

Abstract
Protein kinase CK2 is a ubiquitously expressed serine/threonine kinase composed of two catalytic subunits (α) and/or (α′) and two regulatory (β) subunits. The expression and kinase activity of CK2 is elevated in many different cancers, including glioblastoma (GBM). Brain tumor initiating cells (BTICs) are a subset of cells that are highly tumorigenic and promote the resistance of GBM to current therapies. We previously reported that CK2 activity promotes prosurvival signaling in GBM. In this study, the role of CK2 signaling in BTIC function was examined. We found that expression of CK2α was increased in CD133+ BTICs compared to CD133- cells within the same GBM xenolines. Treatment with CX-4945, an ATP-competitive inhibitor of CK2, led to reduced expression of Sox2 and Nestin, transcription factors important for the maintenance of stem cells. Similarly, inhibition of CK2 also reduced the frequency of CD133+ BTICs over the course of 7 days, indicating a role for CK2 in BTIC persistence and survival. Importantly, using an in vitro limiting dilution assay, we found that inhibition of CK2 kinase activity with CX-4945 or siRNA knockdown of the CK2 catalytic subunits reduced neurosphere formation in GBM xenolines of different molecular subtypes. Lastly, we found that inhibition of CK2 led to decreased EGFR levels in some xenolines, and combination treatment with CX-4945 and Gefitinib to inhibit CK2 and EGFR, respectively, provided optimal inhibition of viability of cells. Therefore, due to the integration of CK2 in multiple signaling pathways important for BTIC survival, CK2 is a promising target in GBM.
Keywords: Glioblastoma, CK2, BTIC, CD133, EGFR
Introduction
Glioblastoma (GBM) (World Health Organization Grade IV astrocytoma), is the most common primary brain malignancy in adults, accounting for 60–70% of malignant gliomas [1]. Malignant gliomas are highly heterogeneous from both histological and molecular standpoints, adding to the complexity of effectively treating patients with GBM [1, 2]. GBM has been classified into four molecular subtypes including, classical, mesenchymal, neural and proneural based on gene expression/aberrations [3]. Recently, efforts have been made to determine the genetic differences between low grade glioma tumors and those that progress to high grade GBM, which will hopefully provide more clinically relevant interventions [4]. Nevertheless, despite current therapy, the median survival is only 12–15 months [1], highlighting the need for new approaches to GBM therapy.
Brain tumor initiating cells (BTICs) are pluripotent, self-renewing and tumorigenic [5]. BTICs are resistant to the current treatment regimen of chemotherapy and radiation [6, 7] and this resistance to treatment is likely responsible for poor patient survival [1]. There is controversy surrounding BTIC markers, but the most well characterized is the surface marker CD133 [8, 9]. The frequency of CD133+ cells is prognostic for overall survival in glioma patients [10] and its expression is important for self-renewal and tumorigenic potential [11]. BTICs are characterized by the expression of stemness factors important in neural stem cell function [5]. Indeed, these stemness markers, such as Sox2 and Nestin, are important for the maintenance of the stem cell phenotype [5].
Protein kinase CK2 (casein kinase 2) is a ubiquitously expressed serine/threonine kinase composed of two catalytic subunits (α) and/or (α′) and two regulatory (β) subunits [12]. CK2 is constitutively active and is essential for viability as evidenced by the embryonic lethality of CK2 knockout mice [13, 14]. Importantly, the expression of CK2α is elevated in many solid tumors [12], including GBM [15–18]. We have previously described a role for CK2 in NF-κB, PI3K/AKT and STAT3 signaling in GBM. Additionally, we and others have found that inhibition of CK2 provides therapeutic efficacy utilizing in vivo models of GBM [16, 17]. In this study, we expand on these results and examine the role of CK2 in BTIC function. We demonstrate that the expression and activity of CK2 are increased in BTICs compared with other GBM cells, and that CK2 has important roles in the expression of stemness factors. Furthermore, we establish that CK2 activity is critical for BTIC survival and neurosphere formation in primary GBM xenolines of multiple molecular subtypes. In addition, we found that inhibition of CK2 led to decreased EGFR levels, and that inhibition of both CK2 and EGFR activation provided the optimal therapeutic response. Consequently, CK2 is an essential molecule with a pivotal role in BTIC function, warranting further investigation as a therapy for GBM.
Materials and methods
Ethics statement
All experiments with mice (female athymic nude, Harlan Laboratories; C57BL/6, Charles River) were performed with the approval of the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Reagents
Antibodies to GAPDH from Abcam; Sox2 from Santa Cruz; and EGFR from BD Transduction Laboratories. EGF and bFGF are from either R&D Systems or Miltenyi Biotec Inc. The Millipore Casein Kinase 2 Assay Kit was used to determine CK2 kinase activity. CX-4945, a CK2 inhibitor, was synthesized and generously provided by Cylene Pharmaceuticals [19].
Human GBM xenolines and cell culture
Human GBM xenolines were isolated from subcutaneous tumors generated from fresh patient samples, which were never cultured on plastic. These flank tumors are maintained by the UAB Brain Tumor Core Facility and have been molecularly characterized as previously described [20]. Culturing the cells as subcutaneous tumors has been shown to better replicate the microenvironment conditions than in vitro cell culture methods. However, this microenvironment is indeed different from the brain. The four xenolines examined represent different molecular subtypes [Proneural (X456), Neural (X1066), Classical (X12 and X6)], have amplifications of EGFR (X12) and/or amplified mutant EGFRvIII (X6), and deletions in p53 (X12, X1066, X456). Xenolines X12, X1066 and X456 also have amplification of CK2α [16]. To generate single cells for culture for each experiment, flank tumors were excised, minced, and digested in the presence of trypsin, collagenase (50 μg/ml) and DNase I (25 μg/ml). Cells were passed over a 40 μm filter, counted, and cultured in neurobasal media (Gibco) containing MACS Neurobrew-21 without vitamin A (Miltenyi), 1% Pen/Strep, 1% L-glutamine, gentamicin sulfate (0.1 mg/ml), EGF (20 ng/ml) and bFGF (20 ng/ml). Images of neurospheres were captured using an EVOS fluorescent microscope at 20× magnification.
Murine NPC preparation
Embryos (E15) were removed from pregnant C57BL/6 mice and dissected to isolate the telencephalon, a method adapted from [21]. The telencephalon was dissociated by trituration using a 1000 μl pipet tip. The dissociated tissue was passed over a 100 μm cell strainer. The resulting single cells were initially plated at a density of 2 × 105 cell/ml in DMEM/F12 media containing MACS Neurobrew-21 without Vitamin A (Miltenyi), 1% Pen/Strep, 1% l-glutamine, EGF (20 ng/ml), bFGF (10 ng/ml) and 0.0002% heparin (Stem Cell). Neurospheres formed over the course of 5–6 days.
Isolation of murine brain tissue
Brain tissue from C57BL/6 mice was harvested on embryonic (E) day 13.5 and then postnatal (P) 1, P5, P10, P20 and P70. RNA was extracted using the mirVana miRNA Isolation Kit (Life Technologies) and cDNA was synthesized as previously described [20].
Flow cytometry
To assay for intracellular expression of antigens, cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Molecular Probes) or Fixable Viability Dye eFluor 780 (eBioscience), fixed, and permeabilized with methanol. To stain for CK2α, 70% methanol was used to permeabilize cells and anti-CK2α (Abcam) and anti-CD133/1 (Miltenyi) were used to stain cells. To detect GFAP, Sox2 and Nestin (antibodies from BD Pharmingen), the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) was used per manufacturer′s recommendations. Additionally, for some experiments, Sox2 was detected using a 90% methanol permeabilization.
To assess CD133/1 expression and viability after prolonged (7 day) culture, cells were first dissociated using gentle trituration and multiple washes with MACS Buffer (Miltenyi) containing 0.5% BSA and 2 mM EDTA. Single cells were then stained with anti-CD133/1. Propidium iodide was added to quantify and exclude dead cells.
All analyses were conducted using FlowJo (Tree Star). Cells were first gated to exclude dead cells, followed by gating on FSC/SSC to exclude fragments and clusters of cells. Stain indices were used to normalize for differences in background autofluorescence between cell types or resulting from culture with CX-4945. A stain index for each antibody was calculated by subtracting the median fluorescent intensity (MFI) of the negative (unstained) population from the MFI of the positive population and dividing by 2 times the robust standard deviation of the negative population [22]. Stain index= [(MFIStained-MFIunstained)/2*SDunstained].
Immunoblotting
Cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of protein (10–20 μg) were loaded onto SDS–PAGE gels as previously described [20]. Antibodies used are specified above.
Quantitative real-time PCR
Total RNA was extracted and cDNA was synthesized as previously described [20]. The Taqman primers/probe sets, CK2α (Mm01243794_g1), CK2α′ (Mm01243455_ m1), CK2β (Mm00487216_m1), Sox2 (Mm03053810_s1), GFAP (Mm01253033_m1), EGFR (Hs01076078_m), CK2α (Hs00751002_s1) and CK2α′ (Hs00176505_m1) were used to quantify gene expression; Eukaryotic 18S rRNA (Hs99999901_s1) served as a reference gene.
Cell proliferation assays
Cells (1.5–2 × 104) were plated in 96 well plates and cultured for 72 h. Cell Proliferation Reagent WST-1 (Roche) was then added, as previously described [16].
RNA interference
X456 cells were transfected with 200 nM of nontarget (NT) siRNA or 100 nM of CK2α and CK2α′ siRNA using Lipofectamine RNAiMax (Life Technologies), as previously described [23]. Cells were transfected 48 h prior to sorting for in vitro limiting dilution assay. Knockdown efficiency was assessed by extracting RNA from transfected cells and using qPCR to quantitate expression of CK2α and CK2α′.
In vitro limiting dilution assay
Xenolines were dissociated and plated into 96 well plates using either dilution plating as indicated on the x-axis (example: 1000, 500, 200, 50, and 10 cells/well; replicates of at least eight per dilution) or by sorting using the BD FACSAria II as described in [24]. When FACS sorted, cells were gated on single cells (SSC-W vs. SSC-H) and propidium iodide staining was used to exclude dead cells. Cells were plated into neurobasal media (Gibco) containing MACS Neurobrew-21 without Vitamin A (Miltenyi), 1% Pen/Strep, 1% l-glutamine, gentamicin sulfate (0.1 mg/ml), EGF (20 ng/ml) and bFGF (20 ng/ml). Cells were monitored daily (by inverted microscope) starting 5–6 days after plating. Once neurospheres were visualized in the wells seeded with the most cells, all wells and conditions were counted. In some experiments cells were plated into 50% conditioned medium from the same xenoline. Conditioned media was harvested after 3–4 days of culture with healthy cells; cells were removed by centrifugation and media was sterile filtered (0.22 μM). Neurospheres were manually counted using an inverted microscope. One technician read all of the plates, which were blinded by a second technician on the day of setup. A neurosphere was defined as a cluster of >8 cells in diameter (80 μM). Extreme limiting dilution analysis was used to analyze data (http://bioinf.wehi.edu.au/software/elda/).
Densitometry and statistical analysis
Densitometric analysis was performed using Image J on scanned blots. GAPDH was used for normalization. Values for DMSO treated cells were then normalized to one for each timepoint.
All statistical analyses were performed using Sigma-Plot. For all analyses, equal variance and normal distribution tests were applied. If both passed, a Student′s t-test was used to compare two samples and a one-way ANOVA with Tukey′s post-hoc test was used to compare three or more samples. If normal distribution failed, then Mann Whitney test was used for two comparisons and Kruskal–Wallis test was used for more than two comparisons. p < 0.05 was considered statistically significant. Error bars represent mean ± SD.
Results
The expression and activity of CK2α is increased in BTICs
CK2 activity is essential for cell viability [13, 14] and CK2 is highly expressed in the brain [25], but little is known about the dynamics of its subunit expression in stem cells compared to more differentiated astrocytes. Initially, we assessed the expression of the CK2 subunits (α, α′, β) during murine neurodevelopment between embryonic day 15 (E15) and postnatal 70 (P70). We found that expression of all three subunits of CK2 was highest at embryonic day 15 (E15) and decreased after birth (P1) (Fig. 1a). Interestingly, the expression pattern of CK2 mirrored that of Sox2, a transcription factor important for late stage cellular reprogramming [26] (Fig. 1a). On the other hand, glial fibrillary acidic protein (GFAP), a marker of differentiation for astrocytes [27], increased following birth (P1) and continued to increase until P5, when the levels remained high until P70 (Fig. 1b). This dynamic between Sox2 and GFAP indicates a transition from a stem-like population where Sox2 expression is high to a more differentiated astrocytic population, as evidenced by increased GFAP expression. Therefore, the finding that CK2 levels are highest in the stem-like population suggests that CK2 may be important for stem cell function.
Fig. 1.
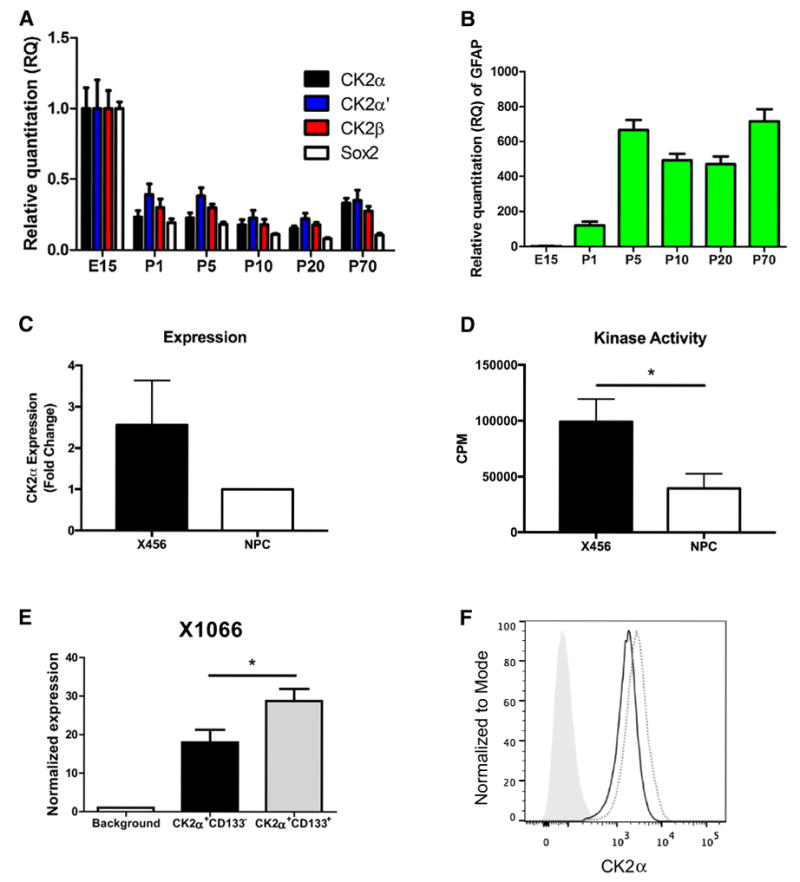
CK2α expression and activity are increased in BTICs. a Expression of CK2 subunits (α, α′, β), SOX2 and b GFAP during murine neurodevelopment. Data represent one mouse per timepoint in replicates of three. c Murine NPCs and human X456 cells were evaluated for CK2α expression by flow cytometry (n = 3). d CK2 kinase activity was assessed in murine NPCs and human X456 cells (n = 3, data represent counts per minute (CPM) with background subtracted for each condition). e CK2α expression in CD133+ and CD133- cells of X1066 xenoline was assessed using flow cytometry (n = 3). f Representative histogram of CK2α expression (gray [solid] represents background control, black line represents CK2α+CD133-, gray [dotted] represents CK2α+CD133+). *p < 0.05
We and others have previously demonstrated that CK2α expression is increased in GBM [15–18]. We extended these findings by assessing the expression and activity of CK2 in BTICs. CK2 protein expression and activity was examined in malignant GBM neurospheres compared to non-transformed murine neural precursor cells (NPCs). Using flow cytometry, we found that protein expression of CK2α, the major catalytic subunit of CK2, is elevated in neurospheres from the X456 GBM xenoline, a pediatric GBM of the Proneural molecular subtype compared with neurospheres from NPCs (Fig. 1c). More importantly, using CK2α and CK2α′ subunits immunoprecipitated from cell lysates, we found that the CK2 kinase activity was significantly elevated in X456 neurospheres compared to NPCs (Fig. 1d). Protein expression of the CK2α subunit and CK2 kinase activity display similar patterns in these cells, suggesting a strong correlation between CK2α protein levels and kinase activity (Fig. 1c, d).
Expression of CK2α is increased in GBM [15–18]; therefore, it is essential to discern if the expression of CK2 is further increased in BTICs, as increased expression of CK2 may render BTICs even more susceptible to CK2 inhibition. As previously mentioned, CD133 is commonly used as a BTIC marker [8, 9]. The validity of the CD133 marker in our xenolines was evaluated, and we observed an enhancement of stemness marker expression (Sox2 and Nestin) in CD133+ cells compared with all live GBM cells (Fig. S1). Flow cytometry was used to determine the pattern of CK2α expression in BTICs (CD133+) compared with non-stem cells (CD133-) within the same GBM xenoline. Indeed, we found that in freshly isolated cells from X1066, a xenoline of the neural subtype, CK2α expression was significantly increased in the stem cell (CD133+) population compared to the CD133- population (Fig. 1e, f). Comparable results were also observed for X12, a xenoline of the classical molecular subtype (data not shown). This indicates that the BTIC populations within the same xenoline have increased expression of the CK2α catalytic subunit.
The expression of stemness factors is reduced by CK2 inhibition
Sox2 is a transcription factor important for proliferation and tumorigenicity of BTICs [28]. To investigate a possible role for CK2 signaling in maintenance of the stem cell phenotype, we treated neurospheres from the GBM xenoline X456 with CX-4945 for 4–24 h and immunoblotted for expression of Sox2. A substantial reduction of Sox2 was observed in cells treated with CX-4945 compared to vehicle control, in a time-dependent manner (Fig. 2a). We further quantified the expression of stemness factors in X456 neurospheres following 24 h of treatment with CX-4945. Using flow cytometry, a significant reduction in the expression of the stemness markers Sox2 and Nestin in cells treated with CX-4945 was observed (Fig. 2b, c). The reduction of Sox2 expression was highly consistent between the immunoblotting and flow cytometry methods (Fig. 2a, b). On the other hand, we did not observe an increase in the expression of GFAP (Fig. 2d). These findings suggest that CK2 signaling is responsible for maintenance of the stem cell phenotype.
Fig. 2.
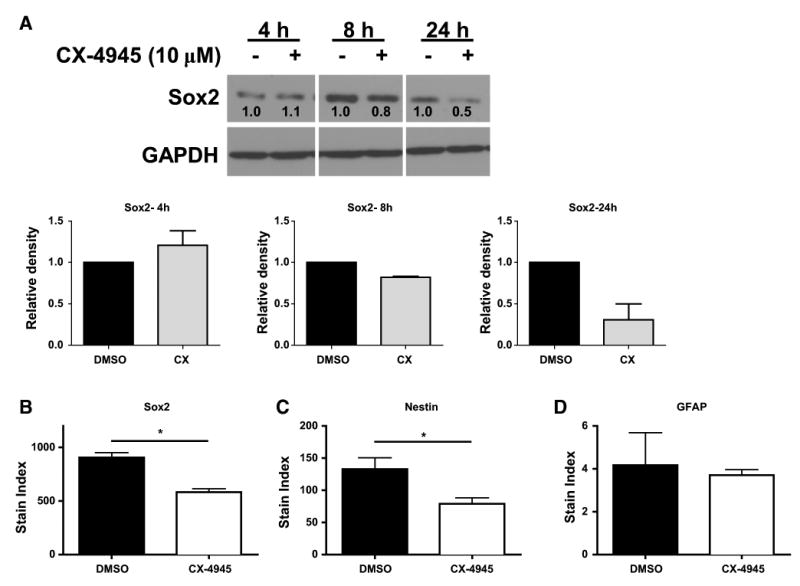
Expression of stemness factors in BTICs is reduced by CK2 inhibition. a X456 cells were assessed for Sox2 expression after 4–24 h culture with CX-4945 (10 μM) or vehicle control. Densitometry (bar graphs) was performed and relative expression is in reference to the DMSO control at each timepoint (n = 2). X456 cells were assessed for expression of b Sox2, c Nestin, and d GFAP by flow cytometry after 24 h culture with CX-4945 (10 μM) or vehicle control (n = 2). *p < 0.05
The viability and frequency of GBM stem cells is dependent on CK2 activity
Protein kinase CK2 has important roles in cell cycle and cell survival [12]. We assessed the effect of CK2 inhibition on neurosphere proliferation/viability using the WST-1 assay. We found that 72 h treatment with CX-4945 effectively reduced proliferation/viability in both X456 and X12 xenolines (Fig. 3a, b). Moreover, the size of neurospheres from X456 and X12 xenolines cultured with CX-4945 was also greatly reduced compared to DMSO control neurospheres (Fig. 3c, d). Together, these findings suggest that inhibition of CK2 reduces proliferation and/or cell viability of BTICs.
Fig. 3.
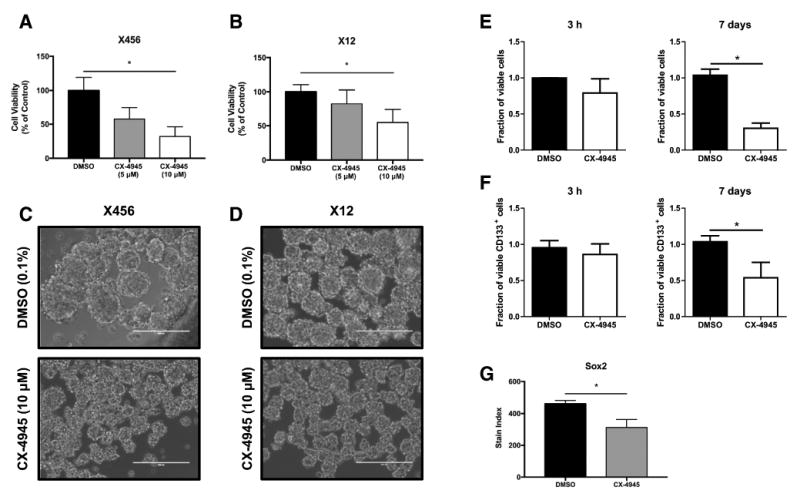
Inhibition of CK2 leads to a reduction in BTIC frequency and proliferation. a X456 (20,000/well) or b X12 (15,000/well) cells were cultured with CX-4945 (5, 10 μM) or vehicle control for 72 h and proliferation assessed using the WST-1 assay (n = 3). Representative photos (20× magnification) of c X456 and d X12 neurospheres 6 days after plating in the absence or presence of CX-4945 (10 μM). White bar equals 200 μM. X12 cells cultured with CX-4945 (10 μM) or vehicle control for 3 h or 7 days were assessed for viability e, frequency of CD133+ stem cells f and total number of CD133+ cells, using flow cytometry. Fractions of viable cells and CD133+ cells were normalized to DMSO control at 3 h or 7 days (n = 3). g X12 cells were assessed for expression of Sox2 by flow cytometry after 7 days of culture with CX-4945 (10 μM) or vehicle control (n = 3). *p < 0.05
To determine if the results from the WST-1 assay were due to a reduction in BTIC viability, cell viability and surface CD133 expression was examined by flow cytometry. After 7 days of culture with CX-4945, there was a significant decrease of viable X12 cells compared to those cultured with DMSO (Fig. 3e, right). There was a modest reduction in viability of CX-4945 treated cells after 3 h of treatment (Fig. 3e, left). Significantly, we also observed a substantial reduction in CD133+ cells following 7 days of CX-4945 treatment when compared to DMSO control (Fig. 3f, right). No reduction in CD133 expression was detected after 3 h of culture with CX-4945 (Fig. 3f, left). Furthermore, the expression of CD133 was assessed after excluding dead cells from analysis. Therefore, the reduction of BTICs following culture with CX-4945 is even more striking considering there are less than 1/3 of cells viable after 7 days. We also confirmed this reduction in BTICs by examining the stemness factor, Sox2, after 7 days of culture with CX-4945. Consistent with our previous results, we observed a significant decrease in Sox2 expression following prolonged culture with CX-4945 (Fig. 3g). Consequently, these findings suggest that inhibition of CK2 has profound effects on BTIC viability and numbers.
CK2 signaling promotes neurosphere formation
The reduction of cell viability and frequency of CD133+ stem cells following CK2 inhibition suggests that CK2 signaling plays an important role in BTIC function. To directly assess the effect of CK2 signaling on BTIC neurosphere formation, we utilized the in vitro limiting dilution assay. This assay is performed by plating exact numbers of cells from a xenoline in a 96 well plate and monitoring for neurosphere formation, allowing for an estimate of the frequency of stem cells. Treatment of three different xenolines (X456, X12 and X6) with CX-4945 led to a significant reduction in neurosphere formation (Fig. 4a–c, f). Additionally, using siRNA knockdown of CK2α and CK2α′, we confirmed that the catalytic subunits of CK2 are critical for neurosphere formation (Fig. 4d–f). These findings highlight the effectiveness of CK2 inhibition on neurosphere formation in multiple molecular subtypes. Importantly, CX-4945 is only added at the onset of the prolonged assay, emphasizing the potency of CK2 inhibition on neurosphere formation.
Fig. 4.
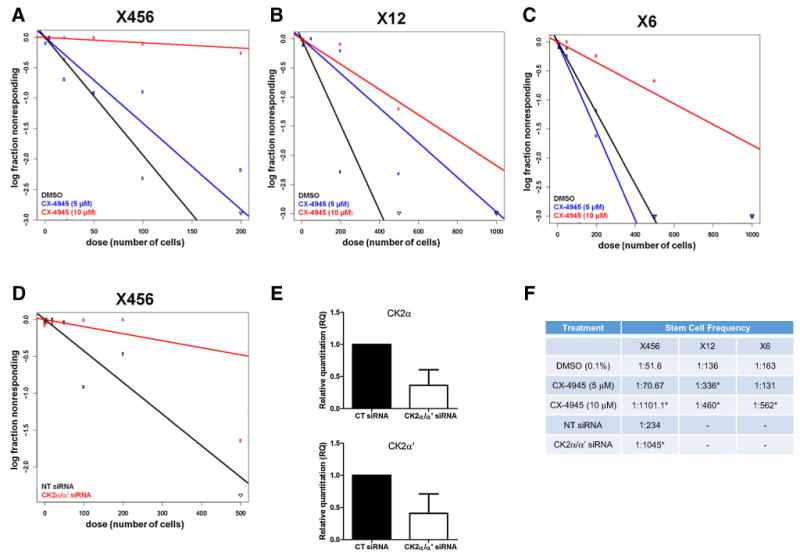
CK2 signaling promotes neurosphere formation. In vitro limiting dilution assay (LDA) was used to assess the neurosphere formation of GBM stem cells on the indicated days. a X456 cells cultured with CX-4945 (0–10 μM) (day 12) or d transfected with siRNA targeting CK2α/α′ (day 10) were assessed for neurosphere formation. e qPCR of CK2α (left) and CK2α′ (right) 48 h post-transfection with non-targeting siRNA or CK2α and CK2α′ siRNA. b X12 cells (day 11) and c freshly isolated cells from xenoline X6 (day 19) were cultured with CX-4945 (0–10 μM) and assessed for neurosphere formation (n = 2). f Stem cell frequency of xenolines following CK2 inhibition with CX-4945 or knockdown of CK2 catalytic subunits. *p < 0.05 comparing to DMSO or NT siRNA
Inhibition of CK2 results in decreased EGFR expression
It has been observed in lung cancer cells that CX-4945 treatment decreases EGFR expression [29]. EGFR amplifications and/or constitutively active mutations (EGFRvIII) are extremely common in GBM. Therefore, we sought to determine if inhibiting CK2 in GBM neurospheres would lead to a decrease in EGFR levels. We found that treatment with CX-4945 significantly reduced EGFR mRNA in X456 cells, but not X6 cells (Fig. 5a–b). The unchanged EGFR levels in X6, which harbors the EGFRvIII constitutively active mutant, could be a potential mechanism of resistance when treated with CX-4945. Therefore, we tested combination CX-4945 and gefitinib treatment to inhibit both CK2 and EGFR signaling in X6 cells. We found that combination treatment led to a significant reduction in viability/proliferation in X6 cells when compared to DMSO or single agent alone (Fig. 5c). These findings suggest that both CK2 and EGFR signaling should be targeted to provide optimal therapeutic response.
Fig. 5.
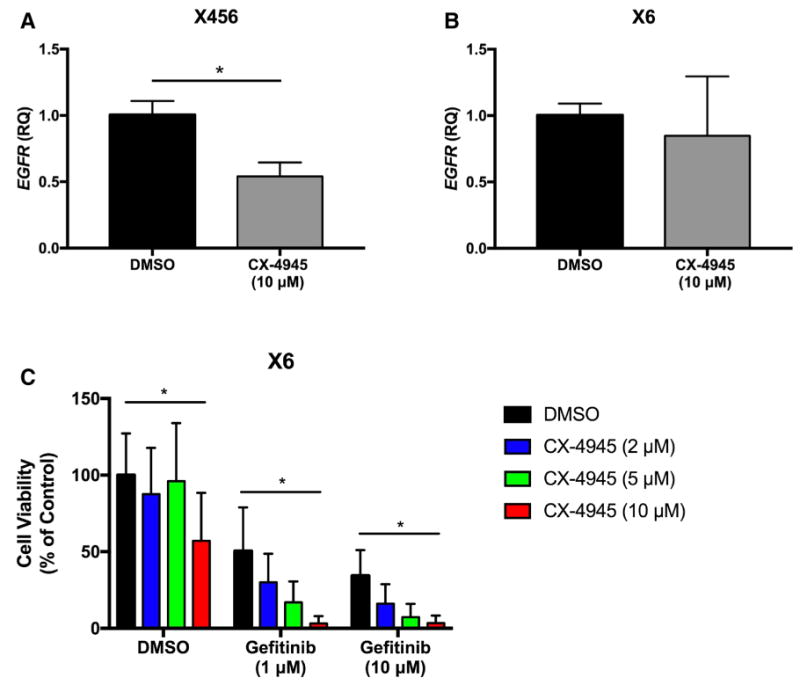
Inhibition of CK2 results in decreased EGFR expression and combination therapies targeting CK2 and EGFR provide optimal response. a–b Xenografts X456 and X6 were treated with CX-4945 (10 μM) for 24 h, and expression of EGFR assessed by qPCR (n = 3). c Xenograft X6 was simultaneously treated with graded concentrations of CX-4945 and gefitinib for 96 h, and cell survival was assessed using the WST-1 assay (n = 3). *p < 0.05
Discussion
We report that expression of CK2α, a CK2 catalytic subunit, is increased in BTICs compared to non-BTICs of the same xenoline. This enhanced expression of CK2 in stemlike populations from xenolines has not been previously described in GBM. Enhanced expression of CK2 catalytic subunits may render these cells more susceptible to inhibition with ATP-competitive inhibitors such as CX-4945. Interestingly, three of the four xenolines tested (X12, X1066, X456) have a gene dosage gain in CSKN2A1, the gene encoding CK2α. We previously reported that this gene dosage gain is found in approximately one-third of GBM tumors, and is enriched in the classical molecular subtype [16]. Notably, we found that the enhanced expression of CK2α in BTICs and reduced neurosphere formation upon CK2 inhibition is not restricted to one molecular subtype. The four xenolines examined represent different molecular subtypes (Proneural, Neural, Classical), have amplifications of EGFR (X12) and/or amplified mutant EGFRvIII (X6), and deletions in p53 (X12, X1066, X456). These findings are important given the extensive heterogeneity of GBM, and indicate that GBMs with amplifications in EGFR, the most common genetic abnormality in GBM [3, 30–32] will likely be responsive to therapeutics directed at CK2 inhibition.
Although examining the role of CK2 in BTIC population in all four TCGA subtypes for all assays would have been desirable, we found this was not always achievable. For instance, the expression of CK2α in CD133+ and CD133- cells was examined in freshly isolated X1066, but these cells were not amenable to longer cultures (e.g. proliferation/viability and limiting dilution assays). Throughout this manuscript, we consistently use X456 and X12 xenolines, representing the proneural and classical subtypes, respectively, to illustrate the critical findings of this manuscript. Furthermore, X6 (classical subtype) was used in the limiting dilution assay and EGFR experiments due to the amplification (VIII) of EGFR as a comparison to X12 and X456, which express wild-type EGFR. Lastly, CD133 expression is undetectable in X456 in our hands, which is why it was not used in experiments where CD133+ cells were characterized.
Recently, the Li group published a paper on CK2 and the role of β-catenin in BTIC function [17]. Our manuscript extends these findings by examining four different xenolines characterized for their molecular signature, representing the Neural, Proneural, and Classical molecular subtypes. Additionally, we confirmed that CD133 expression is reduced following CK2 inhibition. The rationale for examining CD133 expression after 7 days was to assess the long-term effectiveness of CX-4945 in vitro on CD133+ BTICS. Specifically, by culturing the cells over the course of a week, it allowed us to evaluate the viable cell fraction of CD133+ and CD133- cells, giving us further insight into whether CX-4945 would be effective long term or whether a population resistant to the effects of CX-4945 would begin to form. Furthermore, we highlight a reduction in the expression of stemness factors (Sox2, Nestin) in BTICs, as well as a reduction in the overall frequency of BTICs following treatment with CX-4945. This distinction is important as CK2 inhibition not only reduces the expression of stemness factors within individual cells, but also reduces the overall frequency of BTICs. The reduction of the stemness factor Sox2 is particularly meaningful since Sox2 is a reprogramming factor that is important for pluripotency [26]. Additionally, Sox2 expression has been shown to be crucial for proliferation and tumorigenicity of BTICs [28]. The reduction of Sox2 expression following culture with CX-4945 likely contributes to the reduced viability and suppression of neurosphere formation. However, GFAP expression was not induced following culture with CX-4945, suggesting that other stimuli are required to fully transform a cell from a stem-like phenotype into an astrocytic phenotype. The reduction in CD133, Sox2 and Nestin by CK2 inhibition indicates a role for CK2 in BTIC persistence and survival.
Moreover, we demonstrate a functional reduction in neurosphere formation when expression of the catalytic subunits of CK2 are either knocked-down with siRNA or inhibited using CX-4945. The in vitro limiting dilution assay is useful as it does not exclusively rely on particular markers for stem-like cells. We demonstrate the importance of CK2 signaling in BTIC function using three different xenolines, including two of the classical molecular subtype (X12, X6) and one of the proneural subtype (X456). Also, we observe this result in xenolines with a gene dosage gain of CSNK2A1 (X456, X12) as well as no gain (X6). Due to the success of CK2 inhibition in all xenolines tested, our results indicate that inhibition of CK2 would likely be therapeutic in all molecular subtypes.
Lastly, we found that treatment of two different xenolines (X6 and X456) with CX-4945 led to a significant decrease in EGFR mRNA expression, which has also been observed in lung cancer cells [29]. However, only X456 xenoline displayed a decrease in EGFR mRNA expression, while X6, which has the amplified EGFRvIII mutant, did not show a reduction in EGFR mRNA levels. Others have reported that combination CK2 and EGFR inhibition is the most effective method to inhibit tumor growth [33]. Therefore, we tested combination CX-4945 and Gefitinib in the X6 xenoline and found that combination treatment provided optimal inhibition of viability/proliferation when compared to single agent therapy. Therefore, targeting both CK2 and EGFR should be considered to provide optimal therapeutic response.
CK2 signaling has recently been reported to be important in stem-like cells of mesothelioma [34], lung [35], ovarian [36] and cervical [37] cancers, as well as leukemia [38]. This highlights the pervasiveness of CK2 signaling in the biology of stem-like cells. Our findings indicating the dependence of BTICs on CK2 signaling have important implications for GBM therapy, as BTICs represent a highly tumorigenic subset of GBM cells that are radio- and chemoresistant, proangiogenic and highly invasive, and serve as an important target for new therapeutic approaches.
Supplementary Material
Acknowledgments
We thank Dr. G. Yancey Gillespie and Catherine Langford of the UAB Brain Tumor Animal Models Core Facility (NIH P20CA151129) for assistance, and acknowledge the use of the UAB Rheumatic Diseases Core Center-Comprehensive Flow Cytometry Core (P30 AR048311 and P30 A127667). This work was supported by NIH grants R01CA194414 and R01CA1585340 (E.N.B.), T32NS048039 (A.L.R.), T32AI007051 (S.A.G.), R01CA138517 (S.E.N.) and R01CA1515122 (A.B.H.). Additional funding is from American Brain Tumor Association Discovery Grant (B.C.M.), William E. Cash Jr. Memorial Fund in Neuro-Oncology Research (B.C.M. and S.E.N.), Career Transition Award from the National Multiple Sclerosis Society TA3050-A-1 (G.P.M.), UAB Comprehensive Cancer Center (S.E.N.) and the UAB Brain Tumor SPORE Career Development Award Program via P20CA151129 (A.B.H.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11060-017-2378-z) contains supplementary material, which is available to authorized users.
References
- 1.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O′Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Cancer Genome Atlas Research N Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, Zoppoli P, Ling S, Rao AA, Grifford M, Cherniack AD, Zhang H, Poisson L, Carlotti CG, Jr, Tirapelli DP, Rao A, Mikkelsen T, Lau CC, Yung WK, Rabadan R, Huse J, Brat DJ, Lehman NL, Barnholtz-Sloan JS, Zheng S, Hess K, Rao G, Meyerson M, Beroukhim R, Cooper L, Akbani R, Wrensch M, Haussler D, Aldape KD, Laird PW, Gutmann DH, Noushmehr H, Iavarone A, Verhaak RG. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altaner C. Glioblastoma and stem cells. Neoplasma. 2008;55(5):369–374. [PubMed] [Google Scholar]
- 6.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, Yu JS. Analysis of gene expression and chemoresistance of CD133 + cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67–78. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 8.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.Zeppernick F, Ahmadi R, Campos B, Dictus C, Helmke BM, Becker N, Lichter P, Unterberg A, Radlwimmer B, Herold-Mende CC. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14(1):123–129. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 11.Brescia P, Ortensi B, Fornasari L, Levi D, Broggi G, Pelicci G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells. 2013;31(5):857–869. doi: 10.1002/stem.1317. [DOI] [PubMed] [Google Scholar]
- 12.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369(1):1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23(3):908–915. doi: 10.1128/MCB.23.3.908-915.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O′Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28(1):131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271. doi: 10.1038/cddis.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H, Bredel M, Benveniste EN. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res. 2013;19(23):6484–6494. doi: 10.1158/1078-0432.CCR-13-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitta RT, Gholamin S, Feroze AH, Agarwal M, Cheshier SH, Mitra SS, Li G. Casein kinase 2alpha regulates glioblastoma brain tumor-initiating cell growth through the beta-catenin pathway. Oncogene. 2015;34(28):3688–3699. doi: 10.1038/onc.2014.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois N, Willems M, Nguyen-Khac MT, Kroonen J, Goffart N, Deprez M, Bours V, Robe PA. Constitutive activation of casein kinase 2 in glioblastomas: absence of class restriction and broad therapeutic potential. Int J Oncol. 2016;48(6):2445–2452. doi: 10.3892/ijo.2016.3490. [DOI] [PubMed] [Google Scholar]
- 19.Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O′Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70(24):10288–10298. doi: 10.1158/0008-5472.CAN-10-1893. [DOI] [PubMed] [Google Scholar]
- 20.McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y, Nozell SE, Huszar D, Benveniste EN. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Mol Cancer Ther. 2011;10(12):2384–2393. doi: 10.1158/1535-7163.MCT-11-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rietze RL, Reynolds BA. Neural stem cell isolation and characterization. Methods Enzymol. 2006;419:3–23. doi: 10.1016/S0076-6879(06)19001-1. [DOI] [PubMed] [Google Scholar]
- 22.Maecker HT, Frey T, Nomura LE, Trotter J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A. 2004;62(2):169–173. doi: 10.1002/cyto.a.20092. [DOI] [PubMed] [Google Scholar]
- 23.Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, Benveniste EN. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. 2014;34(20):3911–3925. doi: 10.1128/MCB.00980-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16(10):1373–1382. [Google Scholar]
- 25.Guerra B, Siemer S, Boldyreff B, Issinger OG. Protein kinase CK2: evidence for a protein kinase CK2beta subunit fraction, devoid of the catalytic CK2alpha subunit, in mouse brain and testicles. FEBS Lett. 1999;462(3):353–357. doi: 10.1016/s0014-5793(99)01553-7. [DOI] [PubMed] [Google Scholar]
- 26.Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150(6):1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25(9–10):1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 28.Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, Ravetti GL, Zona GL, Daga A, Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27(1):40–48. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 29.So KS, Kim CH, Rho JK, Kim SY, Choi YJ, Song JS, Kim WS, Choi CM, Chun YJ, Lee JC. Autophagosome-mediated EGFR down-regulation induced by the CK2 inhibitor enhances the efficacy of EGFR-TKI on EGFR-mutant lung cancer cells with resistance by T790M. PLoS ONE. 2014;9(12):e114000. doi: 10.1371/journal.pone.0114000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandita A, Aldape KD, Zadeh G, Guha A, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39(1):29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- 31.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 32.Petras M, Lajtos T, Friedlander E, Klekner A, Pintye E, Feuerstein BG, Szollosi J, Vereb G. Molecular interactions of ErbB1 (EGFR) and integrin-beta1 in astrocytoma frozen sections predict clinical outcome and correlate with Akt-mediated in vitro radioresistance. Neuro Oncol. 2013;15(8):1027–1040. doi: 10.1093/neuonc/not046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bliesath J, Huser N, Omori M, Bunag D, Proffitt C, Streiner N, Ho C, Siddiqui-Jain A, O′Brien SE, Lim JK, Ryckman DM, Anderes K, Rice WG, Drygin D. Combined inhibition of EGFR and CK2 augments the attenuation of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer Lett. 2012;322(1):113–118. doi: 10.1016/j.canlet.2012.02.032. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Yang YL, Wang Y, You B, Dai Y, Chan G, Hsieh D, Kim IJ, Fang L, Au A, Stoppler HJ, Xu Z, Jablons DM, You L. CK2a, over-expressed in human malignant pleural mesothelioma, regulates the Hedgehog signaling pathway in mesothelioma cells. J Exp Clin Cancer Res. 2014;33(1):93. doi: 10.1186/s13046-014-0093-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S, Wang Y, Mao JH, Hsieh D, Kim IJ, Hu LM, Xu Z, Long H, Jablons DM, You L. Inhibition of CK2alpha down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS ONE. 2012;7(6):e38996. doi: 10.1371/journal.pone.0038996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang AQ, Cao XC, Tian L, He L, Liu F. Apigenin inhibits the self-renewal capacity of human ovarian cancer SKOV3-derived sphere-forming cells. Mol Med Rep. 2015;11(3):2221–2226. doi: 10.3892/mmr.2014.2974. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Cao XC, Xiao Q, Quan MF. Apigenin inhibits HeLa sphere-forming cells through inactivation of casein kinase 2alpha. Mol Med Rep. 2015;11(1):665–669. doi: 10.3892/mmr.2014.2720. [DOI] [PubMed] [Google Scholar]
- 38.Cheong JW, Min YH, Eom JI, Kim SJ, Jeung HK, Kim JS. Inhibition of CK2{alpha} and PI3K/Akt synergistically induces apoptosis of CD34 + CD38- leukaemia cells while sparing haematopoietic stem cells. Anticancer Res. 2010;30(11):4625–4634. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.