

Abstract
Homologous recombination plays key roles in double-strand break repair, rescue, and repair of stalled replication forks and meiosis. The broadly conserved Rad51/RecA family of recombinases catalyzes the DNA strand invasion reaction that takes place during homologous recombination. We have established single-stranded (ss)DNA curtain assays for measuring individual base triplet steps during the early stages of strand invasion. Here, we examined how base triplet stepping by RecA, Rad51, and Dmc1 is affected by DNA sequence imperfections, such as single and multiple mismatches, abasic sites, and single nucleotide insertions. Our work reveals features of base triplet stepping that are conserved among these three phylogenetic lineages of the Rad51/RecA family and also reveals lineage-specific behaviors reflecting properties that are unique to each recombinase. These findings suggest that Dmc1 is tolerant of single mismatches, multiple mismatches, and even abasic sites, whereas RecA and Rad51 are not. Interestingly, the presence of single nucleotide insertion abolishes recognition of an adjacent base triplet by all three recombinases. On the basis of these findings, we describe models for how sequence imperfections may affect base triplet recognition by Rad51/RecA family members, and we discuss how these models and our results may relate to the different biological roles of RecA, Rad51, and Dmc1.
Keywords: DNA repair, homologous recombination, microscopy, protein-DNA interaction, single-molecule biophysics, Dmc1, Rad51, RecA
Introduction
Homologous recombination allows for the regulated exchange of genetic information between two different DNA molecules of identical or nearly identical sequence composition, and it is a driving force in evolution (1, 2). Homologous recombination contributes to double-strand DNA break (DSB)4 repair (3), the rescue of stalled or collapsed replication forks (4–6), chromosomal rearrangements (7–9), horizontal gene transfer (10), and meiosis (11–13). Defects in recombination compromise genome integrity and lead to the gross chromosomal rearrangements that are a hallmark of cancer (7–9). During recombination, single-stranded DNA (ssDNA), derived from the nucleolytic processing of a DSB or collapsed replication fork, is paired with the complementary strand of a homologous double-stranded DNA (dsDNA), resulting in the displacement of the non-complementary strand from the duplex to generate a D-loop (14, 15). This reaction is referred to as strand invasion, and the resulting intermediate can be channeled through a number of alternative pathways, any of which can allow for the repair of the originally broken DNA molecule using information derived from the template (3, 4).
The homology search and strand invasion reactions are catalyzed by the Rad51/RecA family DNA recombinases (14–16). These recombinases are among the most highly conserved of all DNA repair proteins (17), and prominent family members include bacterial RecA, the archaeal protein RadA, and the eukaryotic recombinase Rad51 (16, 18). In addition to Rad51, most eukaryotes also have the meiosis-specific recombinase Dmc1, although the reason most eukaryotes require a second recombinase for meiosis remains an enduring mystery (11, 19).
RecA is the archetypal recombinase originally identified in genetic screens for Escherichia coli mutants defective in recombination (20), and much of our current understanding of recombination mechanisms can be attributed to studies of this recombinase (15, 21). Rad51/RecA family members are all ATP-dependent DNA-binding proteins that form extended helical filaments on DNA (15, 16). The bound DNA is extended by ∼50% relative to the contour length of B-form DNA (22–26), and crystal structures of RecA-ssDNA and RecA-dsDNA pre- and post-synaptic complexes reveal that the bound DNA is organized into near B-form base triplets separated by ∼8 Å between adjacent triplets (Fig. 1A) (27). We have referred to this unique DNA architecture as recombinase stretched-DNA (RS-DNA) to distinguish it from other forms of mechanically stretched DNA and as a reflection of its unique structural and mechanistic properties (28). To help better understand the mechanisms of genetic recombination, we have established single-molecule assays for studying how pre-synaptic complexes engage dsDNA during the early stages of homologous recombination (28–30). These experiments reveal that strand invasion products are stabilized in 3-nt increments (Fig. 1B), with each 3-nt step exhibiting a characteristic energetic signature that is conserved among the Rad51/RecA family members (28–30).
Figure 1.

Base triplet stepping by the Rad51/RecA family of recombinases. A, crystal structure of the RecA-ssDNA pre-synaptic complex highlighting the base triplet organization of the bound pre-synaptic ssDNA. B, schematic illustration of base triplet stepping, in which each step coincides with complete pairing of all three bases within an RS-DNA triplet. C, experimental assay for quantitating the binding of Atto565-labeled dsDNA fragments to an unlabeled pre-synaptic complex. These panels were adapted with permission from Ref. 28.
Here, we examine how sequence imperfections affect individual base triplet steps by E. coli RecA, Saccharomyces cerevisiae Rad51, and S. cerevisiae Dmc1. We show that RecA, Rad51, and Dmc1 all require perfect Watson-Crick pairing interactions to stabilize base triplet pairing interactions at either the 5′ or 3′ termini of paired intermediates. Single nucleotide insertions completely disrupt base triplet recognition by all three recombinases, underscoring the importance of maintaining proper sequence register during strand exchange. RecA and Rad51 can step over base triplets harboring single mismatches or abasic sites, and Rad51 can step over tandem mismatches, but neither of these recombinases can stabilize the imperfectly paired triplets. In contrast, the meiosis-specific recombinase Dmc1 can step over and stabilize base triplets harboring single mismatches, multiple mismatches, and even abasic sites. Our results are consistent with a model where Dmc1 makes compensatory protein-DNA contacts with lesion-bearing or otherwise mismatched base triplets allowing stabilization of the resulting intermediates.
Results
Assay for base triplet recognition with ssDNA curtains
We used ssDNA curtains and total internal reflection fluorescence microscopy to visualize RecA, Rad51, or Dmc1 pre-synaptic complexes. The ssDNA was generated using M13 as a template for rolling circle replication and then tethered to a lipid bilayer through a biotin-streptavidin linkage and aligned along chromium barriers, as described previously (28, 29, 31, 32). The ssDNA unravels when incubated with RPA-eGFP, and the downstream ends of the RPA-ssDNA are anchored to exposed chromium pedestals through non-specific surface adsorption. Addition of RecA, Rad51, or Dmc1, along with the required nucleotide cofactor, results in the displacement of the fluorescent RPA-eGFP from the ssDNA (28, 29). Once assembled, unbound proteins are flushed away, and dsDNA-binding activity of the pre-synaptic complexes is probed by the addition of short (70-bp) dsDNA oligonucleotides bearing a single tract of sequence microhomology 8–15 nucleotides (nt) in length that is complementary to a unique location on the M13 ssDNA (28, 29). Following a short incubation, the unbound dsDNA is flushed away, and the stability of the remaining dsDNA molecules is measured by survival probability analysis (Fig. 1C) (28, 29, 32).
Microhomology and base triplet recognition
We have previously reported assays in which the internal tract of microhomology was incrementally extended from 8- to 15-nt in the 5′ → 3′ direction relative to the complementary strand within the fluorescent dsDNA (28, 29). These assays, together with accompanying molecular dynamics simulations, revealed that strand invasion intermediates were stabilized in 3-nt increments (28, 29). Here, we sought to determine whether similar results would be obtained if we instead extended the tract of microhomology in the 3′ → 5′ direction (Fig. 2A). To address this question, we used a series of 70-bp oligonucleotides labeled at one end with a single Atto565 dye and bearing an internal tract of microhomology targeted to a specific region on the pre-synaptic ssDNA. The internal tracts of microhomology were flanked by non-homologous sequences that lacked any microhomology exceeding 7 nt in length (supplemental Table S1). E. coli RecA, S. cerevisiae Rad51, or S. cerevisiae Dmc1 pre-synaptic complexes were assembled onto the ssDNA curtains. The fluorescent dsDNA substrates were then injected into the sample chamber and incubated for 10 min at 30 °C, and the unbound DNA was then flushed away while collecting data. Dissociation rates were then obtained from survival probability analysis of the bound dsDNA fragments (supplemental Fig. S1) (28, 29). These experiments revealed that extension of the internal tract of microhomology in the 3′ → 5′ direction yielded changes in dissociation rates for RecA, Rad51, and Dmc1, similar to our previously reported results for 5′ → 3′ extension (Fig. 2, B–D) (28, 29). In addition, each step coincided with an ∼30% change in relative dissociation rates, corresponding to change in free energy (ΔΔG‡) of ∼0.3 kBT (Fig. 2, B–D). From these results, we concluded that 3′ → 5′ extension of an internal tract of microhomology gave rise to periodic changes in dsDNA dissociation rates for RecA, Rad51, and Dmc1. These findings are consistent with our previous experimental observations using substrates in which we incrementally extended the microhomology length in the 5′ → 3′, as anticipated (28, 29). We were careful to note that although we obtained similar results for these two sets of substrates, the findings themselves do directly report upon the polarity of strand exchange because we are only assessing the stability of the reaction intermediates after they have already formed.
Figure 2.

Base triplet stepping observed with incremental extension of the 5′-microhomology. A, schematic illustration of the 70-bp dsDNA substrates with incrementally lengthened 5′-microhomology. B, Atto565-dsDNA dissociation rates (mean ± S.D.) for RecA in the presence of ATPγS; C, S. cerevisiae Rad51 plus ATP; and D, S. cerevisiae Dmc1 plus ATP. All data points were calculated from an average of ∼200 molecules (n = 150–250); the color-coded shading highlights each base triplet; magenta coloring indicates the minimum 8 nt necessary for stable binding, and purple shading corresponds to addition of homologous nucleotides; arrows indicate stepwise reductions in dissociation rates coincident with the third base of each triplet; dashed lines report the mean rate for each step; and the free energy changes (ΔΔG‡) calculated for each triplet step are indicated.
Mismatch discrimination at the 5′ terminus of microhomology
We next asked whether RecA, Rad51, and Dmc1 could discriminate against mismatched bases located within the 5′-terminal triplet of an embedded tract of microhomology. For these assays, a single mismatch was introduced at each of three possible positions at the 5′ end of a 12-nt tract of microhomology (Fig. 3A). These results revealed that a single mismatch at any position within the 5′-terminal triplet abolishes detectable binding of the mismatched triplet by the RecA, Rad51, and Dmc1 (Fig. 3, B–D). These findings are similar to our previous observations for mismatches located at the 3′ end of the internal tract of microhomology (28). Taken together, our results support the conclusion that RecA, Rad51, and Dmc1 are all intolerant of single nucleotide mismatches within terminal base triplets located at either the 5′ or 3′ end of an internal tract of sequence homology.
Figure 3.

Effect of single nucleotide mismatches on 5′-base triplet recognition. A, schematic illustration of dsDNA substrates bearing single nucleotide mismatches within the terminal triplet at the 5′ end of a 12-nt tract of microhomology. Each mutated nucleotide is highlighted in magenta and underlined, and the corresponding sequence of the pre-synaptic ssDNA is indicated. Mismatch substrate-binding data for RecA (B), Rad51 (C), and Dmc1 (D) are as indicated. All indicated ΔΔG‡ values are relative to the step 1 binding data for the substrate bearing 9 nt of microhomology (see Fig. 2). The green and blue dashed lines correspond to the average binding stability for data obtained from the non-mismatched 9–11-nt substrates (reflecting stable association equivalent to three complete triplets) and the 12–14-nt substrates (reflecting stable association equivalent to four complete triplets), respectively. E, schematic illustration of dsDNA substrates bearing single nucleotide mismatches within the penultimate triplet at the 5′ end of a 15-nt tract of microhomology. Corresponding binding data for RecA (F), Rad51 (G), and Dmc1 (H) are as indicated. Green, blue, and purple dashed lines correspond to the averaged binding stability for data obtained from the non-mismatched 9–11-nt substrates (reflecting stable association equivalent to three complete triplets); the 12–14-nt substrates (reflecting stable association equivalent to four complete triplets); and 15-nt substrates (reflecting stable association of 5 complete triplets), respectively, reflecting steps 1–3 in A.
Internal triplets are destabilized within RecA and Rad51 complexes
RecA and Rad51 can step over mismatches located within the penultimate base triplet near the 3′ end of an internal tract of microhomology, but the reduced stability of the resulting intermediates suggests that the mismatched triplet may remain unpaired (28). Dmc1 can also step over internal triplets near the 3′ end of an internal tract of microhomology, but these mismatched triplets still contribute to binding free energy of the resulting intermediate (28). As an extension of these previous findings, we next asked whether RecA, Rad51, and Dmc1 could step over mismatches positioned within the penultimate base triplet near the 5′ end of an internal tract of microhomology (Fig. 3E). Here, a single mismatch was introduced at each of three possible positions within the penultimate base triplet within a 15-nt tract of microhomology (Fig. 3E). For both RecA and Rad51, the binding stability of the resulting intermediate was most consistent with destabilization of the mismatch-bearing base triplet (Fig. 3, F and G). In contrast, the stability of the mismatched intermediates generated by Dmc1 was comparable with the fully paired intermediate (Fig. 3H). We conclude that Dmc1 can stabilize single mismatches located in the penultimate base triplet near either the 5′ or 3′ end of an embedded tract of microhomology. In contrast, RecA and Rad51 were unable to stabilize these mismatched triplets, but both were able to step over the mismatch-bearing triplets to stabilize an adjacent base triplet that is fully homologous to the pre-synaptic ssDNA.
Dmc1 can stabilize triplets bearing multiple mismatches
To explore the potential mechanism of mismatch stabilization by Dmc1 we next sought to determine whether Dmc1 could stabilize base triplets bearing more than one mismatch. For these assays, we utilized a 70-bp dsDNA substrate bearing a 15-nt tract of microhomology in which the penultimate 3′-triplet harbored either two or three nucleotide mismatches with the pre-synaptic ssDNA (Fig. 4A and supplemental Fig. S2). Consistent with the observation that RecA and Rad51 are intolerant of even a single mismatch, neither protein was capable of stabilizing substrates bearing either two or three mismatches (Fig. 4, B and C, and supplemental Fig. S2). In contrast, Dmc1 was able to stabilize each mismatched substrate such that the relative binding energy of the mismatch-bearing substrates was comparable with the fully paired substrate (Fig. 4B).
Figure 4.
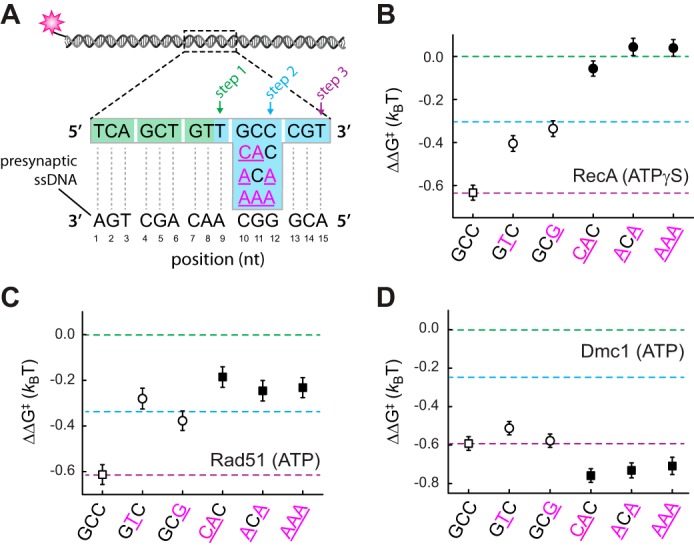
Impact of multiple mismatches on base triplet recognition. A, schematic illustration of dsDNA substrates bearing either two or three nucleotide mismatches within the penultimate triplet at the 3′ end of a 15-nt tract of microhomology. Corresponding binding data for RecA (B), Rad51 (C), and Dmc1 (D) are as indicated. Green, blue, and purple dashed lines correspond to the averaged binding stability for data obtained from the non-mismatched 9–11-nt substrates; the 12–14-nt substrates; and 15-nt substrates, respectively, reflecting steps 1–3 in A. Previously reported data for substrates bearing single mismatches are shown (open circles) for comparison (28).
Interestingly, RecA and Rad51 can both step over triplets bearing single mismatches, but only Rad51 appears to be capable of stepping over triplets bearing either two or three mismatches (Fig. 4, B and C, and supplemental Fig. S2). Based upon the observed changes in binding free energy, RecA appears to be incapable of stabilizing a homologous triplet that lies beyond a triplet bearing either two or three mismatches in reactions using ATPγS. This differential response to substrates harboring multiple mismatches is the only substantive difference we have found between RecA and Rad51 in response to base triplet imperfections. Bulk biochemical assays have revealed that RecA is unable to bypass longer (≥50 bp) heterologous insertions during strand invasion when ATPγS is used as the nucleotide cofactor, but it can bypass these insertions in reactions with ATP (33, 34). Therefore, we sought to determine whether RecA might be able to stabilize the substrates with multiple mismatches in the presence of ATP. These experiments revealed that in the presence of ATP, RecA is capable of stabilizing a homologous triplet that lies beyond an internal triplet bearing multiple mismatches (supplemental Fig. S3). We conclude that in the presence of ATP, RecA and Rad51 respond similarly to internal base triplets bearing multiple mismatches.
Dmc1 can stabilize triplets bearing abasic sites
We next tested substrates bearing a single abasic site at each of the three possible positions within the penultimate base triplet near the 3′ or 5′ end of the microhomology (Fig. 5). Neither RecA nor Rad51 was able to stabilize internal triplets bearing an abasic site, consistent with their intolerance for single base mismatches (Fig. 5, B, C, F, and G). In contrast, Dmc1 was able to stabilize these substrates regardless of the location of the abasic site within the triplet, and similar findings were obtained for abasic sites located near the 3′ or 5′ ends of the internal tract of microhomology (Fig. 5, D and H). These findings provide further support for the hypothesis that Dmc1 does not stabilize mismatched intermediates through a mechanism involving protein-enhanced pairing of non-Watson-Crick interactions, because abasic sites cannot form any type of non-canonical pairing interaction. The finding that Dmc1 can stabilize substrates bearing abasic sites also suggests that the stabilization mechanism likely does not involve direct protein contacts with the nucleotide bases of the suboptimal triplet, but instead it may involve contacts with the ribose-phosphate backbone of the DNA.
Figure 5.

Dmc1 can stabilize triplets bearing abasic sites. A, schematic illustration of dsDNA substrates bearing single abasic sites within the penultimate triplet at the 3′ end of a 15-nt tract of microhomology. Open magenta boxes signify the locations of the abasic sites. Corresponding to binding data for RecA (B), Rad51 (C), and Dmc1 (D) are as indicated. Green, blue, and purple dashed lines correspond to the averaged binding stability for data obtained from the non-mismatched 9-nt substrates; the 12–14-nt substrates; and 15-nt substrates, respectively, reflecting steps 1–3 in A. E, schematic illustration of dsDNA substrates bearing single abasic sites within the penultimate triplet at the 3′ end of a 15-nt tract of microhomology. Open magenta boxes signify the locations of the abasic sites. Corresponding binding data for RecA (F), Rad51 (G), and Dmc1 (H) are as indicated. Green, blue, and purple dashed lines correspond the averaged binding stability for data obtained from the non-mismatched 9–11-nt substrates; the 12–14-nt substrates; and 15-nt substrates, respectively, reflecting steps 1–3 in A.
Non-bridging oxygen modifications do not prevent mismatch stabilization by Dmc1
We next tested whether Dmc1-mediated mismatch stabilization might involve recombinase contacts with the phosphate backbone. To address this possibility, we conducted assays with Dmc1 using mismatched substrates harboring either methylphosphonate or phosphorothioate substitutions within the phosphate backbone of the mismatched triplet (supplemental Fig. S4). Surprisingly, the methylphosphonate substitutions had no appreciable impact upon the ability of Dmc1 to stabilize the mismatched base triplets. These findings suggest that if Dmc1 contacts the phosphate backbone of incoming complementary strand during mismatch stabilization, then these protein-DNA contacts may not involve the non-bridging oxygen atoms, but it would more likely involve other contacts with the ribose-phosphate backbone. Future work will be essential to determine the structural basis for mismatch stabilization by Dmc1.
Single nucleotide insertions disrupt triplet stabilization
We next sought to establish whether RecA, Rad51, or Dmc1 could tolerate single nucleotide insertions. For this purpose, we designed a dsDNA substrate harboring a single base insertion between the penultimate and terminal triplet at either the 3′ or 5′ end of an internal tract of microhomology, which is anticipated to position the terminal base triplets just 1-nucleotide out of register with the pre-synaptic ssDNA (Fig. 6, A and C). Surprisingly, the results revealed that a single base insertion results in a change in binding free energy consistent with the loss of stabilizing interactions involving the terminal base triplets for each of the three recombinases (Fig. 6, B and D). We conclude that RecA, Rad51, and Dmc1 cannot stabilize a homologous 3′- or 5′-terminal base triplet that is moved out of register by the presence of a single nucleotide insertion.
Figure 6.

Single base insertion can disrupt triplet stabilization. A, schematic illustration of dsDNA substrates bearing a single nucleotide insertion adjacent to the penultimate triplet at the 3′ end of a 15-nt tract of microhomology. B, corresponding binding data for Rad51, RecA, and Dmc1 for dsDNA substrates with (closed circles) and without (open squares) the nucleotide insertion. C, schematic illustration of dsDNA substrates bearing a single nucleotide insertion adjacent to the penultimate triplet at the 5′ end of a 15-nt tract of microhomology. D, corresponding binding data for Rad51, RecA, and Dmc1 for dsDNA substrates with (closed circles) and without (open squares) the nucleotide insertion.
Discussion
We have established that the eukaryotic meiosis-specific recombinase Dmc1 is more tolerant of imperfect base triplet-pairing interactions relative to Rad51 and RecA. As discussed below, our findings suggest a model in which Dmc1 stabilizes imperfectly paired triplets through a mechanism involving protein-mediated contacts that compensate for the binding energy that would otherwise be lost due to the unpaired triplet.
Strand exchange takes place in 3-nt steps
Our work suggests that incrementally increasing the length of homology in either the 5′ → 3′ or 3′ → 5′ around a central 8-nt tract of microhomology gives rise to stepwise changes in binding free energy that follow a highly predictable base triplet pattern. In addition, the free energy changes associated with base triplet stabilization appear to be indistinguishable for base triplets located at either end of the internal tract of microhomology. These observations, together with the finding that the recombinases can all step over mismatches or abasic sites positioned within the penultimate triplet at either the 5′ or 3′ ends of the internal tract of microhomology, support a model in which strand exchange might proceed in either the 5′ → 3′ or 3′ → 5′ directions, and they suggest that bidirectional strand exchange may be conserved across the Rad51/RecA family of recombinases (Fig. 7A). Several studies have suggested that strand exchange can take place with a defined polarity (35–38), whereas others have reported that strand exchange can occur in either direction (39, 40). We note that our assays do not directly observe strand exchange steps as they are taking place, but rather assess the stability of these early recombination intermediates only after they have already formed. Therefore, we do not rule out the possibility that there may exist polarity-dependent differences in the rates of strand exchange that cannot be assessed in our experimental system.
Figure 7.

Effects of lesions on base triplet recognition by RecA, Rad51, and Dmc1. Details of these models are presented under “Discussion.”
Responses of RecA, Rad51, and Dmc1 to sequence imperfections
Single mismatches within the terminal base triplet at either the 5′ or 3′ end of the microhomology prevent stabilization of these terminal triplets by RecA, Rad51, and Dmc1, suggesting that this may be a universal response of Rad51/RecA family members to imperfectly paired terminal base triplets (Fig. 7A). This result is surprising for Dmc1 because this recombinase can stabilize all other tested imperfections (i.e. single, double, and triple mismatches, and abasic sites), with the exception of single nucleotide insertions, so long as the imperfections are flanked by at least one fully homologous base triplet (Fig. 7B). These findings raise the question of whether the inability of Rad51/RecA recombinases to stabilize imperfect terminal triplets provides some specific biological advantage for homologous recombination or whether it might be a reflection of the thermodynamic properties of RS-DNA or both. One possibility is that imperfect terminal triplets play an important role in ensuring accurate recombination, perhaps by providing a kinetic barrier to slow the progression of stand invasion, and providing a decision point for the pre-synaptic complex (and associated factors) to either continue strand invasion or dissociate to sample other regions of the genome for a more perfectly matched donor sequence.
Triplet pairing requires perfect alignment
Single base insertions abolish recognition of the adjacent triplet by all three recombinases, suggesting that this may be a conserved feature of base triplet recognition by the Rad51/RecA family of DNA recombinases (Fig. 7A). This finding highlights intolerance of the pre-synaptic complex to what might have been considered a relatively minor structural defect. For instance, one might have assumed that a single base insertion would simply have been accommodated between the two homologous triplets, which would in principle eliminate the need to fully extend the phosphodiester linkage between these two triplets. Instead, the inserted base appears to abolish interactions with the downstream triplet, suggesting that amino acids within the pre-synaptic complex may be positioned to prevent accommodation of extra nucleotides. Indeed, inspection of the RecA crystal structure reveals that the L2 DNA-binding loop lies between adjacent base triplets, suggesting that steric hindrance by L2 may be responsible for preventing accommodation of base insertions (27). Our experiments cannot yet address whether the presence of additional homologous base triplets beyond the nucleotide insertion would eventually allow stabilization, although we favor the idea that they would, given that homologous recombination can take place between longer nucleic acid substrates bearing either nucleotide insertions or deletions. Future work will be necessary to determine how much sequence homology is necessary on either side of a base insertion to allow triplet pairing interactions with both flanking sequences.
Interestingly, RecA and Rad51 can both step over triplets bearing single mismatches at either the 5′ or 3′ ends of the microhomology, but neither Rad51 nor RecA can stabilize the resulting mismatched triplets (Fig. 7B). This finding contrasts with results for Dmc1, which can both step over and stabilize single mismatches at either the 5′ or 3′ ends of the microhomology (Fig. 7B). RecA and Rad51 can also step over abasic sites, but cannot stabilize the resulting triplets bearing the abasic residues. This finding is again in striking contrast with Dmc1, which can step over and stabilize base triplets bearing abasic sites regardless of the location of the abasic site within the base triplet (Fig. 7C). These results have important implications for understanding the mechanistic basis by which Dmc1 stabilizes substrates bearing imperfect triplets (see below).
Effects of multiple mismatches
Experiments looking at multiple mismatches yielded several important findings. First, RecA can step over triplets bearing a single mismatched nucleotide but not two or three mismatches in reactions with ATPγS (Fig. 7D). This contrasts with the findings for Rad51, which is able to step over base triplets bearing two or even three mismatches (Fig. 7D). We speculate that in the presence of ATPγS, RecA might be capable of stepping over these tandem mismatches if there were additional sequence homology beyond the tandem mismatches, perhaps by initiating an independent strand invasion reaction downstream of the mismatches. Interestingly, RecA can step over multiple mismatches in reactions with ATP (Fig. 7D), which is consistent with prior biochemical assays demonstrating that RecA can bypass larger sequence heterologies only in the presence of ATP (33, 34). This requirement for ATP has led to the hypothesis that RecA may have an ATP-dependent motor activity that is coupled to heterology bypass (41). An alternative possibility is that ATP hydrolysis-dependent protein turnover may be necessary to reduce the stiffness of the RecA filament, allowing a separate segment of the pre-synaptic complex to more readily re-engage the dsDNA substrate beyond tandem mismatches.
These experiments also revealed that Dmc1 can stabilize base triplets bearing either one, two, or three mismatches, so long as those triplets are flanked by at least one homologous triplet (Fig. 7D). This finding highlights the marked tolerance of Dmc1 to sequence imperfections at the level of a single base triplet, and it also has crucial implications for understanding how Dmc1 stabilizes imperfectly matched RS-DNA triplets (see below).
Models for base triplet stability
Our results can be considered within the context of two contrasting models describing the physical stability of mismatched RS-DNA base triplets. The first model is that imperfectly matched RS-DNA base triplets are themselves inherently unstable (Fig. 7E). This unstable triplet model would account for all of our observations with RecA and Rad51, indicating that the inability of these two proteins to stabilize imperfect triplets stems from the lesion-bearing triplets themselves being unstable relative to a perfectly paired triplet. The unstable triplet model is also supported by the previous reports that RecA makes very limited contact with the incoming complementary DNA strand, which is instead held in place primarily by Watson-Crick hydrogen bond pairing interactions with the pre-synaptic ssDNA (27). However, if the unstable triplet model is correct, then Dmc1 must somehow stabilize the imperfect triplet, or otherwise compensate for the loss of binding free energy that would be expected if the imperfect triplets were not paired within the Dmc1-dsDNA postsynaptic complex.
An alternative model, which we do not favor, is that imperfect RS-DNA base triplets are themselves inherently stable (Fig. 7E). This model could explain all of our results with Dmc1, but it would then require that RecA and Rad51 have built-in mechanisms for sensing and actively destabilizing imperfect base triplets. Moreover, it is difficult to envision how an RS-DNA base triplet would tolerate single, double, and triple mismatches and even abasic sites without any loss of triplet pairing stability. Indeed, molecular dynamics (MD) simulations of either RS-DNA or isolated base triplets reveal that RS-DNA is highly dynamic relative to B-DNA, and single base mismatches cause rapid destabilization of triplet pairing interactions.5 Recent reports suggest that non-complementary sequences are stable within RecA filaments during a 10-ns MD simulation (42), so future work will be required to more fully understand how mismatched sequences behave during recombination. However, our experimental results seem to argue against a model where mismatched triplets are inherently stable and must be actively destabilized by RecA and Rad51. Specifically, Dmc1 can stabilize base triplets bearing single abasic sites at any of the three possible positions, and it is again difficult to envision that a base triplet harboring an abasic site could be inherently stable within an RS-DNA triplet. Together, we consider our findings to be most consistent with the unstable triplet model, suggesting that RecA and Rad51 need not actively destabilize imperfect RS-DNA base triplets, but instead Dmc1 must somehow compensate for the loss of Watson-Crick binding free energy due to mismatched base triplets.
Possible implications for homologous recombination
Our results suggest that RecA and Rad51 require perfect Watson-Crick pairing to allow stabilization of RS-DNA base triplets, whereas Dmc1 can stabilize non-complementary triplets embedded with longer tracts of homology. We emphasize that we do not yet know the biological implications of these findings. One possibility is that this biophysical difference may reflect the biological specialization of the meiosis-specific recombinase Dmc1, namely the requirement for Dmc1 to promote recombination between polymorphic alleles of different parental origins (see below). Future work will therefore be essential to determine whether or not the differences we observe between Rad51 and Dmc1 in response to mismatches and abasic sites reflect some broader biological difference between these two proteins.
Importantly, our work only reflects the effects of mutations within individual base triplets. The overall pairing interactions are highly stable, with half-lives still exceeding tens of minutes, even when mismatches or basic sites are present. Furthermore, we anticipate that the relative differences in stability for longer strand-exchange products will be very small. A single unpaired triplet within a paired intermediate that is tens or perhaps hundreds of base triplets in length will not in and of itself destabilize pairing interactions. Indeed, experimental and theoretical studies of RecA have recently shown that after initial recognition of an 8-nt homologous sequence, the presence of mismatches outside this region has little impact on overall stability (42). The previous work with RecA by Prentiss and co-workers (42) is in good general agreement with our findings. At this stage, we can only infer that the differential stabilization of imperfectly matched RS-DNA triplets might reflect some broader difference in the overall stability of a complete strand-exchange product, which in turn may manifest as a recombinase- and mismatch-dependent difference in recombination efficiency. Future work will be essential to continue testing for potential relationships between triplet pairing, or mispairing, and recombination efficiencies.
Finally, the results reported here reflect the basal activities of E. coli RecA, S. cerevisiae Rad51, and S. cerevisiae Dmc1. These recombinases acts in concert with many other proteins; for example, a total of ∼20 proteins or protein complex participate in recombination in E. coli (14, 43, 44), and ∼45 distinct proteins or protein complexes participate in recombination in S. cerevisiae (4, 45). Interestingly, recent in vivo studies have shown that Rad51-mediated recombination is unexpectedly tolerant of mismatches (46). Therefore, the crucial next step will be to start understanding how recombination accessory proteins augment the basal base triplet recognition activities of the recombinases during strand invasion.
Experimental procedures
DNA curtains
Single-stranded DNA substrates were generated using M13mp18 (7,249-nt; New England Biolabs) as a template for rolling circle replication (RCR), as described (29, 31). In brief, a biotinylated primer was annealed to the M13mp18 template (∼30 nm) at a 1:1.2 molar ratio (primer, M13mp18) in buffer containing 40 mm Tris-HCl (pH 8.0), 50 mm NaCl, and 10 mm MgCl2. The annealing mixture was heated for 5 min at ∼90 °C and then cooled slowly to room temperature. Annealed products were diluted to a final concentration of 15 nm and stored at 4 °C until use. Replication reactions were prepared in RCR buffer (50 mm Tris-HCl (pH 7.5), 4 mm DTT, 10 mm ammonium sulfate, and 10 mm MgCl2), containing annealed primer/M13mp18 mixture (350 pm), 150 nm φ29 DNA polymerase, and dNTP mixture (200 μm each). Reactions were incubated at 30 °C for 25 min and then diluted 10-fold into buffer containing 40 mm Tris-HCl (pH 7.5), 2 mm MgCl2, 50 mm NaCl, 1 mm DTT, and 0.2 mg/ml BSA prior to use.
Flow cells were made by depositing chromium barriers onto the surface of a fused silica slide by electron beam lithography and then assembled into a sample chamber using double-sided tape and a borosilicate cover slide, as described (47). Bilayers were prepared with 91.5% 1,2-dioleoyl-sn-glycero-3-phosphocholine, 0.5% biotinylated 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl), and 8% mPEG 2000–1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, and the biotinylated ssDNA molecules were attached to the bilayer through a biotin-streptavidin linkage. Anchored ssDNA molecules were then aligned at the barriers by application of flow in buffer containing 40 mm Tris-HCl (pH 7.5), 2 mm MgCl2, 50 mm NaCl, 1 mm DTT, 0.2 mg/ml BSA, and 0.1 nm RPA-eGFP for 15 min at 1 ml/min. All experiments were conducted with a custom-built prism-type total internal reflection fluorescence microscope (Nikon) equipped with a 488-nm blue laser (Coherent Sapphine, 200 milliwatt) and a 561-nm yellow laser (Coherent Sapphine, 200 milliwatt).
Reaction conditions and data analysis
E. coli RecA, S. cerevisiae Rad51, and S. cerevisiae Dmc1 were prepared as described (28, 29). Rad51/RecA pre-synaptic complexes were assembled as described previously (28, 29), using the following reaction buffers: E. coli RecA buffer, 25 mm Tris acetate (pH 7.5), 4 mm magnesium acetate, 10 mm sodium acetate, 1 mm ATPγS, or 2.5 mm ATP plus an ATP-regenerating system (20 mm creatine and 0.04 mg/ml creatine kinase), 1 mm DTT, and 0.2 mg ml−1 BSA; S. cerevisiae Rad51 buffer, 30 mm Tris acetate (pH 7.5), 20 mm magnesium acetate, 50 mm KCl, 1 mm DTT, 2.5 mm ATP, and 0.2 mg ml−1 BSA; S. cerevisiae Dmc1, 40 mm Tris-HCl (pH 7.5), 2 mm MgCl2, 1.5 mm CaCl2, 100 mm KCl, 2.5 mm ATP, 1 mm DTT, and 0.2 mg ml−1 BSA. Note that the identity of the nucleotide cofactor used in each different experiment is specified in each of the corresponding figure panels. All data were collected at 30 °C.
All dsDNA binding data were collected and analyzed as described previously (28, 29). In brief, Atto565-tagged dsDNA oligonucleotides (2–10 nm; supplemental Table S1) were injected into the sample chamber in the same buffers (see above), and reactions were incubated for 10 min at 30 °C in the absence of buffer flow. Flow cells were quickly flushed (40 s at 1.0 ml min−1) with fresh buffer to remove unbound Atto565-dsDNA, and the flow rate was then reduced (0.2 ml min−1) to remove dissociated dsDNA and to replenish free nucleotide cofactor. Data were obtained by acquiring 100-ms frames at 30- or 60-s intervals; collection intervals were optimized relative to the overall lifetime of each dsDNA substrate, and the laser was shuttered between acquired images to minimize photobleaching. Kymographs were generated using Fiji and were analyzed by measuring the time that each Atto565-dsDNA molecule remained bound to the pre-synaptic complexes after flushing unbound DNA from the sample chamber. The probability that a bound molecule survived to a particular time point (t) was determined as the fraction of Atto565-dsDNA molecules that remained bound at time t, and survival probability graphs were constructed from the resulting data. All data points were calculated from an average of ∼200 molecules (n = 150–250). The dissociation kinetics for all dsDNA substrates were well described by single exponential fits to the survival probability data (supplemental Fig. S1).
Free energy calculations were performed as described previously (28, 29). In brief, for dsDNA substrates harboring ≥8 nt of microhomology, the free energy barrier, ΔG‡, for escape from a potential well can be related to the rate as shown in Equation 1,
| (Eq. 1) |
where A is a jump frequency, kb is the Boltzmann constant, and T is the temperature. The difference between the barrier heights between two different escape processes can be compared, leading to Equation 2.
| (Eq. 2) |
All reported ΔΔG‡ values were normalized such that ΔG‡ for the dsDNA containing a single 8-nt tract of microhomology is zero, and the experimentally measured data used to calculate ΔΔG‡ were the kd values for each different substrate obtained from survival probability data.
Author contributions
J. Y. L designed and conducted the single molecule experiments and data analysis. J. B. S. and Z. Q. assisted with single molecule experiments, data analysis, and experimental design. Y. K. expressed and purified Rad51 and Dmc1. E. C. G. supervised the project and wrote the manuscript with input from J. Y. L., J. B. S., Z. Q., and P. S.
Supplementary Material
Acknowledgments
We thank Kyle Kaniecki, Corentin Moevus, Luisina de Tullio, Tsuyoshi Terakawa, Daniel Duzdevich, and Brooks Crickard for comments on the manuscript.
This work was supported in part by National Institutes of Health Grant R35GM118026 (to E. C. G.) and Grants R01ES007061 and P01CA92584 (to P. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supplemental Figs. S1–S4 and Tables S1A–S1C.
T. Terawaka and E. C. Greene, unpublished data.
- DSB
- double-strand DNA break
- ssDNA
- single-stranded DNA
- nt
- nucleotide
- eGFP
- enhanced GFP
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- RS-DNA
- recombinase stretched-DNA
- RCR
- rolling circle replication
- MD
- molecular dynamics.
References
- 1. Coop G., and Przeworski M. (2007) An evolutionary view of human recombination. Nat. Rev. Genet. 8, 23–34 [DOI] [PubMed] [Google Scholar]
- 2. Ortiz-Barrientos D., Engelstädter J., and Rieseberg L. H. (2016) Recombination rate evolution and the origin of species. Trends Ecol. Evol. 31, 226–236 [DOI] [PubMed] [Google Scholar]
- 3. Pâques F., and Haber J. E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Symington L. S., Rothstein R., and Lisby M. (2014) Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., and Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 6. Flores-Rozas H., and Kolodner R. D. (2000) Links between replication, recombination and genome instability in eukaryotes. Trends Biochem. Sci. 25, 196–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kolodner R. D., Putnam C. D., and Myung K. (2002) Maintenance of genome stability in Saccharomyces cerevisiae. Science 297, 552–557 [DOI] [PubMed] [Google Scholar]
- 8. Malkova A., and Haber J. E. (2012) Mutations arising during repair of chromosome breaks. Annu. Rev. Genet. 46, 455–473 [DOI] [PubMed] [Google Scholar]
- 9. Mehta A., and Haber J. E. (2014) Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 6, a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soucy S. M., Huang J., and Gogarten J. P. (2015) Horizontal gene transfer: building the web of life. Nat Rev Genet 16, 472–482 [DOI] [PubMed] [Google Scholar]
- 11. Brown M. S., and Bishop D. K. (2014) DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb. Perspect. Biol. 7, a016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neale M. J., and Keeney S. (2006) Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature 442, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hunter N. (2015) Meiotic recombination: the essence of heredity. Cold Spring Harb. Perspect. Biol. 7, a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kowalczykowski S. C. (2015) An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 7, a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morrical S. W. (2015) DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 7, a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bianco P. R., Tracy R. B., and Kowalczykowski S. C. (1998) DNA strand exchange proteins: a biochemical and physical comparison. Front Biosci. 3, D570–D603 [DOI] [PubMed] [Google Scholar]
- 17. Aravind L., Walker D. R., and Koonin E. V. (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 27, 1223–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin Z., Kong H., Nei M., and Ma H. (2006) Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. U.S.A. 103, 10328–10333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bishop D. K., Park D., Xu L., and Kleckner N. (1992) DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456 [DOI] [PubMed] [Google Scholar]
- 20. Clark A. J., and Margulies A. D. (1965) Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. U.S.A. 53, 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prentiss M., Prévost C., and Danilowicz C. (2015) Structure/function relationships in RecA protein-mediated homology recognition and strand exchange. Crit. Rev. Biochem. Mol. Biol. 50, 453–476 [DOI] [PubMed] [Google Scholar]
- 22. Ogawa T., Yu X., Shinohara A., and Egelman E. H. (1993) Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 259, 1896–1899 [DOI] [PubMed] [Google Scholar]
- 23. Egelman E. H., and Stasiak A. (1986) Structure of helical RecA-DNA complexes. Complexes formed in the presence of ATP-γ-S or ATP. J. Mol. Biol. 191, 677–697 [DOI] [PubMed] [Google Scholar]
- 24. Sheridan S. D., Yu X., Roth R., Heuser J. E., Sehorn M. G., Sung P., Egelman E. H., and Bishop D. K. (2008) A comparative analysis of Dmc1 and Rad51 nucleoprotein filaments. Nucleic Acids Res. 36, 4057–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sung P. (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265, 1241–1243 [DOI] [PubMed] [Google Scholar]
- 26. Yu X., Jacobs S. A., West S. C., Ogawa T., and Egelman E. H. (2001) Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl. Acad. Sci. U.S.A. 98, 8419–8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Z., Yang H., and Pavletich N. P. (2008) Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature 453, 489–494 [DOI] [PubMed] [Google Scholar]
- 28. Lee J. Y., Terakawa T., Qi Z., Steinfeld J. B., Redding S., Kwon Y., Gaines W. A., Zhao W., Sung P., and Greene E. C. (2015) DNA recombination. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qi Z., Redding S., Lee J. Y., Gibb B., Kwon Y., Niu H., Gaines W. A., Sung P., and Greene E. C. (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee J. Y., Qi Z., and Greene E. C. (2016) ATP hydrolysis promotes duplex DNA release by the RecA pre-synaptic complex. J. Biol. Chem. 291, 22218–22230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gibb B., Silverstein T. D., Finkelstein I. J., and Greene E. C. (2012) Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal. Chem. 84, 7607–7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi Z., and Greene E. C. (2016) Visualizing recombination intermediates with single-stranded DNA curtains. Methods 105, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J. I., Cox M. M., and Inman R. B. (1992) On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. I. Bypassing a short heterologous insert in one DNA substrate. J. Biol. Chem. 267, 16438–16443 [PubMed] [Google Scholar]
- 34. Rosselli W., and Stasiak A. (1991) The ATPase activity of RecA is needed to push the DNA strand exchange through heterologous regions. EMBO J. 10, 4391–4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox M. M., and Lehman I. R. (1981) Directionality and polarity in recA protein-promoted branch migration. Proc. Natl. Acad. Sci. U.S.A. 78, 6018–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murayama Y., Tsutsui Y., and Iwasaki H. (2011) The fission yeast meiosis-specific Dmc1 recombinase mediates formation and branch migration of Holliday junctions by preferentially promoting strand exchange in a direction opposite to that of Rad51. Genes Dev. 25, 516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sung P., and Robberson D. L. (1995) DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 82, 453–461 [DOI] [PubMed] [Google Scholar]
- 38. West S. C., Cassuto E., and Howard-Flanders P. (1981) Heteroduplex formation by recA protein: polarity of strand exchanges. Proc. Natl. Acad. Sci. U.S.A. 78, 6149–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Namsaraev E. A., and Berg P. (1998) Branch migration during Rad51-promoted strand exchange proceeds in either direction. Proc. Natl. Acad. Sci. U.S.A. 95, 10477–10481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Namsaraev E. A., and Berg P. (2000) Rad51 uses one mechanism to drive DNA strand exchange in both directions. J. Biol. Chem. 275, 3970–3976 [DOI] [PubMed] [Google Scholar]
- 41. Cox M. M. (2007) Motoring along with the bacterial RecA protein. Nat. Rev. Mol. Cell Biol. 8, 127–138 [DOI] [PubMed] [Google Scholar]
- 42. Danilowicz C., Yang D., Kelley C., Prévost C., and Prentiss M. (2015) The poor homology stringency in the heteroduplex allows strand exchange to incorporate desirable mismatches without sacrificing recognition in vivo. Nucleic Acids Res. 43, 6473–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Persky N. S., and Lovett S. T. (2008) Mechanisms of recombination: lessons from E. coli. Crit. Rev. Biochem. Mol. Biol. 43, 347–370 [DOI] [PubMed] [Google Scholar]
- 44. Camerini-Otero R. D., and Hsieh P. (1995) Homologous recombination proteins in prokaryotes and eukaryotes. Annu Rev. Genet. 29, 509–552 [DOI] [PubMed] [Google Scholar]
- 45. Krogh B. O., and Symington L. S. (2004) Recombination proteins in yeast. Annu Rev. Genet. 38, 233–271 [DOI] [PubMed] [Google Scholar]
- 46. Anand R., Beach A., Li K., and Haber J. (2017) Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544, 377–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greene E. C., Wind S., Fazio T., Gorman J., and Visnapuu M. L. (2010) DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 472, 293–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.