

Building blocks for pharmaceutical/electronic materials have been prepared using a templated C–H bond–breaking approach.
Keywords: metalation, arenes, iodoarenes, X-ray diffraction, NMR Spectroscopy, biphenyl
Abstract
The development of new methodologies to affect non–ortho-functionalization of arenes has emerged as a globally important arena for research, which is key to both fundamental studies and applied technologies. A range of simple arene feedstocks (namely, biphenyl, meta-terphenyl, para-terphenyl, 1,3,5-triphenylbenzene, and biphenylene) is transformed to hitherto unobtainable multi-iodoarenes via an s-block metal sodium magnesiate templated deprotonative approach. These iodoarenes have the potential to be used in a whole host of high-impact transformations, as precursors to key materials in the pharmaceutical, molecular electronic, and nanomaterials industries. To prove the concept, we transformed biphenyl to 3,5-bis(N-carbazolyl)-1,1′-biphenyl, a novel isomer of 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CPB), a compound which is currently widely used as a host material for organic light-emitting diodes.
INTRODUCTION
Until 1938, organometallic reagents were considered as exotic rarities, commanding little application in synthetic chemistry (1). However, the discoveries by Gilman and Bebb (2) and Wittig et al. (3) that alkyllithium reagents could be used to selectively metalate anisole (and other heterosubstituted arenes) at an ortho position opened the doors to the first major utilization of organometallics in synthesis. This discovery was later coined directed ortho-metalation (DoM) (4), and from this juncture, organometallic reagents were seen as “indispensable tools in chemical synthesis” (1). DoM can be used to install new functionalities on arenes, thus allowing the construction of new molecules (5–7), and relies on converting a relatively inert C–H bond in an organic molecule into a more reactive and versatile C–metal bond by treating it with a metalating reagent (normally an organolithium reagent). The beauty of the DoM reaction is it efficiently and selectively metalates the C–H bond adjacent to a directing group (DG), which promotes reaction due to its dual coordination/inductive roles. Since the pioneering works of Gilman and Bebb (2) and Wittig et al. (3), the DoM concept has been advanced using monometallic (8–12) and bimetallic (13–19) systems.
In his influential text, Schlosser stated, “deviations from the ortho-rule do exist but are scarce” (1). Enabling activation of arene C–H bonds at positions other than ortho has become a high interest and impact area of research, with the goal of allowing easy access to key pharmaceuticals, agrochemicals, and novel materials. In the main, new methodologies involve transition metal C–H activation chemistry (20–28). In 2014, we exploited s-block organometallic chemistry and reported directed ortho-meta′– and meta-meta′–dimetalations using a sodium magnesiate reagent [Na4Mg2(TMP)6(nBu)2] 1 (TMP is 2,2,6,6-tetramethylpiperidide) as an alkyl base (29). It functions in a templated manner, allowing double metalations to occur, and has been shown to exhibit regioselective 2,5-double alkyl basicity toward a range of substituted arenes, including amides and carbamates and 3,5-double basicity toward anilines, culminating in the isolation of a group of heterometallic inverse crown molecules (30). Here, we use the template metalation strategy to functionalize unactivated multiaryl compounds (non-DoM substrates) that lack acidity due to the absence of functional groups, regioselectively at meta sites. The biphenyl motif (Fig. 1A) is an important building block in the pharmaceutical, agrochemical, and polymer/materials sciences. It is present in telmisartan, valsartan, and oritavancin and is a key component of molecules for organic light-emitting devices, such as N,N′-di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB) and 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CPB). Biphenyl-based hydrocarbons, such as terphenyls and 1,3,5-triphenylbenzene (tpb) (Fig. 1B), are also cores for molecules used in the devices and for the synthesis of nanomaterials, such as graphene/graphene nanoribbons (31). Novel methods to improve the preparation of functionalized multiarenes for these applications are continuously being sought. Reflecting the importance of developing new methods to access novel polyaryl systems, Suzuki et al. (32) have recently reported the elegant synthesis of a new class of “multitalented” hexaarylbenzenes (33) that contain five or six different arene substituents on a single benzene molecule.
Fig. 1. Prevalence of the biphenyl skeleton in pharmaceutical compounds and organic light-emitting diode materials.
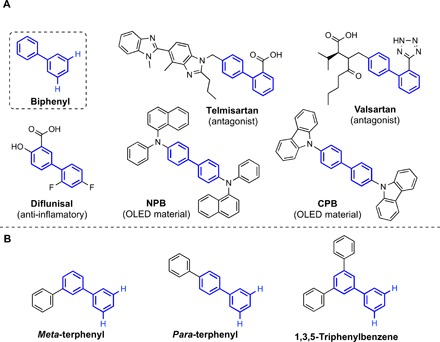
(A) Biphenyl and the structural prevalence of its framework in pharmaceutical agents and host materials for organic light-emitting diodes (OLED). (B) Biphenyl-based polyaryl hydrocarbons.
RESULTS AND DISCUSSION
We targeted the templated metalation of simple polyaryl compounds (namely, biphenyl, para-terphenyl, meta-terphenyl, tpb, and antiaromatic biphenylene) using 1 and subsequently transformed the organometallic intermediates into new versatile organic precursors (34). Because of their lack of DGs (hence, the absence of docking site and strong acidifying group), the substrates studied are difficult to metalate using conventional s-block organometallics, unless a Lewis basic functionality activates them. Inklings that this work may be successful stemmed from the fact that we had previously shown that 1 can be used to doubly metalate naphthalene at the 1 and 4 positions regioselectively, which can be subsequently converted to 1,4-diiodonaphthalene (35).
Substituted biphenyls are often prepared by coupling reactions (36). In terms of metalation chemistry, both rings of biphenyl can be singly deprotonated using two equivalents of n-butyllithium/TMEDA (N,N,N′,N′-tetramethylethylenediamine) (37). Synthetically useful yields of monolithiobiphenyls can be obtained using lithium-halogen exchange reactions (38, 39). Introducing a wholly new situation, by treating biphenyl with a methylcyclohexane solution of 1 in a 1:1 molar ratio, it is possible to regioselectively dimagnesiate biphenyl at the 3 and 5 positions of a single phenyl ring, as evidenced by nuclear magnetic resonance (NMR) spectroscopic analysis of the reaction mixture. [Na4Mg2(TMP)6(3,5-biphenyl-di-ide)] 2 was isolated in 70% yield and characterized by x-ray crystallography (Fig. 2A). When two or more equivalents of 1 were used, 2 was again observed, presumably because of the high steric protection afforded to the second phenyl ring, disfavoring further reactivity. When biphenyl is treated with one or two equivalents of the monometallic reagents NaTMP or TMPMgnBu, intact unreacted biphenyl was obtained (Supplementary Materials). The key bond parameters of 2 (Supplementary Materials) resemble those of other comparable inverse crown molecules (29, 40). When 2 is reacted with an excess of iodine/tetrahydrofuran (THF) solution, 3,5-diiodobiphenyl 3 can be prepared in 64% isolated yield (Fig. 3, i).
Fig. 2. Molecular structures of isolated intermediates.
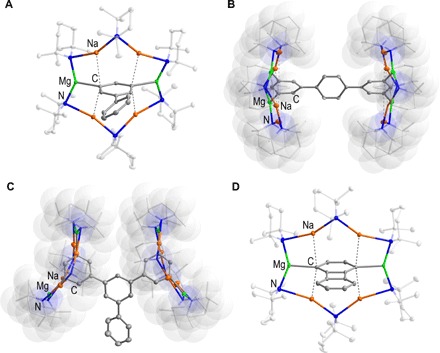
(A) 2. (B) 6. (C) 12. (D) 14. Hydrogen atoms, methylcyclohexane solvent molecules of crystallization, and disordered components of TMP and arene moieties are omitted for clarity. Displacement ellipsoids are displayed at 35% probability with the TMP frameworks pictures as capped sticks with translucent space-filling van der Waals surfaces for a probe of 1.5 Å radius for 6 and 12.
Fig. 3. Synthetic studies for the preparation of multi-iodo polyaryl and antiaromatic compounds.
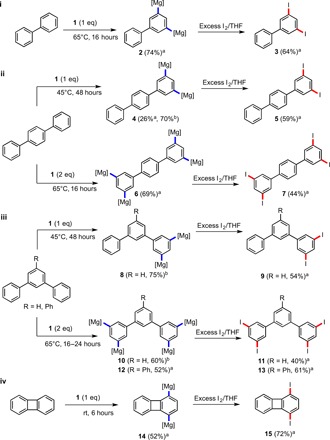
aIsolated yield. bExtent of dimetalation determined by NMR spectroscopic studies based on in situ reaction of 1 with the corresponding polyaryl substrate. [Mg] equates to [Na2MgTMP3]. rt, room temperature.
The next arene molecules studied were meta- and para-terphenyl. Terphenylide anions—prepared via lithium-halogen exchange reactions of 1-halo-2,5-diphenylbenzenes and n-butyllithium—are useful ligands in coordination chemistry because they provide a pocket, which protects the metal from forming oligomers despite occupying only one coordination site. Power exploited this special chemistry using a bulky meta-terphenyl, 2,6-bis-[(2,6-diisopropyl)phenyl]phenyl to isolate the first quintuple bond between two metals, namely, two Cr(I) cations (41). It appears that the parent terphenyls have not thus far been deprotonated using any base. Focusing on para-terphenyl, when treated with an equivalent of 1 in methylcyclohexane solution, NMR spectroscopic analysis showed dimetalation of the arene at 3,5 positions of a single phenyl ring, likely producing [Na4Mg2(TMP)6(3,5-para-terphenyl-di-ide)] 4 (Supplementary Materials). Because of its high solubility, 4 was isolated in low to moderate yield (26%). An in situ mixture was reacted with iodine/THF solution and successfully converted to 3,5-diiodo-para-terphenyl 5 in 59% yield (Fig. 3, ii). As well as characterization by NMR spectroscopy, good quality crystals of 5 were successfully grown from an ethanol solution and were characterized by x-ray crystallography (Supplementary Materials). Increasing the base/substrate ratio (to 2:1) allowed further metalation of the para-terphenyl ligand to occur presumably because of the greater distance between the two “outer” phenyl rings (when compared with biphenyl), as evidenced by NMR spectroscopy. Crystals were obtained, which after x-ray analysis showed fourfold metalation of para-terphenyl culminating in the isolation of {[Na4Mg2(TMP)6]2(3,3″,5,5″-para-terphenyl-tetra-ide)} 6 in 69% yield (Fig. 2B). The outer rings of the para-terphenyl have each been doubly metalated, and each affected “arene dianion” is hosted by separate inverse crown rings. The key metrics of the inverse crown rings in 6 are essentially identical to those in the biphenyl derivative 2. Complex 6 represents the first example of a molecule, which contains two inverse crowns within its molecular structure. Previously, it has been shown that metallocenes can also be tetradeprotonated; however, these tetranions are accommodated within a single 16-membered inverse crown ring (42, 43). When isolated 6 is reacted with iodine/THF, NMR spectroscopy reveals that it is transformed to 3,3″,5,5″-tetraiodo-para-terphenyl 7 in 51% yield (Fig. 3, ii). When an in situ mixture of 6 is reacted with excess iodine/THF to prepare 7, a slightly lower yield (44%) was obtained.
The chemistry observed with meta-terphenyl (to produce 8 to 11) essentially mirrors that of 4 to 7, that is, di- and tetrametalated compounds {[Na4Mg2(TMP)6]2(3,5,-meta-terphenyl-di-ide)} 8 and {[Na4Mg2(TMP)6]2(3,3″,5,5″-meta-terphenyl-tetra-ide)} 10, respectively, were obtained and characterized by NMR spectroscopy, as well as their respective di- and tetraiodo derivatives 3,5-diiodo-meta-terphenyl 9 (54%) and 3,3″,5,5″-tetraiodo-meta-terphenyl 11 (40%).
Turning to tpb, it has emerged in recent years as an important ligand, formally allowing stabilization of Ca(I) cations (44). C–H metalation of tpb is rare, and complexes derived from it generally emerge from the use of a pre–halo-substituted (normally at the central C6 ring) reagent. One example of where a genuine C–H metalation is observed is when 1-bromo-2,4,6-triphenylbenzene is reacted with 2 molar equivalents of n-butyllithium (45). The first equivalent of alkyllithium induces a metal-bromine exchange, and the second metalates an interannular peri-C–H bond, formally giving rise to a dianion of triphenylbenzene (46). Structurally, tpb seemed an intriguing reagent to metalate. If the reaction was in keeping with the aforementioned results, it could be envisaged that di-, tetra-, or perhaps hexadeprotonation would be possible. In reality, when the arene is reacted with 1 to 3 molar equivalents of 1, the sole isolated product is the tetradeprotonated variant {[Na4Mg2(TMP)6]2{3,3″,5,5″-(1′,3′,5′-triphenylbenzene)-tetra-ide]} 12 (Fig. 2C). A twofold deprotonation of tpb was not detected, perhaps because of the more acidic nature of tpb (in comparison with meta-terphenyl) due to the presence of the third phenyl group withdrawing electron density from the other two phenyl rings. When 6 is compared with 12, there is a marked difference in the positions and orientations of the inverse crown rings (Fig. 2B versus Fig. 2C). In 6, they adopt a mutual convex orientation residing in a position toward the periphery of the metalated phenyl rings. Likely because of the extra steric demand of the additional phenyl group in tpb, the inverse crowns adopt a mutual concave orientation in 12, residing in a position more toward the center of the metalated rings. These pronounced structural differences highlight the conformational flexibility of the template, allowing spatially distinct arenes to be accommodated. By reacting 12 with an excess of iodine/THF, it can be smoothly transformed to its tetraiodo relative 3,3″,5,5″-tetraiodo-1′,3′,5′-triphenylbenzene 13 in 61% yield.
Finally, we turned our attention to the prototypical antiaromatic compound biphenylene. Again, reports detailing the direct metalation of biphenylene are very scarce. When reacted with n- or t-butyllithium and quenched with electrophiles, the major species observed are the 1-substituted biphenylenes (47). When biphenylene is reacted with a methylcyclohexane solution of 1, it was doubly metalated; however, metalation occurred simultaneously at the 1 and 4 positions to yield {[Na4Mg2(TMP)6]2(1,4-biphenylene-di-ide)} 14 (Fig. 2D). This metalation pattern is identical to that obtained with naphthalene, presumably because of the similar fused-ring nature of the substrates (thus, each arene ring is initially disubstituted) (35). Complex 14 can be converted to 1,4-diiodobiphenylene 15 in high yield (72%) by reaction with iodine/THF.
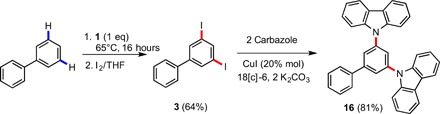
Organometallic species 2, 4, 6, 8, 10, 12, and 14 are soluble in [D12]cyclohexane, allowing multinuclear NMR spectroscopy to be performed. A key finding is that the solid-state structures appear to be maintained in [D12]cyclohexane solution (Supplementary Materials). Iodinated organic molecules 3, 5, 7, 9, 11, 13, and 15 are structurally simple; however, surprisingly, this study appears to represent their first synthetic routes, isolation, and full characterization. Access to these molecules has perhaps been hindered because most of polyaryl species are prepared via coupling reactions [for example, Suzuki-Miyaura cross-coupling (34)], so the C–I bonds in the potential synthons are themselves susceptible to coupling chemistry, thus it is difficult to prevent undesired overcoupling from occurring. The methodology presented avoids transition metal coupling chemistry, thus the iodine atoms remain intact on the arene rings, allowing further exploitation. Given the importance of iodoarenes as coupling partners in C–C and C–heteroatom bond formation chemistry (48, 49) it is likely that these organic molecules will act as vital building blocks for the synthesis of a whole host of new functional materials. As proof of concept and illustrating the synthetic applicability of our approach, we have prepared a novel isomer of the aforementioned material CPB by converting biphenyl to 3,5-bis(N-carbazolyl)-1,1′-biphenyl 16 in high yield (81%; Fig. 4) via copper-catalyzed Ullmann-type coupling of carbazole with 3.
Fig. 4. Utilization of 1,3-diiodobiphenyl for Ullmann coupling reaction with carbazole for the preparation of 16.
MATERIALS AND METHODS
General methods and chemicals
All reactions and manipulations were performed under a protective atmosphere of dry pure argon gas using standard Schlenk techniques unless otherwise stated. n-Hexane was dried by heating it to reflux over sodium benzophenone ketyl and used freshly distilled under nitrogen atmosphere. Methylcyclohexane was distilled over sodium metal under nitrogen and then stored with activated 4 Å molecular sieves under argon before use. Anhydrous N,N′-dimethylformamide (DMF) was purchased from Sigma-Aldrich and used without further purification. [D12]cyclohexane and [D6]benzene were degassed and stored over activated 4 Å molecular sieves under argon before use. 2,2,6,6-Tetramethylpiperidine [TMP(H)] was purchased from Merck KGaA and stored over activated molecular sieves (4 Å). nBuLi (1.6 M in hexanes) and nBu2Mg (1 M in heptane) solutions were purchased from Sigma-Aldrich and titrated before use. Copper iodide, carbazole, potassium carbonate, 18-crown-6, sodium thiosulfate, iodine, anhydrous magnesium sulfate, ammonium chloride, and sodium chloride were purchased from Sigma-Aldrich and used as received. nBuNa (50), NaTMP (51), and TMPMg nBu (29, 52) were prepared according to literature methods and used as solids from a glove box or in situ. Sodium tert-butoxide was purchased from Sigma-Aldrich and dried under vacuum at 150°C in a Schlenk tube for 30 min before use. Biphenyl, meta-terphenyl (1,3-diphenylbenzene), para-terphenyl (1,4-diphenylbenzene), and tpb were purchased from Sigma-Aldrich and used without further purification. Biphenylene was prepared according to the reported method and used as a crystalline solid (37). nBuNa, NaTMP, TMPMg nBu, and the organometallic products 2, 4, 6, 12, and 14 were isolated and handled inside an argon-filled glove box, whereas the derivatives 8 and 10 were in situ prepared and used as methylcyclohexane solutions. Organic products 3, 5, 7, 9, 11, 13, 15, and 16 were isolated without the need of protocols to exclude atmospheric oxygen or moisture. NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer, operating at 400.1 and 100.6 MHz for 1H and 13C NMR, respectively. The NMR assignments were performed using 13C{1H}-DEPT135, 1H,1H-COSY, 1H,13C-HSQC, and 1H,13C-HMBC experiments. 1H and 13C{1H} chemical shifts were expressed in parts per million (δ = ppm) and referenced to residual solvent peaks. All coupling constants (J) are absolute values (hertz), and the description of signals are br for broad, s for singlet, d for doublet, dd for doublet of doublets, t for triplet, tt for triplet of triplets, q for quartet, and m for multiplet. Elemental analyses (triplicate) were carried out using a PerkinElmer 2400 elemental analyzer. Low-resolution mass spectra for products 3, 5, 7, 9, 11, 13, 15, and 16 were recorded on either an Agilent 7890A instrument coupled to an Agilent 5975C mass spectrometer (gas chromatography–mass spectrometry, either chemical or electron ionization mode) with helium as a carrier gas at 1 ml/min and using an Restek Rxi-5Sil-MS column (30 m × 250 μm × 0.25 μm) or an Kratos-Shimadzu Axima CFR MALDI-TOF spectrometer (no matrix) in positive ion mode. Accurate mass spectra were obtained at the University of Swansea in the Engineering and Physical Science Research Council (EPSRC) National Mass Spectrometry Facility, and spectra were recorded using either a Waters GCT Premier benchtop orthogonal acceleration time-of-flight mass spectrometer (electron impact ionization mode) or a Waters Xevo G2-S using the atmospheric solids analysis probe in positive mode. Values are reported as mass/charge ratio (m/z).
Preparation of metalating reagent 1
Solutions of 1 (0.05 and 0.066 M in methylcyclohexane) were prepared in situ according to literature methods (29, 35). Detailed procedures are given for the preparation of 2 to 17 in the Supplementary Materials.
General synthesis of organometallic reagents 2, 4, 6, 8, 10, 12, and 14
In an argon-filled Schlenk tube, freshly prepared nBuNa (640 mg, 8 mmol) was suspended in methylcyclohexane (24 ml), and TMP(H) (2.04 ml, 12 mmol) was then added via syringe to give NaTMP as a pale yellow suspension, which was stirred for 30 min at ambient temperature. Commercial nBu2Mg (4 ml, 1 M solution in n-heptane, 4 mmol) was then added via syringe to give a 0.066 M pale yellow solution of 1 in methylcyclohexane. The metalating reagent 1 was stirred for 30 min at ambient temperature before use. Then, the appropriate polyarene was added, and the resulting reaction mixture was heated at the appropriate temperature. Where possible, crystals suitable for x-ray diffraction were grown (see full details in the Supplementary Materials). The NMR spectra of isolated amorphous solids/crystals in [D12]cyclohexane solution were obtained.
General synthesis of organic compounds 3, 5, 7, 9, 11, 13, and 15
The corresponding organic substrate (biphenyl, para-terphenyl, meta-terphenyl, tpb, and biphenylene; 1 mmol each) was treated with an in situ solution of 1 in methylcyclohexane prepared according to the synthetic organometallic protocols described in the Supplementary Materials for 2, 4, 6, 8, 10, 12, and 14. The 0.05 M solutions of 1 in methylcyclohexane (20 ml) were used for the in situ preparation of 2, 4, 8, and 14, whereas the 0.066 M solutions of 1 in methylcyclohexane (30 ml) were used for the in situ preparation of 6, 10, and 12. All reactions were stirred at different temperatures and for different times and in situ monitored by NMR spectroscopy in either [D12]cyclohexane or [D6]benzene until magnesiation of the corresponding aromatic substrate was achieved to give the corresponding organometallic derivatives (table S1). After the in situ metalation step was achieved, the reaction mixture was cooled to −78°C in a dry ice/acetone bath for 30 min and then added (either via cannula or disposable air-tight syringe) to an iodine solution in THF at the same temperature. For the preparation of 3, 5, 9, and 15, 10 ml of a 1 M solution of iodine in THF was used, whereas for 7, 11, and 13, 20 ml of a 1 M solution of iodine in THF was used. The resulting reaction mixtures were stirred at −78°C for at least 3 hours and then allowed to slowly warm up to ambient temperature for a period of 24 hours. Saturated aqueous NH4Cl solution (~10 ml) was then added, followed by saturated aqueous Na2S2O3 solution (~10 ml) until bleaching occurred. The crude mixture was extracted with EtOAc (3 × 20 ml), and the combined organic layers were washed with brine (20 ml) and dried over anhydrous MgSO4. The solvent was removed in a rotary evaporator, and the crude product was purified by flash column chromatography (silica gel and n-hexane/ethyl acetate) to yield the pure title compound. Alternatively, 7 can be prepared in 51% yield by reacting 6 (1 mmol) with an iodine/THF solution (20 mmol iodine/20 ml THF). The organic derivatives 3, 5, 7, 13, and 15 can be crystallized from either MeOH, EtOH, or EtOAc. Compounds 7, 11, and 13 exhibit low solubility in commonly used organic solvents, which hinders their isolation and purification in the scale used in their preparations.
Synthesis of 16
In an argon-filled Schlenk tube, 3,5-diiodo-1,1′-biphenyl (150 mg, 0.37 mmol) was mixed with carbazole (140 mg, 0.83 mmol), potassium carbonate (114 mg, 0.83 mmol), copper(I) iodide (32 mg, 0.17 mmol), and 18-crown-6 (30 mg, 0.1 mmol). Then, anhydrous DMF was added (5 ml), and the reaction mixture was refluxed for 24 hours under argon atmosphere. After cooling down to ambient temperature, the reaction mixture was filtered through a short pad of Celite, and the solvent was removed under vacuum. The crude product was purified by flash column chromatography (silica gel and n-hexane/ethyl acetate) to form 16 as a pale yellow solid (0.145 g, 81%). Compound 16 can be crystallized from ethyl acetate by slow evaporation to give colorless block-like crystals suitable for an x-ray diffraction study (see the Supplementary Materials for NMR spectroscopic and mass spectral data).
Supplementary Material
Acknowledgments
Funding: We would like to thank R. Mulvey, E. Hevia, and S. Robertson for useful and insightful discussions and the financial support of the EPSRC (Career Acceleration Fellowship; EP/J001872/1 and EP/L001497/1). We are also grateful to the EPSRC UK National Mass Spectrometry Facility, Swansea, for collecting our high-resolution mass spectral data. Author contributions: A.J.M.-M. carried out most of the experimental work, drafted most of the Supplementary Materials, and contributed ideas to the project. A.R.K. and I.D.H.O. ensured that the x-ray crystallography work was carried out to high level and contributed to the drafting of the manuscript. B.J.F. and S.J. carried out the initial experimental work. C.T.O. conceived and developed the project and drafted the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: X-ray crystallographic data for compounds 2, 3, 5, 6, 12, 13, 14, 15, and 16 are freely available at the Cambridge Crystallographic Data Centre (CCDC 1516422-30) via http://www.ccdc.cam.ac.uk/data_request/cif. The research data associated with this paper are openly available at http://dx.doi.org/10.15129/b1d7b580-838c-4621-ad43-fccd2579024d. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700832/DC1
Detailed experimental procedures
Materials and methods
Synthetic procedures
X-ray crystallography
Supplementary text
NMR spectra
fig. S1. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra showing the nonaromatic resonances for biphenylene (top; black), reaction mixture containing two conformers of 14 (middle; blue), and isolated [Na4Mg2(TMP)6(1,4-biphenylene-di-ide)] 14 (bottom; major conformer).
fig. S2. 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum showing the nonaromatic resonances for the two conformers of 14.
fig. S3. Sections of experimental and simulated 1H NMR spectra of 14.
fig. S4. Sections of experimental and simulated 1H NMR spectra of 15.
fig. S5. Molecular structure of 2 showing the contents of the asymmetric unit cell.
fig. S6. Molecular structure of 3 and its extended packing.
fig. S7. Molecular structure of 5 and its extended packing.
fig. S8. Molecular structure of 6 showing atomic connectivity.
fig. S9. Molecular structure of 12 showing the contents of the asymmetric unit cell.
fig. S10. Molecular structure of 13 and its extended packing.
fig. S11. Molecular structure of 14 showing the contents of the asymmetric unit cell.
fig. S12. Molecular structure of 15 and its extended packing.
fig. S13. Molecular structure of 16 showing the contents of the asymmetric unit cell.
fig. S14. 1H NMR study (400.1 MHz; [D12]cyclohexane, 300 K) of biphenyl (top; red) and a control reaction of biphenyl and NaTMP in a 1:2 M ratio in methylcyclohexane after 24 hours at 65°C (bottom; blue).
fig. S15. 1H NMR study (400.1 MHz; [D12]cyclohexane, 300 K) of biphenyl (top; red) and a control reaction of biphenyl and nBuMgTMP in a 1:2 M ratio in methylcyclohexane after 16 hours at 65°C (bottom; blue).
fig. S16. Representative example of 3,5-dimetalation of biphenyl to give 2.
fig. S17. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S18. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S19. Sections of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S20. Sections of the phase-sensitive 1H,13C-HSQC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S21. Sections of the 1H,13C-HMBC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S22. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of biphenyl (top; red) and 2 (bottom; blue) showing the aromatic resonances.
fig. S23. Sections of the 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectra of biphenyl (top; red) and 2 (bottom; blue) showing the aromatic resonances.
fig. S24. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S25. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 3.
fig. S26. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S27. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S28. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S29. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of an in situ sample of 4.
fig. S30. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S31. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 5.
fig. S32. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S33. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S34. Sections of the 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S35. 1H NMR (400.1 MHz; [D12]cyclohexane/C7H14, 300 K) spectrum of [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6.
fig. S36. Section of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane/C7H14, 300 K) spectrum of [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6 showing the cross peaks for the aromatic resonances.
fig. S37. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of para-terphenyl (top; green), 4 (middle; red), and [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6 (bottom; blue).
fig. S38. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 7.
fig. S39. 13C NMR (100.6 MHz; CDCl3, 300 K) spectrum of 7.
fig. S40. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8.
fig. S41. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8.
fig. S42. 1H NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S43. 13C{1H} NMR (100.6 MHz; 300 K, CDCl3) spectrum of 9.
fig. S44. 1H,1H-COSY NMR (400.1 MHz; 300 K, CDCl3) spectrum of 3,5-diiodo-meta-terphenyl 9.
fig. S45. 1H,13C-HSQC NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S46. 1H,13C-HMBC NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S47. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of [Na8Mg4TMP12(3,3′,5,3′-meta-terphenyl-tetra-ide)] 10.
fig. S48. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of meta-terphenyl (top; green), [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8 (middle; red), and [Na8Mg4TMP12(3,3′,5,3′-meta-terphenyl-tetra-ide)] 10 (bottom; blue) showing the aromatic resonances.
fig. S49. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 11.
fig. S50. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 11.
fig. S51. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S52. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S53. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S54. 1H NMR (400.1 MHz; [D6]benzene, 300 K) spectrum of {Na8Mg4[TMP]12[3,3″,5,5″-(1′,3′,5′-triphenylbenzene-tetra-ide)]} 12.
fig. S55. Section of the 1H,1H-COSY NMR (400.1 MHz; [D6]benzene, 300 K) spectrum of 12 showing the cross peaks for the aromatic resonances.
fig. S56. 1H NMR (400.1 MHz; [D6]benzene, 300 K) spectra of tpb (top; red) and 12 (bottom; blue).
fig. S57. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3,3″,5,5″-tetraiodo-5′-phenyl-benzene 13.
fig. S58. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 13.
fig. S59. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 13.
fig. S60. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 13.
fig. S61. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum sections of 13.
fig. S62. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S63. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S64. Sections of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S65. Sections of the phase-sensitive 1H,13C-HSQC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S66. Sections of the 1H,13C-HMBC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S67. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) section of the spectra of biphenylene (top; red) and isolated 14 (bottom; blue, major conformer).
fig. S68. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S69. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 15.
fig. S70. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S71. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S72. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S73. 1H NMR (400.1 MHz; CDCl3, 300 K) spectra of biphenylene (top; red) and 15 (bottom; blue).
fig. S74. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectra of biphenylene (top; red) and 15 (bottom; blue).
fig. S75. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S76. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 16.
fig. S77. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S78. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S79. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
table S1. Metalation conditions and scope.
REFERENCES AND NOTES
- 1.Organometallics in Synthesis: Third Manual, M. Schlosser, Ed. (John Wiley & Sons, 2013). [Google Scholar]
- 2.Gilman H., Bebb R. L., Relative reactivities of organometallic compounds. XX. Metalation. J. Am. Chem. Soc. 61, 109–112 (1939). [Google Scholar]
- 3.Wittig G., Pockels U., Droge H., Über die Austauschbarkeit von aromatisch gebundenem Wasserstoff gegen Lithium mittels Phenyl-lithiums. Ber. Dtsch. Chem. Ges. 71, 1903–1912 (1938). [Google Scholar]
- 4.Snieckus V., Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev. 90, 879–933 (1990). [Google Scholar]
- 5.Beak P., Snieckus V., Directed lithiation of aromatic tertiary amides: An evolving synthetic methodology for polysubstituted aromatics. Acc. Chem. Res. 15, 306–312 (1982). [Google Scholar]
- 6.T. K. Macklin, V. Snieckus, in Handbook of C-H Transformations, G. Dyker, Ed. (Wiley-VCH, 2005), vol. 1, pp. 106–118. [Google Scholar]
- 7.Whisler M. C., MacNeil S., Snieckus V., Beak P., Beyond thermodynamic acidity: A perspective on the complex-induced proximity effect (CIPE) in deprotonation reactions. Angew. Chem. Int. Ed. 43, 2206–2225 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Macklin T. K., Snieckus V., Directed ortho metalation methodology. The N,N-dialkyl aryl O-sulfamate as a new directed metalation group and cross-coupling partner for Grignard reagents. Org. Lett. 7, 2519–2522 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Metallinos C., Nerdinger S., Snieckus V., N-cumyl benzamide, sulfonamide, and aryl O-carbamate directed metalation groups. Mild hydrolytic lability for facile manipulation of directed ortho metalation derived aromatics. Org. Lett. 1, 1183–1186 (1999). [Google Scholar]
- 10.Beak P., Brown R. A., The ortho lithiation of tertiary benzamides. J. Org. Chem. 42, 1823–1824 (1977). [Google Scholar]
- 11.Kauch M., Hoppe D., Synthesis of halogenated phenols by directed ortho-lithiation and ipso-iododesilylation reactions of O-aryl N-isopropylcarbamates. Synthesis 1578–1589 (2006). [Google Scholar]
- 12.Mortier J., Moyroud J., Bennetau B., Cain P. A., The carboxylic acid group as an effective director of ortho-lithiation. J. Org. Chem. 59, 4042–4044 (1994). [Google Scholar]
- 13.Krasovskiy A., Krasovskaya V., Knochel P., Mixed Mg/Li amides of the type R2NMgCl.LiCl as highly efficient bases for the regioselective generation of functionalized aryl and heteroaryl magnesium compounds. Angew. Chem. Int. Ed. 45, 2958–2961 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Haag B., Mosrin M., Ila H., Malakhov V., Knochel P., Regio- and chemoselective metalation of arenes and heteroarenes using hindered metal amide bases. Angew. Chem. Int. Ed. 50, 9794–9824 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Marquise N., Bretel G., Lassagne F., Chevallier F., Roisnel T., Dorcet V., Halauko Y. S., Ivashkevich O. A., Matulis V. E., Gros P. C., Mongin F., Deproto-metallation using mixed lithium–zinc and lithium–copper bases and computed CH acidity of 2-substituted quinolines. RSC Adv. 4, 19602–19612 (2014). [Google Scholar]
- 16.Snégaroff K., Komagawa S., Yonehara M., Chevallier F., Gros P. C., Uchiyama M., Mongin F., Lithium cadmate-mediated deprotonative metalation of anisole: Experimental and computational study. J. Org. Chem. 75, 3117–3120 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Armstrong D. R., Crosbie E., Hevia E., Mulvey R. E., Ramsay D. L., Robertson S. D., TMP (2,2,6,6-tetramethylpiperidide)-aluminate bases: Lithium-mediated alumination or lithiation–alkylaluminium-trapping reagents? Chem. Sci. 5, 3031–3045 (2014). [Google Scholar]
- 18.Kondo Y., Shilai M., Uchiyama M., Sakamoto T., TMP−zincate as highly chemoselective base for directed ortho metalation. J. Am. Chem. Soc. 121, 3539–3540 (1999). [Google Scholar]
- 19.Uchiyama M., Matsumoto Y., Nobuto D., Furuyama T., Yamaguchi K., Morokuma K., Structure and reaction pathway of TMP-zincate: Amido base or alkyl base? J. Am. Chem. Soc. 128, 8748–8750 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Cheng C., Hartwig J. F., Rhodium-catalyzed intermolecular C–H silylation of arenes with high steric regiocontrol. Science 343, 853–857 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Dong Z., Wang J., Dong G., Simple amine-directed meta-selective C–H arylation via Pd/norbornene catalysis. J. Am. Chem. Soc. 137, 5887–5890 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Larraufie M.-H., Maestri G., Beaume A., Derat É., Ollivier C., Fensterbank L., Courillon C., Lacôte E., Catellani M., Malacria M., Exception to the ortho effect in palladium/norbornene catalysis. Angew. Chem. Int. Ed. 50, 12253–12256 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Catellani M., Frignani F., Rangoni A., A complex catalytic cycle leading to a regioselective synthesis of o,o′-disubstituted vinylarenes. Angew. Chem. Int. Ed. 36, 119–122 (1997). [Google Scholar]
- 24.Luo J., Preciado S., Larrosa I., Overriding ortho–para selectivity via a traceless directing group relay strategy: The meta-selective arylation of phenols. J. Am. Chem. Soc. 136, 4109–4112 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Leow D., Li G., Mei T.-S., Yu J.-Q., Activation of remote meta-C–H bonds assisted by an end-on template. Nature 486, 518–522 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang R.-Y., Li G., Yu J.-Q., Conformation-induced remote meta-C–H activation of amines. Nature 507, 215–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bag S., Patra T., Modak A., Deb A., Maity S., Dutta U., Dey A., Kancherla R., Maji A., Hazra A., Bera M., Maiti D., Remote para-C–H functionalization of arenes by a D-shaped biphenyl template-based assembly. J. Am. Chem. Soc. 137, 11888–11891 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Maji A., Bhaskararao B., Singha S., Sunoj R. B., Maiti D., Directing group assisted meta-hydroxylation by C–H activation. Chem. Sci. 7, 3147–3153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Martinez A. J., Kennedy A. R., Mulvey R. E., O’Hara C. T., Strong bases. Directed ortho-meta- and meta-meta-dimetalations: A template base approach to deprotonation. Science 346, 834–837 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Mulvey R. E., Modern ate chemistry: Applications of synergic mixed alkali-metal–magnesium or –zinc reagents in synthesis and structure building. Organometallics 25, 1060–1075 (2006). [Google Scholar]
- 31.Gharagozloo-Hubmann K., Müller N. S., Giersig M., Lotze C., Franke K. J., Reich S., Requirement on aromatic precursor for graphene formation. J. Phys. Chem. C 120, 9821–9825 (2016). [Google Scholar]
- 32.Suzuki S., Segawa Y., Itami K., Yamaguchi J., Synthesis and characterization of hexaarylbenzenes with five or six different substituents enabled by programmed synthesis. Nat. Chem. 7, 227–233 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Vij V., Bhalla V., Kumar M., Hexaarylbenzene: Evolution of properties and applications of multitalented scaffold. Chem. Rev. 116, 9565–9627 (2016). [DOI] [PubMed] [Google Scholar]
- 34.J. Mortier, Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds (John Wiley & Sons Inc., 2016). [Google Scholar]
- 35.Martinez-Martinez A. J., Armstrong D. R., Conway B., Fleming B. J., Klett J., Kennedy A. R., Mulvey R. E., Robertson S. D., O’Hara C. T., Pre-inverse-crowns: Synthetic, structural and reactivity studies of alkali metal magnesiates primed for inverse crown formation. Chem. Sci. 5, 771–781 (2014). [Google Scholar]
- 36.A. Jutand, G. Lefèvre, Transition metal-catalyzed carbon–carbon cross-coupling, in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, J. Mortier, Ed. (John Wiley & Sons Inc., 2016), 511–545. [Google Scholar]
- 37.Schaub T., Radius U., A convenient synthesis of biphenylene. Tetrahedron Lett. 46, 8195–8197 (2005). [Google Scholar]
- 38.Liu Z., Tan H., Wang L., Fu T., Xia Y., Zhang Y., Wang J., Transition-metal-free intramolecular carbene aromatic substitution/Büchner reaction: Synthesis of fluorenes and [6,5,7]benzo-fused rings. Angew. Chem. Int. Ed. 54, 3056–3060 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Bähr G., Gelius R., Organozinnverbindungen mit raumfüllenden Arylgruppen, I. Chem. Ber. 91, 812–818 (1958). [Google Scholar]
- 40.Blair V. L., Carrella L. M., Clegg W., Conway B., Harrington R. W., Hogg L. M., Klett J., Mulvey R. E., Rentschler E., Russo L., Tuning the basicity of synergic bimetallic reagents: Switching the regioselectivity of the direct dimetalation of toluene from 2,5- to 3,5-positions. Angew. Chem. Int. Ed. 47, 6208–6211 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen T., Sutton A. D., Brynda M., Fettinger J. C., Long G. J., Power P. P., Synthesis of a stable compound with fivefold bonding between two chromium(I) centers. Science 310, 844–847 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Clegg W., Henderson K. W., Kennedy A. R., Mulvey R. E., O’Hara C. T., Rowlings R. B., Tooke D. M., Regioselective tetrametalation of ferrocene in a single reaction: Extension of s-block inverse crown chemistry to the d-block. Angew. Chem. Int. Ed. 40, 3902–3905 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Andrikopoulos P., Armstrong D. R., Clegg W., Gilfillan C. J., Hevia E., Kennedy A. R., Mulvey R. E., O’Hara C. T., Parkinson J. A., Tooke D. M., A homologous series of regioselectively tetradeprotonated group 8 metallocenes: New inverse crown ring compounds synthesized via a mixed sodium−magnesium tris(diisopropylamide) synergic base. J. Am. Chem. Soc. 126, 11612–11620 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Krieck S., Görls H., Yu L., Reiher M., Westerhausen M., Stable “inverse” sandwich complex with unprecedented organocalcium(I): Crystal structures of [(thf)2Mg(Br)-C6H2-2,4,6-Ph3] and [(thf)3Ca{μ-C6H3-1,3,5-Ph3}Ca(thf)3]. J. Am. Chem. Soc. 131, 2977–2985 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Girolami G. S., Riehl M. E., Suslick K. S., Wilson S. R., Rare example of a monomeric aryllithium complex. X-ray crystal structure of (2,4,6-triphenylphenyl)lithium-bis(diethyl ether). Organometallics 11, 3907–3910 (1992). [Google Scholar]
- 46.Jenkins H. A., Abeysekera D., Dickie D. A., Clyburne J. A. C., Double lithiation of 2,4,6-triphenylbromobenzene: Synthesis of bis-amidines and an aluminium bis-amidinate complex. J. Chem. Soc. Dalton Trans. 3919–3922 (2002). [Google Scholar]
- 47.Beck R., Johnson S. A., Mechanistic implications of an asymmetric intermediate in catalytic C–C coupling by a dinuclear nickel complex. Chem. Commun. 47, 9233–9235 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Lahrood A. R., Björk J., Heckl W. M., Lackinger M., 1,3-Diiodobenzene on Cu(111)—An exceptional case of on-surface Ullmann coupling. Chem. Commun. 51, 13301–13304 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Zhang G., Auer-Berger M., Gehrig D. W., Blom P. W. M., Baumgarten M., Schollmeyer D., List-Kratochvil E. J. W., Müllen K., Blue light emitting polyphenylene dendrimers with bipolar charge transport moieties. Molecules 21, E1400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schade C., Bauer W., Schleyer P. V. R., n-Butylsodium: The preparation, properties and NMR spectra of a hydrocarbon- and tetrahydrofuran-soluble reagent. J. Organomet. Chem. 295, C25–C28 (1985). [Google Scholar]
- 51.Gehrhus B., Hitchcock P. H., Kennedy A. R., Lappert M. F., Mulvey R. E., Rodger P. J. A., Synthesis and crystal structure of trimeric sodium 2,2,6,6-tetramethylpiperidide (NaTMP). J. Organomet. Chem. 587, 88–92 (1999). [Google Scholar]
- 52.Hevia E., Kennedy A. R., Mulvey R. E., Weatherstone S., Synthesis and crystal structure of [{nBuMg(μ-TMP)}2] and of a homometallic inverse crown in tetranuclear [{nBuMg2[μ-N(H)Dipp]2(μ3-OnBu)}2]. Angew. Chem. Int. Ed. 43, 1709–1712 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Sheldrick G. M., Crystal structure refinement with SHELXL. Acta Cryst. C 71, 3–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hubschle C. B., Sheldrick G. M., Dittrich B., ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 44, 1281–1284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spek A. L., PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C 71, 9–18 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Katritzky A. R., Akhmedov N. G., Güven A., Doskocz J., Akhmedova R. G., Majumder S., Hall C. D., gNMR simulated 1H and proton-coupled 13C NMR spectra of substituted 3-nitropyridines. GIAO/DFT calculated values of proton and carbon chemical shifts and coupling constants. J. Mol. Struct. 787, 131–147 (2006). [Google Scholar]
- 57.Bringmann G., Price Mortimer A. J., Keller P. A., Gresser M. J., Garner J., Breuning M., Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem. Int. Ed. 44, 5384–5427 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Clayden J., Moran W. J., Edwards P. J., LaPlante S. R., The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. 48, 6398–6401 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Ruzziconi R., Spizzichino S., Mazzanti A., Lunazzi L., Schlosser M., The biphenyl-monitored effective size of unsaturated functional or fluorinated ortho substituents. Org. Biomol. Chem. 8, 4463–4471 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Toyota S., Iida T., Kunizane C., Tanifuji N., Yoshida Y., Rotational isomerism involving an acetylenic carbon IV: Synthesis and structure of bis(1,1′;3′,1″-terphenyl-2′-yl)ethynes: Molecular design of sterically congested alkynes toward restricted rotation about acetylenic axis. Org. Biomol. Chem. 1, 2298–2302 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Modjewski M., Lindeman S. V., Rathore R., A versatile preparation of Geländer-type p-terphenyls from a readily available diacetylenic precursor. Org. Lett. 11, 4656–4659 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Lunazzi L., Mazzanti A., Minzoni M., Anderson J. E., Structure, conformation, and dynamic processes of the stereolabile atropisomers of hindered terphenyl hydrocarbons. Org. Lett. 7, 1291–1294 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700832/DC1
Detailed experimental procedures
Materials and methods
Synthetic procedures
X-ray crystallography
Supplementary text
NMR spectra
fig. S1. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra showing the nonaromatic resonances for biphenylene (top; black), reaction mixture containing two conformers of 14 (middle; blue), and isolated [Na4Mg2(TMP)6(1,4-biphenylene-di-ide)] 14 (bottom; major conformer).
fig. S2. 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum showing the nonaromatic resonances for the two conformers of 14.
fig. S3. Sections of experimental and simulated 1H NMR spectra of 14.
fig. S4. Sections of experimental and simulated 1H NMR spectra of 15.
fig. S5. Molecular structure of 2 showing the contents of the asymmetric unit cell.
fig. S6. Molecular structure of 3 and its extended packing.
fig. S7. Molecular structure of 5 and its extended packing.
fig. S8. Molecular structure of 6 showing atomic connectivity.
fig. S9. Molecular structure of 12 showing the contents of the asymmetric unit cell.
fig. S10. Molecular structure of 13 and its extended packing.
fig. S11. Molecular structure of 14 showing the contents of the asymmetric unit cell.
fig. S12. Molecular structure of 15 and its extended packing.
fig. S13. Molecular structure of 16 showing the contents of the asymmetric unit cell.
fig. S14. 1H NMR study (400.1 MHz; [D12]cyclohexane, 300 K) of biphenyl (top; red) and a control reaction of biphenyl and NaTMP in a 1:2 M ratio in methylcyclohexane after 24 hours at 65°C (bottom; blue).
fig. S15. 1H NMR study (400.1 MHz; [D12]cyclohexane, 300 K) of biphenyl (top; red) and a control reaction of biphenyl and nBuMgTMP in a 1:2 M ratio in methylcyclohexane after 16 hours at 65°C (bottom; blue).
fig. S16. Representative example of 3,5-dimetalation of biphenyl to give 2.
fig. S17. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S18. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S19. Sections of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S20. Sections of the phase-sensitive 1H,13C-HSQC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S21. Sections of the 1H,13C-HMBC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 2.
fig. S22. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of biphenyl (top; red) and 2 (bottom; blue) showing the aromatic resonances.
fig. S23. Sections of the 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectra of biphenyl (top; red) and 2 (bottom; blue) showing the aromatic resonances.
fig. S24. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S25. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 3.
fig. S26. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S27. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S28. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3.
fig. S29. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of an in situ sample of 4.
fig. S30. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S31. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 5.
fig. S32. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S33. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S34. Sections of the 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 5.
fig. S35. 1H NMR (400.1 MHz; [D12]cyclohexane/C7H14, 300 K) spectrum of [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6.
fig. S36. Section of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane/C7H14, 300 K) spectrum of [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6 showing the cross peaks for the aromatic resonances.
fig. S37. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of para-terphenyl (top; green), 4 (middle; red), and [Na8Mg4(TMP)12(3,3′,5,5′-para-terphenyl-tetra-ide)] 6 (bottom; blue).
fig. S38. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 7.
fig. S39. 13C NMR (100.6 MHz; CDCl3, 300 K) spectrum of 7.
fig. S40. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8.
fig. S41. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8.
fig. S42. 1H NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S43. 13C{1H} NMR (100.6 MHz; 300 K, CDCl3) spectrum of 9.
fig. S44. 1H,1H-COSY NMR (400.1 MHz; 300 K, CDCl3) spectrum of 3,5-diiodo-meta-terphenyl 9.
fig. S45. 1H,13C-HSQC NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S46. 1H,13C-HMBC NMR (400.1 MHz; 300 K, CDCl3) spectrum of 9.
fig. S47. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of [Na8Mg4TMP12(3,3′,5,3′-meta-terphenyl-tetra-ide)] 10.
fig. S48. Sections of the 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectra of meta-terphenyl (top; green), [Na4Mg2(TMP)6(3,5-meta-terphenyl-di-ide)] 8 (middle; red), and [Na8Mg4TMP12(3,3′,5,3′-meta-terphenyl-tetra-ide)] 10 (bottom; blue) showing the aromatic resonances.
fig. S49. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 11.
fig. S50. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 11.
fig. S51. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S52. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S53. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 9.
fig. S54. 1H NMR (400.1 MHz; [D6]benzene, 300 K) spectrum of {Na8Mg4[TMP]12[3,3″,5,5″-(1′,3′,5′-triphenylbenzene-tetra-ide)]} 12.
fig. S55. Section of the 1H,1H-COSY NMR (400.1 MHz; [D6]benzene, 300 K) spectrum of 12 showing the cross peaks for the aromatic resonances.
fig. S56. 1H NMR (400.1 MHz; [D6]benzene, 300 K) spectra of tpb (top; red) and 12 (bottom; blue).
fig. S57. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 3,3″,5,5″-tetraiodo-5′-phenyl-benzene 13.
fig. S58. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 13.
fig. S59. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 13.
fig. S60. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 13.
fig. S61. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum sections of 13.
fig. S62. 1H NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S63. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S64. Sections of the 1H,1H-COSY NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S65. Sections of the phase-sensitive 1H,13C-HSQC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S66. Sections of the 1H,13C-HMBC NMR (400.1 MHz; [D12]cyclohexane, 300 K) spectrum of 14.
fig. S67. 13C{1H} NMR (100.6 MHz; [D12]cyclohexane, 300 K) section of the spectra of biphenylene (top; red) and isolated 14 (bottom; blue, major conformer).
fig. S68. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S69. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 15.
fig. S70. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S71. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S72. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 15.
fig. S73. 1H NMR (400.1 MHz; CDCl3, 300 K) spectra of biphenylene (top; red) and 15 (bottom; blue).
fig. S74. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectra of biphenylene (top; red) and 15 (bottom; blue).
fig. S75. 1H NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S76. 13C{1H} NMR (100.6 MHz; CDCl3, 300 K) spectrum of 16.
fig. S77. 1H,1H-COSY NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S78. 1H,13C-HSQC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
fig. S79. 1H,13C-HMBC NMR (400.1 MHz; CDCl3, 300 K) spectrum of 16.
table S1. Metalation conditions and scope.