple comparison tests. The probability level < 0.05 (P < 0.05) were considered as significant. Results were expressed as Mean ± SEM.
3. Results
3.1. Plasma enzymatic and nonenzymatic antioxidants level
Fig. 1 shows the plasma superoxide dismutase (SOD) level in patients of CKD with and without cognitive dysfunction. The level was significantly decreased in patients of CKD with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction and normocytic normochromic anemia. In overall also, plasma SOD levels of above experimental groups were significantly decreased when compared with healthy subjects.
Fig. 1.

The values were expressed as Mean ± SEM of Healthy subjects (n = 30), Normocytic normochromic Anemia (n = 30), Alzheimer's disease (n = 10), CKD with cognitive dysfunction (n = 30) and CKD without cognitive dysfunction (n = 30). Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups.
Fig. 2 shows the plasma catalase (CAT) level in patients of CKD with and without cognitive dysfunction. The level was significantly decreased in patients of CKD with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction and normocytic normochromic anemia. In overall also, plasma CAT level of above experimental groups were significantly decreased when compared with healthy subjects like SOD.
Fig. 2.

The values were expressed as Mean ± SEM of Healthy subjects (n = 30), Normocytic normochromic Anemia (n = 30), Alzheimer's disease (n = 10), CKD with cognitive dysfunction (n = 30) and CKD without cognitive dysfunction (n = 30). Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups.
Fig. 3 shows the plasma Glutathione peroxidase (GPx) level in patients of CKD with and without cognitive dysfunction. The level was significantly decreased in patients of CKD with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction and normocytic normochromic anemia. In overall also, plasma GPx level of above experimental groups were significantly decreased when compared with healthy subjects like SOD and CAT.
Fig. 3.

The values were expressed as Mean ± SEM of Healthy subjects (n = 30), Normocytic normochromic Anemia (n = 30), Alzheimer's disease (n = 10), CKD with cognitive dysfunction (n = 30) and CKD without cognitive dysfunction (n = 30). Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups.
Fig. 4 shows the plasma reduced glutathione (GSH) level in patients of CKD with and without cognitive dysfunction. The level was significantly decreased in patients of CKD with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction and normocytic normochromic anemia. Like enzymatic antioxidant parameters, in overall also, plasma GSH level of above experimental groups was significantly decreased when compared with healthy subjects.
Fig. 4.

The values were expressed as Mean ± SEM of Healthy subjects (n = 30), Normocytic normochromic Anemia (n = 30), Alzheimer's disease (n = 10), CKD with cognitive dysfunction (n = 30) and CKD without cognitive dysfunction (n = 30). Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups.
3.2. Plasma LPO level and amyloid β expression
Fig. 5(a) shows the plasma lipid peroxidation (LPO) level in patients of CKD with and without cognitive dysfunction. The level was significantly increased in patients of CKD with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction and normocytic normochromic anemia. In overall also, LPO level of above experimental groups were significantly increased when compared with healthy subjects.
Fig. 5.

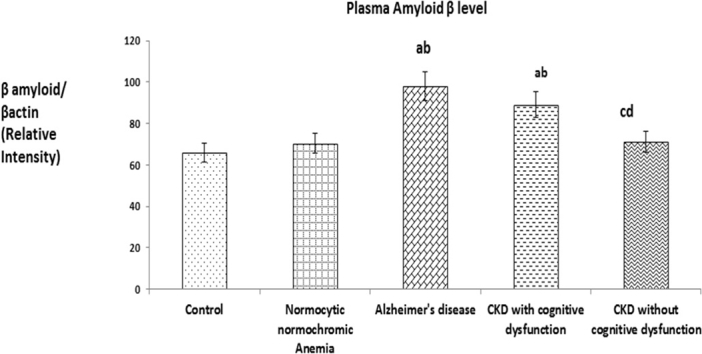
(a). The values were expressed as Mean ± SEM of Healthy subjects (n = 30), Normocytic normochromic Anemia (n = 30), Alzheimer's disease (n = 10), CKD with cognitive dysfunction (n = 30) and CKD without cognitive dysfunction (n = 30). Letters a, b, c and d denote the statistical significance of the data at the level of P < 0.05. (a) denote comparison of control Vs. other groups; (b) denote comparison of normocytic normochromic anemia Vs. Alzheimer's disease (AD), CKD with and without cognitive dysfunction groups; (c) denote comparison of AD Vs. CKD with and without cognitive dysfunction groups; (d) denote comparison between CKD with and without cognitive dysfunction groups. (b). Expression of plasma Aβ (1–42). Representative Western blots illustrating bands of amyloid β (1–42) in plasma from healthy subjects, normocytic normochromic anemia, Alzheimer's disease, CKD with and without cognitive dysfunction groups.
Fig. 5(b) shows representative Western blots illustrating bands of plasma amyloid β from healthy subjects, normocytic normochromic anemia, Alzheimer's disease, CKD with and without cognitive dysfunction groups. The plasma Aβ (1–42) expression was significantly increased in CKD patients with cognitive dysfunction like AD patients when compared to patients of CKD without cognitive dysfunction, normocytic normochromic anemia and healthy subjects.
3.3. Correlation between plasma LPO and Aβ42 expression
Cognitive dysfunction has been increasingly recognized in chronic kidney disease (CKD) patients. The major component of plaques is the amyloid β (Aβ) peptide released from proteolytic cleavage of amyloid precursor protein (APP). Oxidative stress is prevalent in CKD and it plays an important role in cognitive dysfunction. Increased oxidative stress leads to cause cleavage of APP and Aβ production. We examined relationship between plasma LPO levels and Aβ42 expressions. The linear regression analysis was carried out on a scatter plot of plasma LPO levels against Aβ42 expressions of the respective patients, and Pearson's correlation coefficient was determined. The significantly positive correlation found between plasma LPO levels and Aβ42 expression in patients with (Fig. 6A) Healthy subjects (r = 0.962, P ≤ 0.0001), (Fig. 6B) Normocytic normochromic Anemia (r = 0.9765, P ≤ 0.0001), (Fig. 6C) Alzheimer's disease (r = 0.9856, P ≤ 0.0001), (Fig. 6D) CKD with cognitive dysfunction (r = 0.9713, P ≤ 0.0001), and (Fig. 6E) CKD without cognitive dysfunction (r = 0.9706, P ≤ 0.0001).
Fig. 6.

Correlation between plasma lipid peroxidation and β amyloid-42 expression in (A) Healthy subjects (n = 30), (B) Normocytic normochromic Anemia (n = 30), (C) Alzheimer's disease (n = 10), (D) CKD with cognitive dysfunction (n = 30) and (E) CKD without cognitive dysfunction (n = 30). Correlations were investigated using simple linear regression analysis.
4. Discussion
CKD is a serious and rapidly growing health problem in world wide. Neurological complications occur in almost all patients with severe CKD, potentially affecting all levels of the nervous system, from the central nervous system through to the peripheral nervous system. Cognitive dysfunction has long been recognized as complication of CKD [30], and its prevalence is more than double compared to the general population and is dependent on the severity of CKD.
Mitochondria are an essential cause of ROS within most mammalian cells and the initial ROS obtained by one electron reduction of molecular oxygen is the superoxide anion (O2−*). Over 90% of ROS production by mitochondria can lead to oxidative damage to mitochondrial proteins, membranes and DNA, impairing the capability of mitochondria to synthesize adenosine triphosphate (ATP) and to carry out their wide range of intermediary metabolic functions, including the tricarboxylic acid (TCA) cycle, fatty acid oxidation, urea cycle, amino acid metabolism, heme synthesis and iron-sulfur (FeS) centre assembly that are vital to the regular process of most cells [31]. Many clinical studies have confirmed the key role played by Reactive oxygen species (ROS) in acute and chronic kidney diseases. Renal failure is frequently associated with oxidative stress, as levels of different markers including plasma F2-isoprostanes, advanced oxidation protein products and malonyldialdehyde (MDA) are increased in patients with varying degrees of renal function, including CKD patients [32], [33], [34], [35], [36], [37].
In CKD patients enhanced ROS production is underlain mainly by inflammation, malnutrition and presence of endogenous stable oxidants in the uremic plasma [38], [39]. The kidney is greatly energetic and therefore relies heavily on aerobic metabolism for the production of ATP by oxidative phosphorylation. The decrease in molecular O2 along the ETC within mitochondria is vital for renal cellular function, yet potentially devastating long-term effect. Studies show, the impaired mitochondrial respiration was recorded in CKD patients and deregulation of the mitochondrial respiratory machinery in CKD patients, closely associated with enhanced oxidative stress [40]. Uremic toxins especially uric acid may also be a source of oxidative stress in CKD patients directly through the activity of xanthine oxidoreductase. Malondialdehyde (MDA) is another end-product generated by lipid peroxidation and has been used to demonstrate increased oxidative stress during CKD.
Several studies show the importance of oxidative stress in the pathogenesis of cognitive impairment. Study also clearly demonstrates that oxidative stress damage occurs in patients with mild cognitive impairment (MCI) and some enzymatic markers of oxidative stress are similar in MCI and Alzheimer's disease (AD) patients, suggesting that oxidative damage could be one important aspect for the onset of severe cognitive impairment [41]. It is generally recognized that biological systems possess integrated responses to detect and control the various forms of stress. This is accomplished by a complex network of the so-called longevity assurance processes, which are composed of several genes termed Vitagenes and it produce molecules such as heat shock proteins, glutathione, bilirubin endowed with antioxidant and anti-apoptotic activities [16], [17]. The antioxidant function of GSH is accomplished largely by GSH peroxidase (GPx)- catalyzed reactions, which reduce hydrogen peroxide and lipid peroxide as GSH is oxidized to GSSG. GSSG in turn is reduced back to GSH by GSSG reductase at the expense of NADPH, forming a redox cycle. Organic peroxides can also be reduced by GPx and GSH S-transferase. Catalase can also reduce hydrogen peroxide but it is present only in peroxisome. This makes GSH particularly important in the mitochondria in defending against both physiologically and pathologically generated oxidative stress. A diminished GSH content below the normal level may indicate the disturbance of cellular redox status and change in the redox dependent regulation of genes and the risk of oxidative damage. It has been shown that interruption in the regulation of GSH level is observed in a wide range of pathologies including neurodegenerative diseases. Studies also suggest that high lipid peroxidation and decreased antioxidant defenses may be present early in cognitive disorders [42]. In the present study, (Fig. 1, Fig. 2, Fig. 3, Fig. 4) both enzymatic (SOD, CAT, GPx) and non enzymatic antioxidant (GSH) parameters were decreased while LPO level and Aβ expression (Fig. 5a & b) were increased in plasma of patients of CKD with cognitive dysfunction like AD subjects when compared to CKD without cognitive dysfunction.
Studies show, increased oxidative stress leads to cause cleavage of APP and Aβ production [19], [20]. In this study (Fig. 6A, B, C, D & E), we observed significant positive correlation between plasma LPO levels and Aβ42 expression in subjects of all groups by Pearson's correlation coefficient analysis (P ≤ 0.0001, scatter plot analysis) and from this we demonstrate that the elevated plasma LPO level is associated with increased plasma Aβ42 expression. Based on literatures and the result from this study also, the increased oxidative stress may be one of the factors for cognitive dysfunction in CKD patients when compared to CKD without cognitive dysfunction.
Deposits of Aβ (39–43 amino acid sequence) are the characteristic pathological hallmarks of neurodegenerative disorder and the most common form of senile dementia. Aβ is released by the proteolytic cleavage of APP and aggregates to form oligomers and estimation of plasma Aβ is considered as a promising diagnostic tool for AD and cognitive impairment. In this study, the elevated plasma Aβ was observed in AD patients and this finding is consistent with previous studies that have demonstrated increasing level of the same [12], [13], [14], [15]. In the present study, the increased Aβ was found in plasma of CKD patients with cognitive dysfunction like AD groups when compared to CKD patients without cognitive dysfunction, normocytic normochromic anemic and healthy subjects. Studies show that oxidative stress and Aβ production are proportionally linked to each other because Aβ induces oxidative stress in vivo and in vitro [19], and oxidative stress increases the production of Aβ by proteolytic cleavage of APP [43], [44] and Methionine 25 has been critical residue in Aβ (1–42) mediated oxidative stress [45]. In this study, increased Aβ in plasma may be due to increased level of LPO in CKD subjects with cognitive decline. Study in animal models also shown increased LPO precedes amyloid plaque formation in a mice model of Alzheimer amyloidosis [46]. Some studies also shown abnormal and increased plasma Aβ levels seen in patients associated with cognitive decline and dementia [9], [10], [47], [48].
5. Conclusion
In the present study, we demonstrated that decreased enzymatic, nonenzymatic antioxidant parameters while increased LPO level were seen in plasma of CKD patients with cognitive dysfunction when compared to other groups. Abnormal increased expression of Aβ was also seen in same trend. In conclusion, based on results from this study, we proved that abnormal plasma Aβ expression along with altered enzymatic and nonenzymatic antioxidant status are also the factors for cognitive decline in CKD patients apart from the accumulation uremic toxins. This determination is a good diagnostic accuracy for the identification and confirmation of cognitive decline in CKD patients. Among the cellular pathways conferring protection against oxidative stress, a key role is played by vitagenes, which include HSPs Heme oxygenase-1 (HO-1) and HSP70, as well as thioredoxin/thioredoxin reductase system. In future, analysis of those genes may also provide therapeutic benefit in oxidative stress associated cognitive dysfunction in CKD patients.
Conflicts of interest
All authors have no conflicts of interest to declare.
Transparency document
Transparency document.
Acknowledgements
This work was financially supported by a grant from the Indian Council of Medical Research (No. GIA/77/2014-DHR), New Delhi, India. Also, we gratefully acknowledge Ms. Riya, Department of Clinical psychology, SRM Medical college hospital and Research centre, SRM University, for her help in the assessment of cognitive function in CKD patients.
Footnotes
The Transparency document associated with this article can be found, in the online version.
References
- 1.Hossain M.P., Goyder E.C., Rigby J.E., Nahas M.E.I. CKD and poverty: a growing global challenge. Am. J. Kidney Dis. 2009;53:166–174. doi: 10.1053/j.ajkd.2007.10.047. [DOI] [PubMed] [Google Scholar]
- 2.Babitt J.L., Lin H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012;23:1631–1634. doi: 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray A.M. Cognitive impairment in the aging dialysis and chronic kidney disease populations: an occult burden. Adv. Chronic Kidney Dis. 2008;15:123–132. doi: 10.1053/j.ackd.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seliger S.L., Siscovick D.S., Stehman-Breen C.O., Gillen D.L., Fitzpatrick A., Bleyerm A., Kuller L.H. Moderate renal impairment and risk of dementia among older adults: the cardiovascular health cognition study. J. Am. Soc. Nephrol. 2004;15:1904–1911. doi: 10.1097/01.asn.0000131529.60019.fa. [DOI] [PubMed] [Google Scholar]
- 5.Kurella M., Yaffe K., Shlipak M.G., Wenger N.K., Chertow G.M. Chronic kidney disease and cognitive impairment in menopausal women. Am. J. Kidney Dis. 2005;45:66–76. doi: 10.1053/j.ajkd.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 6.Fukunishi I., Kitaoka T., Shirai T., Kino K., Kanematsu E., Sato Y. Psychiatric disorders among patients undergoing hemodialysis therapy. Nephron. 2002;91:344–347. doi: 10.1159/000058418. [DOI] [PubMed] [Google Scholar]
- 7.Butterfield D.A., Reed T., Newman S.F., Sultana R. Roles of amyloid beta peptide associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic. Biol. Med. 2007;43(5):658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gronewold J., Klafki H.W., Baldelli E., Kaltwasser B., Seidel U.K., Todica O., Volsek M., Haußmann U., Wiltfang J., Kribben A., Bruck H., Hermann D.M. Factors responsible for plasma β-amyloid accumulation in chronic kidney disease. Mol. Neurobiol. 2016;53:3136–3145. doi: 10.1007/s12035-015-9218-y. [DOI] [PubMed] [Google Scholar]
- 9.LladoSaz S., Atienza M., Cantero J.L. Increased levels of plasma amyloid-beta are related to cortical thinning and cognitive decline in cognitively normal elderly subjects. Neurobiol. Aging. 2015;36(10):2791–2797. doi: 10.1016/j.neurobiolaging.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 10.Poljak A., Sachdev P.S. Plasma amyloid beta peptides: an Alzheimer's conundrum or a more accessible Alzheimer's biomarker? Expert. Rev. Neurother. 2017;17(1):3–5. doi: 10.1080/14737175.2016.1217156. [DOI] [PubMed] [Google Scholar]
- 11.Figurski M.J., Waligórska T., Toledo J., Vanderstichele H., Korecka M., Lee V.M.Y., Trojanowski J.Q., Shaw L.M. Improved protocol for measurement of plasma β-amyloid in longitudinal evaluation of Alzheimer's Disease Neuroimaging Initiative study patients. Alzheimers Dement. 2012;8(4):250–260. doi: 10.1016/j.jalz.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schupf N., Patel B., Pang D., Zigman W.B., Silverman W., Mehta P.D., Mayeux R. Elevated plasma-amyloid peptide A_42 levels, incident dementia, and mortality in down syndrome. Arch. Neurol. 2007;64(7):1007–1013. doi: 10.1001/archneur.64.7.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biere A.L., Ostaszewski B., Stimson E.R., Hyman B.T., Maggio J.E., Selkoe D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 14.Cosentino S.A., Stern Y., Sokolov E., Scarmeas N., Manly J.J., Tang M.X., Schupf N., Mayeux R.P. Plasma-amyloid and cognitive decline. Arch. Neurol. 2010;67(12):1485–1490. doi: 10.1001/archneurol.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho S.M., Kim H.V., Lee S., Kim H.Y., Kim W., Kim T.S., Kim D.J., Kim Y.S. Correlations of amyloid-b concentrations between CSF and plasma in acute Alzheimer mouse model. Sci Rep. 2014;4:6777. doi: 10.1038/srep06777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Iavikoli I., Paola R.D., Koverech A., Cuzzocrea S., Rizzarelli E., Calabrese E.J. Cellular stress responses, hermetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta. 2012;1822:753–783. doi: 10.1016/j.bbadis.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Calabrese V., Cornelius C., Leso V., Trovato-Salinaro A., Ventimiglia B., Cavallaro M., Scuto M., Rizza S., Zanoli L., Neri S., Castellino P. Oxidative stress, glutathione status, sirtuin and cellular stress response in type 2 diabetes. Biochim. Biophys. Acta. 2012;1822:729–736. doi: 10.1016/j.bbadis.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield D.A. Beta amyloid associated free radical oxidative stress and neurotoxicity: implications for Alzheimer's disease. Chem. Res. Toxicol. 1997;10:495–506. doi: 10.1021/tx960130e. [DOI] [PubMed] [Google Scholar]
- 19.Tabner B.J., El-Agnaf O.M., Turnbull S., German M.J., Paleologou K.E., Hayashi Y., Copper L.J., Fullwood N.J., Allsop D. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer's disease and familial British dementia. J. Biol. Chem. 2005;280:35789–35792. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- 20.Tamagno E., Guglielmotto M., Aragno M., Borghi R., Autelli V., Giliberto L., Muraca G., Danni O., Zhu X., Smith M.A., Perry G., Jo D.G., Mattson M.P., Tabaton M. Oxidative stress activates a positive feedback between the c- and β-secretase cleavages of the β-amyloid precursor protein. J. Neurochem. 2008;104(3):683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cockrell J.R., Folstein M.F. Mini mental state examination (MMSE) Psychopharmacol. Bull. 1988;24:689–692. [PubMed] [Google Scholar]
- 22.Wechsler D. Psychological. Corporation; San Antonio: 1987. Wechsler Memory Scale-Revised Manual. [Google Scholar]
- 23.Riccio C.A., Wolfe M.E., Romine C., Davis B., Sullivan J.R. The tower of London and neuropsychological assessment of ADHD in adults. Arch. Neuropsychol. 2004;19:661–671. doi: 10.1016/j.acn.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Marklund S., Marklund G. Involvement of superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 25.Sinha A.K. Colorimetric assay of catalase. Anal. Biochem. 1972;47:389–394. doi: 10.1016/0003-2697(72)90132-7. [DOI] [PubMed] [Google Scholar]
- 26.Rotruck J.T., Pope A.L., Ganther H.E., Saunson A.B., Hafeman G., Hoek-straw W.G. Selenium; biochemical role as a component of glutathione peroxidase. Science. 1973;179:588–590. doi: 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- 27.Moron M.S., Depierre J.W., Mannervik B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochem. Biophys. Acta. 1979;582:67–78. doi: 10.1016/0304-4165(79)90289-7. [DOI] [PubMed] [Google Scholar]
- 28.Devasagayam T.P., Tarachand U. Decreased lipid peroxidation in rat kidney during gestation. Biochem. Biophys. Res. Commun. 1987;145:134–138. doi: 10.1016/0006-291x(87)91297-6. [DOI] [PubMed] [Google Scholar]
- 29.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the folin-phenol reagent. J. Biol. Chem. 1951;193:265–270. [PubMed] [Google Scholar]
- 30.Sehgal A.R., Grey S.F., DeOreo P.B. Prevalence, recognition, and implications of mental impairment among hemodialysis patients. Am. J. Kidney Dis. 1997;30:41–49. doi: 10.1016/s0272-6386(97)90563-1. [DOI] [PubMed] [Google Scholar]
- 31.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dounousi E., Papavasiliou E., Makedou A. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006;48:752–760. doi: 10.1053/j.ajkd.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 33.Massy Z.A., Stenvinkel P., Drueke T.B. The role of oxidative stress in chronic kidney disease. Semin. Dial. 2009;22(4):405–408. doi: 10.1111/j.1525-139X.2009.00590.x. [DOI] [PubMed] [Google Scholar]
- 34.Modaresi A., Nafar M., Sahraei Z. Oxidative stress in chronic kidney disease. Iran. J. Kidney Dis. 2015;9(3):165–179. [PubMed] [Google Scholar]
- 35.Forbes J.M., Soulis T., Thallas V. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia. 2001;44(1):108–114. doi: 10.1007/s001250051587. [DOI] [PubMed] [Google Scholar]
- 36.Goh S., Cooper M.E. The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrin. Metab. 2008;93(4):1143–1152. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- 37.Meenakshi Sundaram S.P., Nagarajan S., Manjula Devi A.J. Chronic kidney disease-effect of oxidative stress. Chin. J. Biol. 2014:1–6. [Google Scholar]
- 38.Roselaar S.E., Nazhat N.B., Winyard P.G., Jones P., Cunningham J., Blake D.R. Detection of oxidants in uremic plasma by electron spin resonance spectroscopy. Kidney Int. 1995;48:199–206. doi: 10.1038/ki.1995.285. [DOI] [PubMed] [Google Scholar]
- 39.Locatelli F., Canaud B., Eckardt K.U., Stenvinkel P., Wanner C., Zoccali C. Oxidative stress in end-stage renal disease: an emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003;18:1272–1280. doi: 10.1093/ndt/gfg074. [DOI] [PubMed] [Google Scholar]
- 40.Kao M.P., Ang D.S., Pall A., Struthers A.D. Oxidative stress in renal dysfunction: mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010;24(1):1–8. doi: 10.1038/jhh.2009.70. [DOI] [PubMed] [Google Scholar]
- 41.Padurariu M., Ciobica A., Hritcu L., Stoica B., Bild W., Stefanescu C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer's disease. Neurosci. Lett. 2010;469:6–10. doi: 10.1016/j.neulet.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 42.Torres L.L., Quaglio N.B., de Souza G.T., Garcia R.T., Dati L.M., Moreira W.L., Loureiro A.P., de Souza-Talarico J.N., Smid J., Porto C.S., Bottino C.M., Nitrini R., Barros S.B., Camarini V., Marcourakis T. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer's disease. J. Alzheimers Dis. 2011;26(1):59–68. doi: 10.3233/JAD-2011-110284. [DOI] [PubMed] [Google Scholar]
- 43.Tong Y., Zhou W., Fung V., Christensen M.A., Qing H., Sun X., Song W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- 44.Tamagno E., Guglielmotto M., Monteleone D., Tabaton M. Amyloid-b production: major link between oxidative stress and BACE1. Neurotox. Res. 2012;22:208–219. doi: 10.1007/s12640-011-9283-6. [DOI] [PubMed] [Google Scholar]
- 45.Butterfield D.A., Swomley A.M., Sultana R. Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013;19(8):823–835. doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Praticò D., Uryu K.S., Leight, Trojanoswki J.Q., Lee V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001;21(12):4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Assini A., Cammarata S., Vitali A., Colucci M., Giliberto L., Borghi R., Inglese M.L., Volpe S., Ratto S., Dagna-Bricarelli F., Baldo C., Argusti A., Odetti P., Piccini A., Tabaton M. Plasma levels of amyloid-protein 42 are increased in women with mild cognitive impairment. Neurology. 2004;63:828–831. doi: 10.1212/01.wnl.0000137040.64252.ed. [DOI] [PubMed] [Google Scholar]
- 48.Coppus A.M., Schuur M., Vergeer J., Janssens A.C., Oostra B.A., Verbeek M.M., Van Duijn C.M. Plasma β amyloid and the risk of Alzheimer's disease in Down syndrome. Neurobiol. Aging. 2012;33(9):1988–1994. doi: 10.1016/j.neurobiolaging.2011.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.