

Abstract
Despite numerous studies on chemical and thermal stability of metal–organic frameworks (MOFs), mechanical stability remains largely undeveloped. To date, no strategy exists to control the mechanical deformation of MOFs under ultrahigh pressure. Here, we show that the mechanically unstable MOF-520 can be retrofitted by precise placement of a rigid 4,4′-biphenyldicarboxylate (BPDC) linker as a “girder” to afford a mechanically robust framework: MOF-520-BPDC. This retrofitting alters how the structure deforms under ultrahigh pressure and thus leads to a drastic enhancement of its mechanical robustness. While in the parent MOF-520 the pressure transmitting medium molecules diffuse into the pore and expand the structure from the inside upon compression, the girder in the new retrofitted MOF-520-BPDC prevents the framework from expansion by linking two adjacent secondary building units together. As a result, the modified MOF is stable under hydrostatic compression in a diamond-anvil cell up to 5.5 gigapascal. The increased mechanical stability of MOF-520-BPDC prohibits the typical amorphization observed for MOFs in this pressure range. Direct correlation between the orientation of these girders within the framework and its linear strain was estimated, providing new insights for the design of MOFs with optimized mechanical properties.
Short abstract
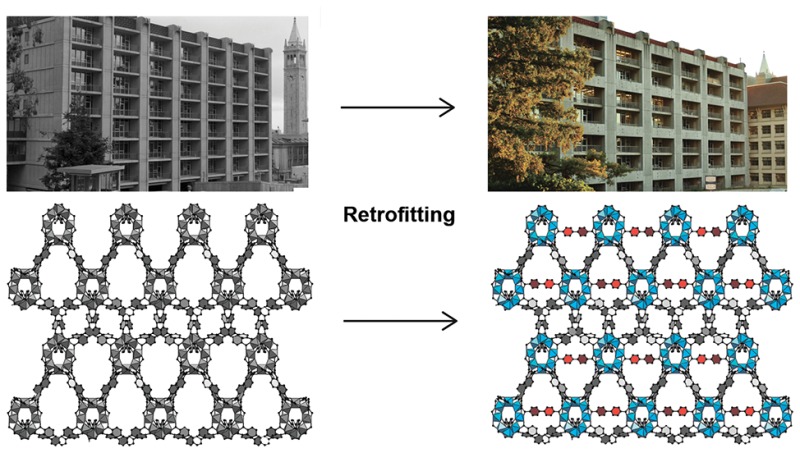
The concept of molecular retrofitting demonstrates how to adapt the metal−organic framework to extreme stress. The visualization of the retrofitting approach is shown in the architecture of Latimer Hall at UC Berkeley and on the molecular level for MOF-520.
Introduction
A unique feature of metal–organic frameworks (MOFs) is that they have “pores without walls”, where the internal space is encompassed by multimetallic junctions and organic linkers rather than continuous impenetrable walls as in traditional porous materials. This junction-and-linker arrangement maximally exposes the adsorptive sites and increases their number, leading to ultrahigh surface areas for MOFs.1−3 Thus, having pores without walls couples the high storage capacity with the facile diffusion of molecules in and out of the pores, making MOFs useful in natural gas storage, separation of gas mixtures, and selective catalysis.4−7 It is remarkable that, even with the vast openness of MOF structures, they have been shown to be architecturally, thermally, and chemically stable: properties that are essential for their development from basic science to applications and commercialization.8 As more applications come online, the next challenge is to show how these open structures withstand mechanical stress, to which they inevitably will be subjected during their operation and long-term use. In this context, the question of what is the breaking point of a given framework becomes paramount. Here, we show how applying extreme pressure (ca. 10,000 atm, 1 gigapascal) onto MOF crystals provides means for identifying the weakest component of the framework. We also introduce the concept of molecular retrofitting and demonstrate its use in adapting such a framework to extreme mechanical stress (Figure 1). Specifically, we examined the mechanical response of porous MOF-520 crystal structure, Al8(μ-OH)8(HCOO)4(BTB)4 (BTB = 1,3,5-benzenetribenzoate), to hydrostatic pressures from ambient up to 3 gigapascal (GPa).9 We observed expansion of the framework along two crystallographic axes as a function of pressure, and thereby identified the most vulnerable element of this MOF. We proceeded to introduce 4,4′-biphenyldicarboxylate (BPDC) linkers as molecular “girders” of ideal size and shape to covalently fit into the backbone of MOF-520. In this way, the original fragile MOF-520 was retrofitted by BPDC to become a mechanically robust framework as evidenced by the fact that it remains crystalline up to 5.5 GPa and on subsequent decompression.
Figure 1.

Visualization of the retrofitting approach in architecture (top) and on a molecular level (bottom). The images of Latimer Hall at UC Berkeley before and after retrofitting are shown for conceptual clarity. The single-crystal X-ray diffraction structures of pristine MOF-520 in gray versus the retrofitted MOF-520-BPDC with BPDC girders shown in red.
Typically, the extreme pressure regime is achieved by loading of single-crystalline or powder sample between the two minuscule culets of opposing diamonds of a diamond-anvil cell (DAC).10 A pressure transmitting medium (PTM) uniformly surrounds the sample to provide hydrostaticity.11 Usually the crystal structures of MOFs under these conditions experience significant distortions, both in the multimetallic SBUs and in the organic linkers, and even undergo reversible/irreversible phase transitions.12−18 The possibility of having the pressure medium molecules diffuse into the MOF pores is unique as it allows study and evaluation of framework deformity. In most cases, the penetration of solvent molecules under extreme pressure inside the MOF (termed overhydration effect) creates internal pressure, which expands the already filled MOF.19−22 At initial compression, the framework expands, but then contracts at higher pressures. The behavior of the framework under these conditions provides an opportunity to identify its weakest points. However, the overhydration effect usually results in reduction of long-range order in the crystal, and the sample becomes irreversibly amorphous. Typically, this lack of crystallinity under pressure has prohibited the study of framework distortions by means of single-crystal X-ray diffraction techniques. These challenges are addressed in the present study.
Results and Discussion
Mechanical Damage of MOF-520 under Extreme Pressure
First, we studied the mechanical response of the crystal structure of MOF-520 toward the increase of the hydrostatic pressure in methanol/ethanol pressure transmitting media in a DAC. MOF-520 is composed of Al-based SBUs linked together with BTB linkers. The secondary building units in MOF-520 are octametallic rings with Al octahedra sharing corners through eight −OHs and four formate ligands. Every SBU is linked by 12 BTBs, resulting in a (12,3)-connected net with fon topology (Figure 2).23 MOF-520 crystallizes in the non-centrosymmetric space group P42212 with unit cell parameters of 18.3754(6) Å (a) and 37.6893(12) Å (c) and a unit cell volume of 12726.0(9) Å3.
Figure 2.

MOF-520 is built from Al-based octametallic secondary building units and organic BTB linkers. Introduction of H2BPDC by the CAL method into MOF-520 leads to the new retrofitted MOF-520-BPDC possessing new skl topology. BTB organic linkers are reduced to gray triangles, BPDC girders to orange links, and Al-based SBU to blue polyhedral. Atom color scheme: C, black; O, red; Al, blue polyhedra. H atoms are omitted for clarity. Yellow balls indicate the space in the octahedral arrangement of building units in the framework.
The single-crystalline sample of MOF-520 was placed into a DAC with a culet size of 500 μm, a tungsten gasket, and a methanol/ethanol mixture (4:1) as a penetrating PTM at room temperature. Several rubies were also placed next to the MOF crystal to monitor the pressure using the ruby fluorescence method.24 The sample was immersed in the PTM for solvent exchange for at least a week prior to single-crystal X-ray data collection. High-pressure data was collected at the 12.2.2 high-pressure beamline at the Advanced Light Source synchrotron (LBNL). The crystal structure was solved and refined at 10–4, 0.15(2), 0.86(2), 1.47(2), 2.24(2), and 2.82(2) GPa upon compression of MOF. At pressures higher than 2.82 GPa, the sample turned amorphous, and the deterioration of the data prevented the structure solution and refinement. Upon further decompression, the data collection was unsuccessful due to complete degradation of the sample: the crystal cracked and disintegrated into several pieces (Figure S9). The crystallographic data and the pore environment information are shown in Table 1. The pore volume and electron count of unassigned electron density within the pore were calculated using the SQUEEZE algorithm.25
Table 1. Crystallographic Data and Pore Metrics of MOF-520 before Retrofitting as a Function of Hydrostatic Pressure.
| pressure/GPa | a/Å | c/Å | unit cell vol/Å3 | pore vol/Å3 | electron count/e– | electron count per pore vol/e– Å–3 |
|---|---|---|---|---|---|---|
| 0.0001 | 18.920(3) | 37.190(7) | 13313(5) | 9391.5 | 9172 | ∼0.98 |
| 0.15(2) | 19.070(3) | 36.930(7) | 13430(5) | 9653.7 | 15064 | ∼1.56 |
| 0.86(2) | 19.196(3) | 36.569(7) | 13475(4) | 9629.5 | 10558 | ∼1.10 |
| 1.47(2) | 19.182(3) | 36.534(7) | 13443(4) | 9495.8 | 13890 | ∼1.46 |
| 2.24(2) | 19.130(3) | 36.480(7) | 13350(4) | 9528.4 | 12753 | ∼1.33 |
| 2.82(2) | 19.070(3) | 36.409(7) | 13241(5) | 9441.1 | 11252 | ∼1.19 |
The bulk strain of MOF-520 crystal structure in the range from 10–4 to 3 GPa was found to be non-monotonic (Figure 3). Upon initial compression up to 0.86 GPa, the crystal structure of MOF expands by almost 1% due to the overhydration effect. A similar observation was reported before for prototypical frameworks, such as MOF-5, ZIF-8, HKUST-1, and UiO-67.19−22 The amount of unassigned electron density, corresponding to PTM molecules, increases by almost 40% from 9299 electrons after DAC loading to 15064 at 0.15(2) GPa, signaling the penetration of the solvent inside the pore. Despite the uncertainty in the calculation of the electron density by SQUEEZE algorithm caused by the poor quality of the experimental data (low resolution, lack of low-angle reflections, overlap with the reflections from two diamonds), this observation is consistent with the pore volume increase by almost 300 Å3 due to internal pressure. The analysis of anisotropy of compression reveals that the structural expansion along a and b crystallographic axes prevails the contraction along the c direction. Upon further increase of pressure, MOF-520 starts to compress along all directions, resulting in the overall decrease in the unit cell volume. Thus, the relative change in volume reaches −0.5% at 2.82(2) GPa. The pore shrinks in size monotonically, leading to the “activation” of the porous material: the number of methanol molecules inside the pore at 2.82(2) GPa is even smaller than such at 0.15(2) GPa. The analogous “activation” was previously reported for ZIF-8 under pressure.21
Figure 3.

Relative changes in unit cell parameters of MOF-520 (left) and MOF-520-BPDC (right) with pressure. Filled and open symbols correspond to increasing pressure and decompression data, respectively. The standard deviations of values are smaller than the size of symbols. The two different regimes of compression in MOF-520 are highlighted with yellow and blue, respectively. The ellipsoids of strain in red are calculated for both structures using PASCal software;28 only the regime of compression was used for the calculation of ellipsoid for MOF-520.
Introduction of the 4,4′-Biphenyldicarboxylate (BPDC) Linkers as Girders
The formate ligands at the SBU of MOF-520 were fully replaced with carboxylic functionalities following the protocol of the coordinative alignment (CAL) method.26 The distance between formate ligands of two adjacent SBUs is ca. 9.5 Å in MOF-520, which allows the precise fitting of the 4,4′-biphenyldicarboxylate of comparable length (10.1 Å). Thus, by soaking MOF-520 single crystals in a saturated solution of H2BPDC, it was possible to replace two formates, linking two SBUs by a rigid molecular girder. The loading rate of this new organic linker was monitored by digested 1H NMR. After 4 days, the crystals of the resulting MOF-520-BPDC, Al8(μ-OH)8(BPDC)2(BTB)4, were washed with anhydrous N,N-dimethylformamide, and the SXRD data were collected at 100 K at ambient pressure. Noteworthy, the introduction of the new linker does not decrease the crystallinity of the sample: the resolution of the data collected at 100 K for MOF-520-BPDC is comparable with such for pristine MOF-520 (Tables S1 and S2). Since the NMR spectra represent the whole batch of the material but not the individual single crystal, during the structure solution and refinement, the occupancy of BPDC girder was estimated by means of SXRD. The sample was fully characterized in terms of surface area, thermal stability, and composition after activation in order to directly compare the MOFs before and after girder installation [for details see the Supporting Information].
The introduction of the 4,4′-biphenyldicarboxylate girder does not change the symmetry of the MOF, and the space group of MOF-520-BPDC remains P42212. At the same time, the structure is expanded along the crystallographic axes a and b by 0.8 Å but compressed along the longest axis c by 1 Å (Table 2). This structural modification leads to the overall expansion of the unit cell by more than 700 Å3. As expected, the crystallographic density of the framework is increased after modification by just 0.05 g/cm3 due to the expansion of the unit cell. The pore volume estimated from the nitrogen adsorption measurement at 77 K is decreased by almost 25% from 1.28 cm3/g for MOF-520 to 0.91 cm3/g for MOF-520-BPDC. Similar reduction of the BET surface area was observed: 3630 and 2548 m2/g for MOF-520 and MOF-520-BPDC, respectively; attributable to the addition of the girder. Despite of the overall decrease in porosity, the pores in the MOF-520-BPDC are still accessible for the methanol/ethanol mixture as a pressure transmitting medium.
Table 2. Crystallographic Data and Pore Metrics of MOF-520 after Retrofitting with BPDC, MOF-520-BPDC, as a Function of Hydrostatic Pressure.
| pressure/GPa | a/Å | c/Å | unit cell vol/Å3 | pore vol/Å3 | electron count/e– | electron count per pore vol/e– Å–3 |
|---|---|---|---|---|---|---|
| 0.0001 | 19.215(4) | 36.779(4) | 13580(6) | 9104.1 | 8612 | ∼0.95 |
| 0.32(2)a | 19.2263(19) | 36.708(3) | 13569(3) | 8992.7 | 6989 | ∼0.77 |
| 0.64(2) | 19.1965(10) | 36.699(2) | 13523.9(16) | 9031.8 | 9455 | ∼1.04 |
| 1.12(2) | 19.185(2) | 36.720(2) | 13515(2) | 9080.1 | 9464 | ∼1.04 |
| 1.67(2) | 19.156(2) | 36.710(2) | 13471(3) | 9054.4 | 9792 | ∼1.08 |
| 2.26(2) | 19.123(2) | 36.648(3) | 13401(3) | 9023.6 | 10519 | ∼1.16 |
| 2.45(2)a | 19.083(4) | 36.725(7) | 13374(6) | 8888.5 | 12365 | ∼1.39 |
| 2.86(2) | 19.086(2) | 36.593(3) | 13330(3) | 8942 | 11046 | ∼1.23 |
| 3.32(5)a | 19.0287(13) | 36.5400(17) | 13230.8(19) | 8825.8 | 9673 | ∼1.09 |
| 4.20(5) | 18.955(4) | 36.464(4) | 13101(5) | 8712.1 | 9756 | ∼1.12 |
| 4.71(5)a | 18.933(3) | 36.424(4) | 13056(4) | 8640.2 | 9979 | ∼1.15 |
| 5.33(5) | 18.857(3) | 36.302(7) | 12909(4) | 8572.3 | 10360 | ∼1.21 |
Measured upon decompression.
In the retrofitted MOF, each SBU is now linked by four BPDCs in addition to 12 BTBs, resulting in a new (16,3)-connected net. While the overall symmetry of the unit cell is preserved during the modification, the retrofitted MOF possesses the new and thus far unreported skl topology (Figure 2). In MOF-520-BPDC the girders are directed along the crystallographic axes a and b, while the c direction remains unchanged. The distance between two adjacent SBUs is increased by almost 0.6 Å due to the incorporation of the BPDC linker.
Mechanical Robustness and Retention of Crystallinity of MOF-520-BPDC under Extreme Pressure
Our next step was to study the mechanical response of the new modified framework to variations in hydrostatic pressure. Using theoretical calculations, it was hypothesized before that the introduction of the new linker may increase the rigidity of MOF toward the extreme pressure.27 The single-crystalline sample of MOF-520-BPDC was placed into a DAC filled with a methanol/ethanol pressure medium, and single-crystal X-ray diffraction data were collected up to 5.5 GPa. It is worth noting that we did not collect the data at even higher pressures because of the size of the crystal: the distance between two culets of diamonds was almost equal to the longest dimension of the sample, and further compression could result in cracking of the MOF crystal. Additionally, the SXRD data were also collected on the subsequent decompression.
Notably, the crystal structure of retrofitted MOF experiences only one regime of distortion. The installation of rigid girder removes the overhydration effect upon initial compression but also increases the stiffness of the framework. The relative decrease in unit cell volume at 1 GPa is just 0.3%, which is almost five times smaller than in the case of pristine MOF-520. More importantly, the existence of the girder between two SBUs does not hinder the solvent molecules from diffusion inside the pore. The amount of unassigned electron density within the pore increases from 8612 electrons after closing the DAC to 9455 electrons at 0.64(2) GPa. Despite the penetration of pressure medium into the MOF, the internal pressure created by these molecules is not enough to expand the framework from the inside. This is not surprising considering that, for expansion of the unit cell, the carbon–carbon bonds of the BPDC girder must be stretched. Further compression of the MOF crystal leads to the reduction of the pore volume by ∼500 Å3, and the amount of unassigned electron density related to resident molecules of PTM within the pore increases from 9455 electrons at 0.34(2) GPa to 10360 electrons at 5.33(5) GPa.
The anisotropy of compression of MOF-520-BPDC is shown in Figure 3. One can see that the structure compresses along all crystallographic directions monotonically. The linear strain reaches almost 2% along the a (and b) crystallographic axis and 1.5% along the c axis at 5.3 GPa. The relative volume change at 5.3 GPa is about 5.5%. Based on the collected data, the ellipsoid of strain was calculated, showing the median compressibility coefficients of 3 TPa–1 along all three crystallographic directions.28 These values are almost two times smaller than those of parent MOF-520, indicating the corresponding increase of stiffness due to the placement of the girders.
The reason for the drastic difference of MOFs’ response to mechanical stimuli is the removal of distortion of the unit cell between two SBUs along the shortest axes due to the girder. For example, the distance between carbon atoms of the formate ligands of two neighboring SBUs is 9.470(4) Å at 100 K, 10.25(2) Å after closing the DAC, 10.31(2) Å in MOF-520 at 0.86 GPa (regime of expansion), and 9.64(4) Å at 2.82 GPa (regime of compression). In contrast, in the retrofitted MOF, the distance between carboxylate carbon atoms in the girder only shortens by 0.37(2) Å at 5.33 GPa.
While in previously studied MOFs, such as MOF-5 and HKUST-1, the weakest element of the structures is the metal–linker bonds in the metal–oxide SBU, in the MOF-520 and retrofitted MOF-520 the rotated phenylene units of the BTB linkers are the weak points responsible for the deformation of the framework (Supporting Information, section S5). In other words, the structure of MOF-520 suffers from mechanical instability due to the organic linker rather than the inorganic SBU. This study showed how this framework instability can be turned into robustness by molecular retrofitting of MOF-520 with BPDC.
Acknowledgments
Support for the synthesis by BASF SE (Ludwigshafen, Germany, Award No. 20153470) and the characterization of compounds by King Abdulaziz City for Science and Technology (Center of Excellence for Nanomaterials and Clean Energy Applications, Award No. 036931) and the high-pressure single-crystal X-ray diffraction studies by the Center for Gas Separations Relevant to Clean Energy Technologies, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0001015. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The high-pressure facilities at the Advanced Light Source are supported by COMPRES, the Consortium for Materials Properties Research in Earth Sciences under NSF Cooperative Agreement EAR 11-57758. We thank Dr. Christine Beavers for the synchrotron X-ray diffraction data acquisition support at the beamline 12.2.2 (Advanced Light Source, Lawrence Berkeley National Laboratory); Dr. Markus Kalmutzki and Christian Diercks for discussions of editing of this manuscript. Data reported in this paper are tabulated in the Supporting Infortmation and archived at the Cambridge Crystallographic Data Centre database under the following reference numbers: 1552341−1552360. We acknowledge the collaboration and valuable input of Prince Dr. Turki bin Saud bin Mohammed Al-Saud (President of KACST).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00169.
The authors declare no competing financial interest.
Supplementary Material
References
- Lin X.; et al. High capacity hydrogen adsorption in Cu(II) tetracarboxylate framework materials: the role of pore size, ligand functionalization, and exposed metal sites. J. Am. Chem. Soc. 2009, 131, 2159–2171. 10.1021/ja806624j. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; et al. Ultrahigh porosity in metal–organic frameworks. Science 2010, 329, 424–428. 10.1126/science.1192160. [DOI] [PubMed] [Google Scholar]
- Farha O. K.; et al. Metal–organic framework materials with ultrahigh surface areas: is the sky the limit?. J. Am. Chem. Soc. 2012, 134, 15016–15021. 10.1021/ja3055639. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Cordova K. E.; Yaghi O. M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- Seo J. S.; et al. A homochiral metal-organic porous material for enantioselective separation and catalysis. Nature 2000, 404, 982–986. 10.1038/35010088. [DOI] [PubMed] [Google Scholar]
- Zhao X.; et al. Hysteretic adsorption and desorption of hydrogen by nanoporous metal-organic frameworks. Science 2004, 306, 1012–1015. 10.1126/science.1101982. [DOI] [PubMed] [Google Scholar]
- Nugent P.; et al. Porous materials with optimal adsorption thermodynamics and kinetics for CO2 separation. Nature 2013, 495, 80–84. 10.1038/nature11893. [DOI] [PubMed] [Google Scholar]
- Faust T. MOFs move to market. Nat. Chem. 2016, 8, 990–991. 10.1038/nchem.2656. [DOI] [PubMed] [Google Scholar]
- Gándara F.; Furukawa H.; Lee S.; Yaghi O. M. High methane storage capacity in aluminum metal-organic frameworks. J. Am. Chem. Soc. 2014, 136, 5271–5274. 10.1021/ja501606h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill L.; Bassett W. A. Miniature diamond anvil pressure cell for single-crystal X-ray diffraction studies. Rev. Sci. Instrum. 1974, 45, 290–294. 10.1063/1.1686607. [DOI] [Google Scholar]
- Angel R. J.; Bujak M.; Zhao J.; Gatta G. D.; Jacobsen S. D. Effective hydrostatic limits of pressure media for high-pressure crystallographic studies. J. Appl. Crystallogr. 2007, 40, 26–32. 10.1107/S0021889806045523. [DOI] [Google Scholar]
- Gagnon K.; Beavers C. M.; Clearfield A. MOFs under pressure: the reversible compression of a single crystal. J. Am. Chem. Soc. 2013, 135, 1252–1255. 10.1021/ja311613p. [DOI] [PubMed] [Google Scholar]
- Bennett T. D.; Cheetham A. K. Amorphous metal-organic frameworks. Acc. Chem. Res. 2014, 47, 1555–1562. 10.1021/ar5000314. [DOI] [PubMed] [Google Scholar]
- Gandara F.; Bennett T. D. Crystallography of metal–organic frameworks. IUCrJ 2014, 1, 563–570. 10.1107/S2052252514020351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudert F.-X. Metal-organic frameworks: the pressure is on. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2015, 71, 585–586. 10.1107/S2052520615020934. [DOI] [PubMed] [Google Scholar]
- McKellar S. C.; Moggach S. A. Structural studies of metal-organic frameworks under high pressure. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2015, 71, 587–607. 10.1107/S2052520615018168. [DOI] [PubMed] [Google Scholar]
- Cai W.; et al. Giant negative area compressibility tunable in soft porous framework. J. Am. Chem. Soc. 2015, 137, 9296–9231. 10.1021/jacs.5b03280. [DOI] [PubMed] [Google Scholar]
- Im J.; Yim N.; Kim J.; Vogt T.; Lee Y. High-pressure chemistry of a zeolitic imidazolate framework compound in the presence of different fluids. J. Am. Chem. Soc. 2016, 138, 11477–11480. 10.1021/jacs.6b07374. [DOI] [PubMed] [Google Scholar]
- Graham A. J.; Allan D. R.; Muszkiewicz A.; Morrison C. A.; Moggach S. A. The effect of high pressure on MOF-5: guest-induced modification of pore size and content at high pressure. Angew. Chem., Int. Ed. 2011, 50, 11138–11141. 10.1002/anie.201104285. [DOI] [PubMed] [Google Scholar]
- Graham A. J.; Tan J. C.; Allan D. R.; Moggach S. A. The effect of pressure on Cu-btc: framework compression vs. guest inclusion. Chem. Commun. 2012, 48, 1535–1537. 10.1039/C1CC16045A. [DOI] [PubMed] [Google Scholar]
- Moggach S. A.; Bennett T. A.; Cheetham A. K. The effect of pressure on ZIF-8: increasing pore size with pressure and the formation of a high-pressure phase at 1.47 GPa. Angew. Chem., Int. Ed. 2009, 48, 7087–7089. 10.1002/anie.200902643. [DOI] [PubMed] [Google Scholar]
- Hobday C. L.; et al. A computational and experimental approach linking disorder, high-pressure behavior, and mechanical properties in UiO frameworks. Angew. Chem. 2016, 128, 2447–2451. 10.1002/ange.201509352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keeffe M.; Yaghi O. M. Deconstructing the crystal structures of metal-organic frameworks and related materials into their underlying nets. Chem. Rev. 2012, 112, 675–702. 10.1021/cr200205j. [DOI] [PubMed] [Google Scholar]
- Piermarini G. J.; Block S.; Barnett J. D.; Forman R. A. Calibration of the pressure dependence of the R1 ruby fluorescence line to 195 kbar. J. Appl. Phys. 1975, 46, 2774–2780. 10.1063/1.321957. [DOI] [Google Scholar]
- Spek A. L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 9–18. 10.1107/S2053229614024929. [DOI] [PubMed] [Google Scholar]
- Lee S.; Kapustin E. A.; Yaghi O. M. Coordinative alignment of molecules in chiral metal-organic frameworks. Science 2016, 353, 808–811. 10.1126/science.aaf9135. [DOI] [PubMed] [Google Scholar]
- Ortiz A. U.; Fuchs A. H.; Coudert F.-X. Metal-organic frameworks with wine-rack motif: What determines their flexibility and elastic properties?. J. Chem. Phys. 2013, 138, 174703. 10.1063/1.4802770. [DOI] [PubMed] [Google Scholar]
- Cliffe M. J.; Goodwin A. L. PASCal: a principal axis strain calculator for thermal expansion and compressibility determination. J. Appl. Crystallogr. 2012, 45, 1321–1329. 10.1107/S0021889812043026. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.