

Abstract
Recent advances in optogenetics have opened new routes to drug discovery, particularly in neuroscience. Physiological cellular assays probe functional phenotypes that connect genomic data to patient health. Optogenetic tools, and in particular tools for all-optical electrophysiology, now provide means to probe cellular disease models with unprecedented throughput and information content. These techniques promise to identify functional phenotypes associated with disease states and to identify compounds that improve cellular function regardless of whether the compound acts directly on a target or through a bypass mechanism. This review discusses opportunities and unresolved challenges in applying optogenetic techniques throughout the discovery pipeline, from target identification and validation, to target-based and phenotypic screens, to clinical trials.
Keywords: Optogenetics, neuroscience, optopatch, phenotypic screening, drug discovery
Optogenetic tools contribute throughout the development pipeline
The essence of drug discovery is to find compounds that modulate a specific physiological process in a particular subset of cells. The challenge is that minimalist high-throughput assays often have little physiological relevance, while richer phenotypic assays are slow and expensive. Typically, hundreds of thousands, or sometimes millions, of compounds fed into a screening campaign are whittled down to a few leads. A discovery team is fortunate if one lead molecule survives clinical trials. The high attrition arises because each step of the development pipeline challenges compounds against new aspects of biology which upstream assays did not address. Recently developed and emerging optogenetic tools provide a means to improve this tradeoff between throughput and realism at each step.
Optogenetics—optical perturbation and optical measurement of physiological processes—has several attributes that make it a useful tool for drug discovery. Optical assays have the potential for high throughput and low cost. This feature has been appreciated at least since the introduction of the Fluorescent Imaging Plate Reader (FLIPR) (see Glossary) system more than 20 years ago.[1] Where optogenetics goes beyond traditional cell-based assays is in (1) the ability to deliver temporally and spatially precise stimuli to elicit defined patterns of molecular and cellular activity, (2) the rapid proliferation of fluorescent protein reporters for a huge diversity of molecular and physiological signals, and (3) the ability to use genetic techniques to target stimulus and readout to specified cell-types within a possibly complex multicellular milieu. These capabilities give optogenetic assays throughput and information content that transcend the limits of more traditional approaches.
An optogenetic disease signature can aid discovery throughout the development process, from target identification to clinical trials (Figure 1, Key Figure). For instance, optogenetic measures of neuronal excitability can be used first to screen for targets via CRISPR-based knockout or knockdown, then to validate the efficacy and specificity of a screening hit via comparison of its effect on knockout and wild-type cells, and finally in a clinical trial with patient stem cell-derived neurons to segregate likely responders from likely non-responders.
Figure 1. Key Figure. Optogenetics throughout the discovery pipeline.
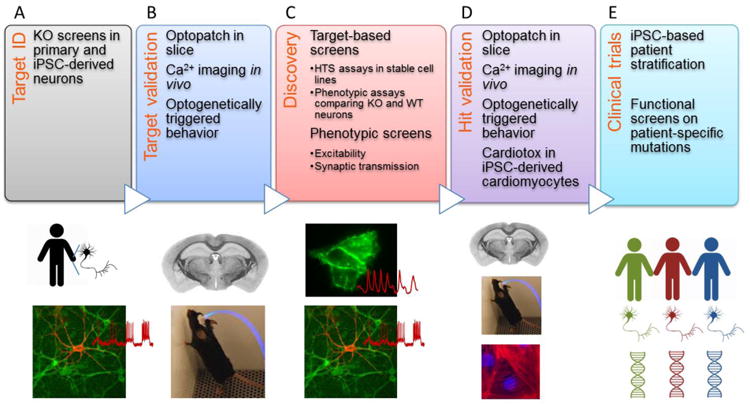
Optogenetic assays can contribute at each stage of drug discovery. A) Comparative measures on rodent or human neurons +/- a disease-causing mutation can establish a screenable phenotype. Alternatively, optogenetic screens of genetic knockouts can identify novel targets which can then be the subject of optogenetic or conventional drug screens. B) Targets identified in culture-based assays should be validated via knockout and functional measurements in acute slice, and ideally in vivo. Optogenetically triggered behaviors can provide a low-variance phenotype for testing the effects of mutations. C) Target-based optogenetic screens can be performed in a wide variety of cell-based assays. Alternatively, phenotypic optogenetic screens can identify compounds that modulate a disease-relevant functional phenotype regardless of mechanism. D) Screening hits should be validated in the same assays used for target validation. In addition, optogenetic assays in human iPSC-derived cardiomyocytes can help with cardiotoxicity assessment. E) Functional optogenetic measurements in human iPSC-derived neurons can seek to distinguish likely responders from non-responders prior to a clinical trial; or to match genetically heterogeneous patients with existing medications.
Optogenetics in neuroscience drug discovery
The impact of optogenetics will likely be greatest in drug discovery for neuroscience. On the one hand, genetic studies are implicating an ever widening array of mutations, ranging from rare large effect-size mutations in the epilepsies [2, 3] to common individually weak mutations in psychiatric disease [4, 5]. On the other hand, the core functions of neurons are electrical spiking and synaptic transmission. Due to the complex nonlinear interactions of multiple ion channels within cells, and of multiple cell types within circuits, the neurophysiological consequences of newly identified mutations can almost never be predicted a priori, nor can one reliably predict how pharmacological modulation of a particular target will affect brain function overall. Even as more targets are identified, neuroscience drug discoverers are turning toward a cell- and circuit-centric phenotypic approach [6, 7], rather than focusing on specific targets.
Cell-based models provide a means to instantiate newly identified mutations in a physiological yet pharmacologically addressable format and thus are an attractive substrate for phenotypic assays. Human induced pluripotent stem cell (hiPSC) technology provides a direct link between in vitro models and real patients. Genome editing technologies provide the ability to introduce and correct targeted mutations in human and rodent neuronal models. These rapidly improving cellular substrates hold the promise of connecting human genetics to the underlying physiology.
The weak link in this chain is the measurement: how to determine the effects of mutations and candidate therapeutics on neural function. The challenge of functional assays in neurons is severe. Electrical spiking and synaptic transmission cannot be purified or measured post-hoc; they must be measured in live cells in situ. Neural firing occurs on the millisecond timescale, beyond the reach of most screening tools. State-dependent drugs bind to ion channels with affinities that vary by orders of magnitude depending upon the patterns of neural activity [8]. Feed-forward and feedback mechanisms can produce circuit-level effects that are opposite to the effects of a compound on isolated cells [9].
Optogenetic approaches are starting to address these challenges. Recent reviews have discussed optogenetic actuators [10, 11] and reporters [12-14] in detail. In this review, we discuss how optogenetics can facilitate target discovery and validation, high-throughput screening, functional phenotypic assays, safety pharmacology and toxicity, and patient stratification for clinical trials.
Optogenetics for high-throughput screening
Drugs and ion channels perform an intricate dance, wherein the affinity of the drug depends on the state of the channel, and the dynamics of the channel depend on the binding of the drug. For instance, activity-dependent block of NaV channels is crucial for targeting analgesic action to hyperactive neurons, while sparing less active neighbors. Ideally, a targeted screen for ion channel modulators would allow one to define state-dependent selection criteria, but conventional fluorescence screens are insensitive to the details of this dance (Box 1).
Box 1. Ion channel screening tools.
Several fluorescence assays have been developed for screening ion channel modulators. Typically, a static membrane voltage is set via the ionic content of the extracellular medium. Sometimes a tool compound is applied to force the channels into a particular state (e.g. veratridine for NaV channels). The response to a test compound is then monitored either via fluorescence of a voltage-sensitive dye or via modulation of the flux of a readily detected ion (e.g. Ca2+ for CaV channels [15], Tl+ for NaV [16] and KV [17] channels, I- for chloride channels [18]). Screens of this sort cannot select for particular binding modes or kinetics.
Automated patch clamp electrophysiology [19] provides accurate programming of membrane potential and readout of membrane current, and is becoming widely used in ion channel screening[20]. However, this approach becomes expensive for large-scale screens and still suffers from technical limitations including a ∼17% drop-out rate of individual wells [21] and difficulty probing channels with small overall currents. For instance, the voltage-gated sodium channels NaV 1.8 and NaV 1.9, which play an important role in mediating pain transmission, are difficult to express at sufficiently high levels for automated patch clamp measurements [22, 23]. Automated patch clamp measurements are typically restricted to stable cell lines, though applications in cardiomyocytes have been demonstrated [24].
Optogenetic control of membrane potential
Light-gated ion channels and pumps provide a robust means to modulate membrane potential rapidly and reversibly. The light-gated cation channelrhodopsins are the exemplars of this class [25, 26]. Upon illumination, they mediate modestly inward-rectifying currents, with reversal potential around 0 mV. Many channelrhodopsins have been discovered and engineered, with differing action spectra,[27-31] kinetics,[32, 33] and ionic selectivities [34-36].
To modulate membrane potential optically, one can pair an optogenetic actuator with an inwardly rectifying potassium channel, such as Kir2.1 or Kir2.3. In the dark, the Kir channel sets the cell resting potential to near the K+ reversal potential, typically ∼-80 mV. Illumination depolarizes the cell toward 0 mV. Channels that activate within this window can be modulated in temporal patterns governed by the time-course of illumination.
To hyperpolarize a membrane one can use light-powered inward chloride pumps (e.g. halororhodopsin [37]), outward proton pumps (e.g. Archaerhodopsin 3 [38]) or outward sodium pumps (e.g. KR2[35]). Light-gated chloride channels (ChloC [39, 40] and recently discovered gtACR[36]) also provide hyperpolarizing drive, if the chloride reversal potential is negative. In HEK cells, the chloride reversal potential is typically ∼-50 mV.
Fluorescent reporters compatible with optogenetic stimulation
An optical readout is required to monitor the activity of a target protein. The key challenge is to avoid optical crosstalk between the measurement and the optogenetic actuation. Near infrared QuasAr proteins provide a fast and sensitive, but dim, readout of membrane voltage, and it avoids spectral crosstalk with blue-excited channelrhodopsins [41]. A recently developed red-shifted voltage-sensitive dye, BeRST1, is also spectrally compatible with channelrhodopsin activation and shows excellent speed, sensitivity and brightness [42]. For Ca2+-based readouts, red-shifted Ca2+-reporter dyes (e.g. CaRuby-Nano[43]) or proteins (e.g. RCaMP2[44], jRGECO1a, jRCaMP1a,b[45]) have recently been introduced. Unfortunately most protein-based fluorescent reporters are based on GFP or spectrally similar fluorophores, and thus suffer optical crosstalk with most optogenetic actuators, i.e. the light used to excite the reporter spuriously activates the actuator, or vice versa. Development of far-red fluorescent reporters remains a pressing need.
Examples of optogenetically enabled screens
Voltage-gated sodium (NaV) channels are a natural target for screening via all-optical electrophysiology (Figure 2a), and are of interest as targets in pain and epilepsies [46]. In HEK293 cells heterologously expressing a Kir channel and a NaV channel, a depolarizing influence can activate the NaV channel, causing a sharp voltage spike to the Na+ reversal potential, followed by NaV channel inactivation and return to baseline driven by the Kir channel. Electrically spiking HEK cells have been demonstrated with NaV 1.3[47], NaV 1.5[48], NaV 1.7[49] and NaV 1.9. A recent all-optical screen of 320 FDA-approved compounds for activity-dependent block of NaV1.7 yielded results in close concordance with manual patch clamp (Fig. 3).[49] By varying the intensity and duration of the optical stimulus pulses, one could probe subtle features of state-dependent pharmacology in a high-throughput format.
Figure 2. Optogenetic high-throughput target-based screens.
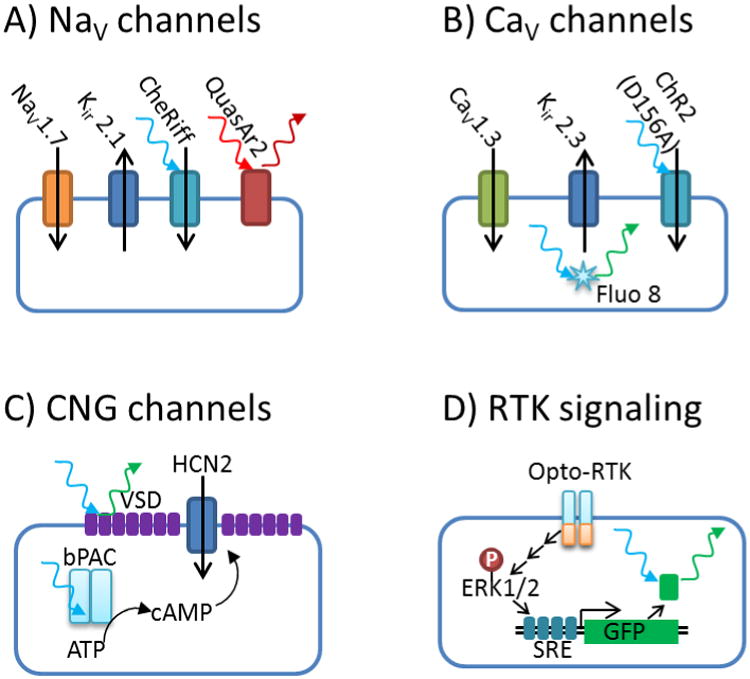
A) Screens for state-dependent modulators of NaV channels can be carried out in Optopatch spiking HEK cells [49]. B) Screens for state-dependent modulators of CaV channels use the gradual recovery of a step function channelrhodopsin, ChR2(D156A), [33] to induce a slowly varying membrane voltage [50]. C) Screens for modulators of cyclic nucleotide gated (CNG) channels use a photo-activated adenylyl cyclase, bPAC, [53] to induce a step in cAMP concentration. D) Screens for modulators of receptor tyrosine kinase (RTK) signaling use a light-activated RTK and a transcriptional readout. [54]
Figure 3. Optical high-throughput screen of sodium channel modulators.
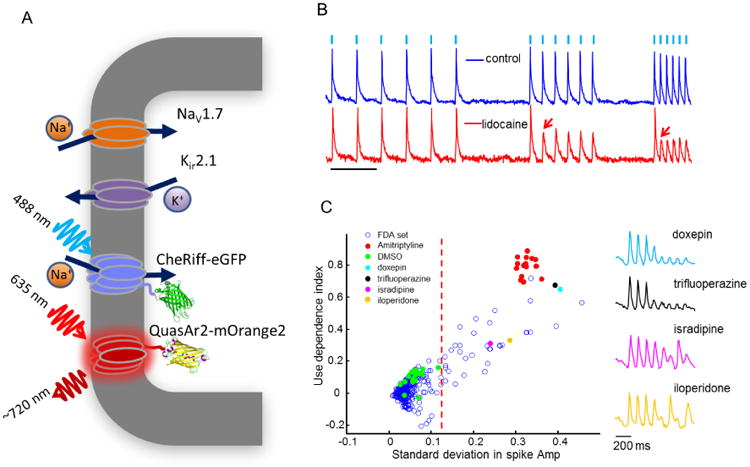
A) HEK293 cell engineered for optogenetic studies on NaV1.7. The cell stably expressed the test channel, NaV1.7, and Kir2.1 to lower the resting potential to near the K+ reversal potential. The blue light-activated ion channel CheRiff induced action potentials upon blue illumination. Fluorescence of the voltage indicator QuasAr2 reported the dynamics. B) Optically induced and optically recorded spikes followed the stimulus pattern in the absence of drug, but in the presence of a state-dependent blocker, lidocaine, the spikes failed at high stimulus frequency (arrows). Scale bar 1 s. C) All-optical screen of 320 FDA-approved compounds for use-dependent block. Each response was characterized by the overall decay in the spike amplitude (use-dependence index) and the standard deviation in the spike amplitude. State-dependent blockers showed clear functional clustering. Image adapted from [49].
A similar approach has been used to probe the CaV1.3 voltage-gated calcium channel (Fig. 2b)[50], a potential target in Parkinson's disease [51]. Here the channel was paired with Kir2.3 to set theresting potential and a “step-function” channelrhodopsin variant, ChR2(D156A)[33] was used to inducea slowly varying membrane potential. Channel activity was probed via a small-molecule Ca2+ indicator,Fluo-8. Although the Ca2+ reporter was excited at a blue wavelength that also drove the channelrhodopsin, crosstalk was minimized by performing the fluorescence measurement on a timescale much shorter than that of Ca2+ influx. The activity of CaV channels could likely also be probed with red-shifted Ca2+ reporters to avoid optical crosstalk.
Cyclic nucleotide-gated (CNG) cation channels are a possible target for neuropathic pain [52]. Figure 2c illustrates an optogenetic screen for the HCN2 channel [50]. Upon blue illumination, a light-activated adenylyl cyclase, bPAC [53], produced cAMP, which then activated the HCN channel. Channel activation was probed via a slow voltage-sensitive dye. Although the wavelength used to excite the dye also activated the bPAC, the dye measurement happened fast enough that the measurement-triggered bPAC activation did not affect the measurement. The HCN channels are also activated by membrane hyperpolarization, so optogenetic approaches could be used, in principle, to probe voltage-dependent pharmacology of these channels.
Receptor tyrosine kinases (RTKs) are interesting targets in oncology, metabolic disorders, and neurodegeneration. However, a lack of known ligands presents a problem for screening against some RTKs. Fig. 2d shows an optogenetic screen for modulators of RTK signaling.[54] Light-sensitive “Opto-RTKs” were formed by incorporation of a light-oxygen-voltage-sensing (LOV) domain which caused dimerization and thereby activation of the RTK in the light. Downstream activation of the MAPK/ERK pathway drove expression of a gene for a fluorescent reporter protein, and the pharmacological modulation of this response was readily detected in a high-throughput format. Remarkably, this approach worked even for RTKs with no known ligand. As in the screens of the CaV and HCN channels, crosstalk between stimulation and readout was avoided by using a slow reporter mechanism, in this case GFP expression.
In these examples, optogenetics offers several key advantages over ligand-based activation of the target pathways. First, for rapid processes, one can achieve precise temporal synchronization across a well, and correspondingly high time resolution in whole-well fluorescence measurements. Second, it is possible to mix cell lines with different actuators paired with spectrally distinct reporters to provide in-well controls. For instance, one could screen for compounds that have differential activation of structurally similar targets. Third, one can vary the illumination intensity across a well to perform in-well dose-response measurements. While sophisticated instrumentation is needed to produce precise optical gradients, a simple shadow mask can divide a well between optogenetically activated and un-activated cells for an in-well reference. These benefits could be applied to many novel optogenetic screens that have not yet been implemented (Box 2).
Box 2. Optogenetics for novel target-based screens.
The power of optogenetically enabled high-throughput screens is just beginning to be realized. Here we give several examples of prospective screening approaches.
Gap junctions
Gap junctions, which are electrical conduits between neighboring cells, are an interesting target in epilepsy [55]. When HEK cells are grown to confluence, they form gap junction-mediated electrical contacts. Overexpression of gap junctions can further strengthen this electrotonic coupling [56]. In syncytial monolayers of spiking HEK cells, optogenetically induced depolarizations lead to propagating waves which are sensitive to gap junction modulators.[48] Mixed co-culture of HEK cells expressing either an optogenetic actuator or a fluorescent voltage indicator would provide a robust means to probe gap junction-mediated electrical conduction.
Transporters and pumps
Membrane voltage is important in the dynamics of many transmembrane proteins beyond ion channels [57]. Indeed, every transmembrane protein that binds a charged ligand, undergoes conformational shifts, or transports charged moieties across the membrane is, in principle, susceptible to changes in membrane voltage [58]. Electrogenic transporters are sensitive to membrane voltage, as are some GPCRs[59, 60]. Voltage affects the activity of the multidrug resistance proteins MRP4 and MRP5 [61], though there has been controversy over whether voltage also affects the multi-drug efflux transporter P-gp (also known as MDR1) [62, 63]. Indeed, changes in membrane voltage have been implicated in regulating the cell cycle[64] and in the progression of cancer[65]. For these targets, optogenetic modulation of membrane potential could augment screening pipelines.
GPCRs and other signaling pathways
Optogenetics can also augment drug discovery for targets where membrane voltage is not a factor. Optogenetic control has been exerted over the three major classes of GPCR-based signaling (Gs[66], Gi/o[67], and Gq/11[68]). Chimeras of bovine rhodopsins and endogenous receptors have been used to make light-activated mimics of the adrenergic receptor (optoXR)[68] and the serotonin receptor (Rh-CT5-ht1a)[67]. Light-responsive soluble proteins have been coupled to the MAPK pathway, PI3K pathway, Rho GTPase signaling, and RTK signaling [69]. Broadly applicable strategies have been developed for controlling protein localization, activity, oligomerization, and secretion with light [69]. Recently, the optogentic tool box was further expanded with a light-controlled CRISPRa activator, which can control endogenous gene expression with spatial and temporal precision.[70] These powerful capabilities are just beginning to enter drug discovery pipelines.
Target-based screening in primary cells
Target proteins can behave very differently in heterologous expression systems and in native cells, due to alternative splicing, post-translational modifications, variations in trafficking, clustering, and degradation, and interactions with modulatory subunits. For instance, dynamic clustering of the soma-localized potassium channel Kv2.1 regulates its activity [71], while trafficking of the sodium channel NaV1.2 along the axon initial segment regulates neuronal excitability[72]. Thus, even for target-based screens, there is merit to assessing target function in its native cellular context.
A few optogenetic tools have been developed to probe function of specific targets in a complex cellular milieu. Fluorescently labeled toxins with state-dependent binding probe activation of native Kv2.1 ion channels [73]. FRET-based reporters can probe activation of specific kinases, GPCRs, and other targets [13, 14]. With these tools, one can perform target-based screens in native cells. There remains a strong need for improved fluorescent reporters for tracking the location and activity of specific drug targets within a complex cellular milieu.
Optogenetics for phenotypic screening
Following the maxim, “you get what you screen for,” [74] target-based screens identify modulators of specific targets. But a modulator of a disease-relevant target might still make a poor drug for several reasons: (1) the hit compounds might also modulate other targets. Indeed, many neuro-active drugs developed around a nominal target turned out later to modulate other ion channels.[75] (2) Selective modulation of a specific target can be a poor therapeutic strategy. Dysregulated neural circuits might not contain a unique target whose modulation will restore the defective function; and selective modulation of a single broadly distributed target might still cause undesirable effects in other brain regions or organs. (3) Many effective drugs were developed without a clear target, or modulate multiple targets. For instance, gabapentin, pregabalin,[76] and levetiracetam,[77] all widely prescribed anti-epileptic drugs, were developed without a clear understanding of mechanism. Screens that directly probe the effects of compounds on neuronal firing or synaptic transmission select for mechanisms relevant to disease biology, agnostic to molecular target. Optogenetic assays now enable phenotypic measurements across a range of context relevant to neuroscience drug discovery (Figure 4).
Figure 4. Optogenetics-enabled phenotypic assays.
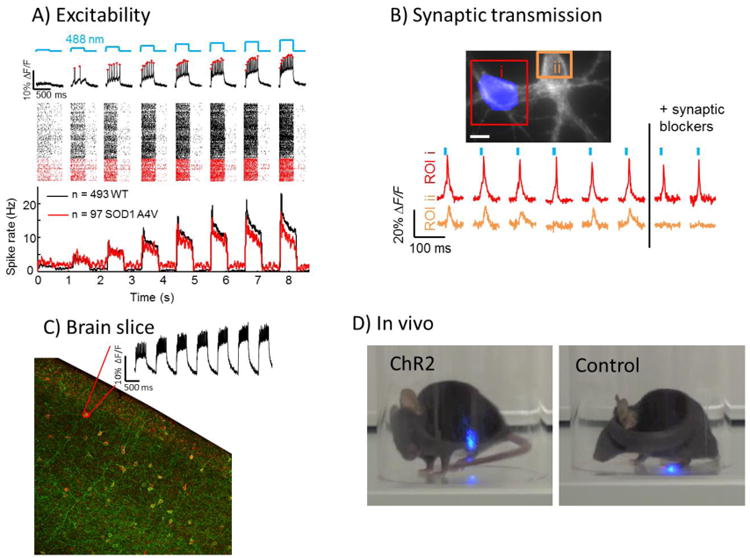
A) High-throughput measures of excitability in cultured neurons can characterize rodent and human iPSC-derived neurons. Here Optopatch recordings are shown from human iPSC-derived motor neurons comparing a wild-type genotype with cells containing an ALS-causing mutation, SOD1(A4V). The mutation causes differences in spontaneous and optogenetically induced firing patterns. Figure adapted from [82]. B) All-optical measures of synaptic transmission can identify mutations or compounds that modulate excitatory or inhibitory signaling in cultured neurons. Here a single rat hippocampal neuron is stimulated with patterned illumination, and excitatory post-synaptic potentials are recorded in a neighboring cell. Figure adapted from [41]. C) All-optical measures of excitability in acute brain slice can identify characteristic firing patterns in genetically defined neuronal sub-types in defined brain regions. Here an Optopatch recording from a somatostatin-positive interneuron is overlaid on an image of a brain slice expressing Optopatch under control of somatostatin-Cre. Figure adapted from [81]. D) A mouse expressing channelrhodopsin 2 in its sensory neurons exhibits nocifensive behavior upon blue light illumination of its left hindpaw (left), while a control mouse does not (right). Figure adapted from [98].
Neuronal excitability
Neuronal firing patterns arise from complex interactions of many ion channels and metabolic factors; and neuronal firing converts these molecular interactions into sensations and behavior. Thus neuronal excitability is a powerful integrative phenotype for disease modeling and drug discovery. Optogenetic techniques now provide high-throughput access to this phenotype.
Optogenetic measures of neuronal excitability can be used in several contexts (Fig. 5): (1) Target identification. Genetic perturbations, e.g. via CRISPR/Cas9 technology, can identify prospective drug targets that modulate neuronal excitability, or validate targets suggested by other means, e.g. genetic studies; (2) Target-based screens. Comparisons of drug effects on excitability of neurons +/- a specific target can distinguish on-target from off-target mechanisms of action; (3) Purely phenotypic screens. Comparisons of drug effects on different neuronal sub-types, e.g. excitatory vs. inhibitory neurons, can identify compounds that have differential effects on excitability, regardless of mechanism of action; (4) Patient stratification. Comparisons of drug effects on patient iPSC-derived neurons and on wild-type controls can seek to match patients with therapeutics or to stratify patients for clinical trials—even when the underlying genetic cause of the patient's illness is not known or is poorly understood.
Figure 5. Optogenetic excitability assays can be used throughout the discovery pipeline.
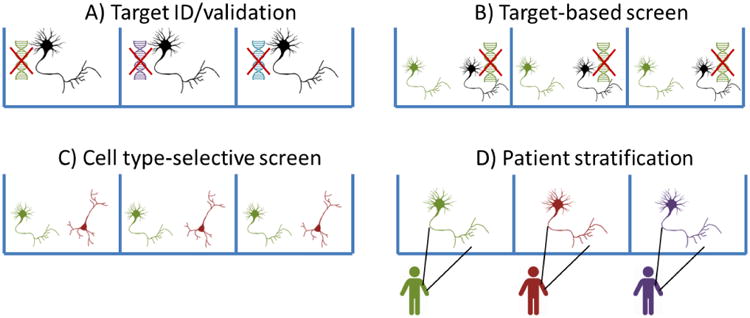
A) Target identification. Each well contains neurons with a distinct genetic perturbation (e.g. gene knockout). Optogenetic measurements can identify genetic modulators of neuronal excitability. B) Target-based screen. Each well contains a mixture of cells expressing and not expressing a specific drug target, and a fluorescent marker to distinguish the populations (green). Optogenetic excitability measurements are performed with single-cell resolution. Compounds that modulate the excitability only of the cells with the target are judged to be functionally selective for the target in the native cellular milieu. C) Cell type-selective screen. Each well contains a mixture of two or more cell types with fluorescent markers identifying the cell type (green vs. red). Optogenetic excitability measurements are performed with single-cell resolution. Compounds that modulate the excitability of only one cell type are judged to have cell type-selectivity, regardless of mechanism of action. D) Patient stratification. Each well contains iPSC-derived neurons from a different patient. Optogenetic excitability measurements in the presence of a test compound can determine how the patient's cells will respond to the compound.
A paired stimulus and readout are needed to perform high-throughput measures of neuronal excitability. Electric field stimulation (EFS) is a convenient means of whole-well stimulation that leaves the visible spectrum free for a variety of optogenetic readouts. EFS has been paired with whole-well measures of Ca2+ dynamics (via GCaMP reporters) in screening format for CNS [6] and DRG [78] neurons and with voltage-sensitive dyes [75]. By using genetically encoded reporters, one could probe the effect of test compounds on neural activity-dependent changes in other parameters as well, such as cAMP (via ICUE3[79]), ATP (via Percival[80]), or other metabolic factors.
When EFS is applied in an imaging format, cell type-specific markers in one color channel, and broadly expressed activity markers in another channel, could be used to probe for cell-type-specific effects of compounds on excitability. Such an approach could be useful, for instance, to detect compounds that differentially suppress firing in nociceptive vs. non-nociceptive sensory neurons; or to detect compounds that differentially affect excitability in excitatory vs. inhibitory CNS neurons.
While EFS leaves the entire optical spectrum available for functional measurements, this technique does not allow targeted stimulation of specific subsets of cells, or stimulation in precisely controlled temporal waveforms. Devoting the blue part of the spectrum to optogenetic actuation still leaves the yellow and red parts of the spectrum for imaging voltage, Ca2+, or other functional reporters.
Simultaneous optogenetic stimulation and readout of membrane voltage has been achieved through pairing a blue-shifted channelrhodopsin variant, CheRiff, with near-IR QuasAr voltage indicators.[41] These paired ‘Optopatch’ constructs enabled wide-field all-optical stimulation and readout of neural activity. Optopatch measurements have been used to probe the excitability of primary rodent and human iPSC-derived CNS [41] and sensory DRG [81] neurons. A recent application probed the excitability of human iPSC-derived motor neurons containing a mutation causal for ALS.[82]
Synaptic transmission
Synaptic dysfunction is implicated in many neuropsychiatric and neurodegenerative diseases. The immense morphological and biochemical complexity of the synapse presents a forbidding obstacle to target-based drug development in this domain. Recently developed tools are beginning to enable optogenetic interrogation of synaptic function. Calcium indicators targeted to presynaptic boutons [83] report Ca2+ entry at the site of vesicle release. pH indicators targeted to the lumen of synaptic vesicles report the de-acidification that occurs during vesicle fusion.[84] A recently developed glutamate indicator, iGluSnfr, reports glutamate release.[85] On the postsynaptic side, spine-targeted Ca2+ indicators are in development[86]. By pairing optogenetic stimulation of a channelrhodopsin (CheRiff) in a presynaptic cell with fluorescence imaging of membrane voltage in a postsynaptic cell expressing QuasAr2, one can detect excitatory and inhibitory postsynaptic potentials. By adding selective blockers of AMPA, NMDA, or glutamate receptors one can isolate the contributions to the post-synaptic potential from specific classes of receptors. Furthermore, genetic targeting of presynaptic actuator and, when appropriate, postsynaptic reporter enables targeted measurements of synaptic transmission between genetically defined neuronal sub-types.
Several challenges must be addressed before synaptic assays can transition from the research domain to screening applications. Homeostatic feedbacks render synaptic strengths highly sensitive to density of neurons and glia, and to the past history of activity.[87] It is technically challenging to achieve sufficiently tight control over these parameters for screening. Assays that measure the same cells pre- and post-compound are likely to be more useful in the near term because one can null-out cell-to-cell variability. Microchannel devices that compartmentalize pre- and post-synaptic cells offer an intriguing possibility for assays in a standardized geometry [88], but scale-up engineering will be needed before these devices are useful for screening. Given the transformative promise of synaptic assays, one can anticipate rapid progress in this domain.
Measurements in brain slice and tissue
Due to circuit-level feedbacks, modulation of cell-autonomous excitability or monosynaptic transmission can have counterintuitive effects at the level of microcircuit function in intact tissue. Furthermore, one important goal is to identify compounds that selectively modulate function of particular microcircuits. This modulation could arise from pleiotropic molecular mechanisms. Thus there is strong demand for optogenetic tools to map neuronal function in the context of intact tissue, e.g. in acute brain slice.
Recent advances in ultrawide-field imaging have opened the possibility of two-photon Ca2+ or glutamate imaging over areas almost covering a complete brain slice [89, 90]. The challenge with passive imaging in acute slice is that under typical conditions, there is little spontaneous activity. Recently, Optopatch measurements of excitability were demonstrated in acute slice, using a transgenic Cre-dependent Optopatch mouse to target Optopatch expression to genetically specified subsets of neurons [81]. It has not yet been possible to perform Optopatch measurements of synaptic transmission in acute slice because postsynaptic potentials in tissue are ∼20-fold smaller than in culture [91]. Due to the low throughput of measurements slice-based measurements, these assays will likely find most use in target validation, e.g. via studies on genetically modified mice, and on hit validation or prioritization, where a small number of molecules can be tested for effects on the target circuit and on other brain regions. Despite these limitations on throughput, optogenetic mapping of compound effects on circuit excitability is a promising frontier for drug discovery.
Measurements in vivo
Most drugs go through tests of efficacy in rodent models before human clinical trials. Optogenetic activation of specific circuits can elicit a wide range of disease-relevant behaviors [92, 93], including fear [94], aggression [95], feeding [96], pain[97, 98] and sleep [99]. The temporal precision and repeatability with which these behaviors can be evoked via optogenetic stimulation provides a robust baseline against which to test the effect of perturbations in candidate target genes or to test application of candidate therapeutics, though one should keep in mind that patterns of neural activity induced by synchronous activation of large ensembles are likely to differ substantially from naturally occurring patterns.
Implantable microscopes[100] and ultra-wide-field imaging preparations[101] further enable optogenetic recording of Ca2+ dynamics in large populations of cells in vivo, which can be used to validate the effect of a test compound on circuit dynamics. Recently developed implants that combine optogenetic stimulation with drug delivery provide a means to test compound effects in a minimally invasive manner.[102] As with measurements in acute brain slice, the low throughput of optogenetic measurements in vivo will most likely restrict applications to target and hit validation.
Optogenetics in safety and toxicology
Safety remains a key hurdle in drug development, and cardiac safety is of paramount concern due to the potential severity of cardiac side-effects. Traditional safety assessments require testing against the cardiac hERG channel, a common cause of off-target cardiotoxicity, followed by tests in dog and human.[103] However, hERG testing may reject some potentially promising candidates. Indeed, several drugs known to be safe and approved before hERG testing became standard would not pass current safety tests. The Cardiac in vitro Pro-Arrhythmia (CiPA) initiative aims to develop more accurate in vitro assays of cardiac risk.[104] Tests in human iPSC-derived cardiomyocytes (hiPSC-CM) provide one arm of this new approach. Optogenetic measurements of voltage[105] and calcium[106], combined with optogenetic pacing[107], provide a rich functional phenotype for exploring drug effects on human cardiac activity. Similarly, the all optical electrophysiology method can also be used for neuro-toxicity study to evaluate compound effect on neuron intrinsic excitability and synaptic transmission.
Future directions: Optogenetics in patient stratification and precision medicine
For many diseases of the nervous system, symptoms alone poorly predict a patient's response to a candidate therapeutic. Despite some notable successes of genetic stratification [108], in many instances genetics provides little additional guidance. Optogenetic measurements on patient iPSC-derived neurons provide a means to instantiate a patient's entire genome in a physiologically relevant and pharmacologically accessible format. A likely direction of future progress will be toward patient stratification based on functional cell-based measurements. Initially this work will occur in the presence of stratification for clinical trials, though ultimately the approach may be extended to individual patients seeking to select among many possible therapeutic options. Patient iPSC-derived neurons are most likely to replicate disease pathology for monogenic channelopathies, where one can reasonably expect a robust cell-autonomous phenotype. The extent to which these cells can recapitulate complex neurodevelopmental and neurodegenerative disease processes remains a topic of active research.
Challenges for optogenetics-enabled drug discovery
Optogenetic techniques offer great promise for accelerating drug discovery. But technical challenges remain (see Outstanding Questions). The set of spectrally compatible optogenetic actuators and reporters is limited, due to the very broad action spectra of microbial rhodopsin-based actuators and the reliance of most reporters on GFP or spectrally similar proteins. Development of far red or near infrared genetically encoded reporters would greatly expand the scope of possible applications. One would also like to see reporters for more analytes than are currently available, particularly for neurotransmitters and neuromodulators, e.g. GABA, glycine, dopamine, serotonin, acetylcholine.
A further challenge is quantification. In conventional patch clamp measurements, one knows the accurate value of the membrane current and voltage. For both optogenetic stimulation and measurement, the strength of the signal depends on the expression level and trafficking of the transducer, the membrane area, and the distribution of illumination intensity. These parameters are challenging to know precisely, though there are several promising remedies. Stable transgenic cell lines can have more homogeneous expression compared to transiently transfected cells, leading to less cell-to-cell variability. Co-expression of a stable fluorescent tag can further provide a means to estimate expression level ratiometrically. The electronic excited state lifetime of some fluorescent reporters can also provide an expression level-independent measure of activation. These techniques have been applied for voltage [109, 110] and calcium[111, 112], but not yet in a high-throughput format. Ultimately, quantitative measurements come from careful experimental design, including e.g. mixed positive and negative control cells in the same well, pre- and post-drug measurements on the same cells, or tool pharmacology for post-measurement calibration of signal magnitudes.
We also need better instrumentation and software. Existing screening tools lack the capabilities for spatially and spectrally patterned illumination, and lack the temporal resolution, spatial resolution and sensitivity needed for high-speed measurements of neuronal firing. While optical stimulation capabilities are starting to be added to existing screening platforms [113], these capabilities remain rudimentary. This challenge will likely be addressed through a series of instruments with differing tradeoffs in throughput and information content. A key divide is whether the instrument provides cellular resolution in a single-well format, or averages over the whole well and probes multiple wells in parallel. For truly high-throughput screens (> 105 compounds), the latter approach will be essential. Whole-well average measurements are most appropriate for screens on heterologously expressed proteins, e.g. in spiking HEK cells, or for measures of neuronal activity where the population-level response is expected to be homogeneous. Follow-up lower throughput assays can use instruments with single-cell resolution and more complex stimulation, but with serial scanning over multiple wells. Both types of measurements have been demonstrated academically, but no company has yet undertaken the engineering to make these tools commercially available.
Finally, we need better software. Imaging a wide area at high speed and high resolution leads to prodigious data rates, typically ∼1 GByte/s. Specialized software pipelines are needed to store, segment, and process these data. Each discovery project must address the challenge of converting terabytes of movies into actionable screening data.
Despite these challenges, optogenetic tools are already making headway in drug discovery. Further technical advances offer the promise to bring in a new age of functional screening for diseases of cellular excitability. Traditional screening approaches have led to high rates of expensive attrition during clinical trials. By testing drugs for functional modulation of neurons, circuits, and patient-derived cells, there is reason for optimism that the initial assays will more closely approximate the patient-relevant biology.
Trends Box.
Optogenetic approaches have potential to contribute throughout the drug discovery pipeline, from target identification to clinical trials.
Optogenetic assays can probe specific targets and cell-types within a rich physiological context.
Optogenetic measurements in patient-derived neurons can elucidate genotype-phenotype relationships and probe disease mechanisms.
The integration of optogenetics with high-throughput screening requires advances in molecular tools, instrumentation and data analytics.
Outstanding Questions Box.
Can we develop red-shifted reporters for neurotransmitters and neuromodulators, which are compatible with optogenetic stimulation?
Can optogenetic measurements on human iPSC-derived neurons be used to predict patient response to drugs?
How best to parameterize and classify the complex stimulus-dependent firing patterns of neuronal disease models in vitro?
How can one attain optogenetic stimuli of precisely calibrated magnitude, and fluorescence measurements of accurately calibrated concentrations or voltages?
Glossary
- Bouton
a swelling in a neuronal axon that contains synaptic vesicles. A synapse comprises a junction of a presynaptic bouton and a postsynaptic dendritic spine
- Channelrhodopsin
a class of transmembrane proteins that transport cations across the plasma membrane in response to light stimulation
- CheRiff
a type of channelrhodopsin that shows good sensitivity to blue light illumination and rapid opening and closing kinetics
- Depolarization
In the resting state of most cells, the inside of the cell is electrically negative relative to the outside (“polarized”). Depolarization refers to a shift to a less negative voltage, often caused by a flow of positive ions into the cell
- Fluorescent Imaging Plate Reader (FLIPR)
a commercially available instrument that monitors the simultaneous fluorescence dynamics of each well in a multi-well plate. This instrument is widely used for high-throughput screening to identify modulators of GPCR and ion channel targets
- GCaMP
a class of protein-based calcium indicators that emit green fluorescence. The brightness of these reporters is highly sensitive to local Ca2+ concentration
- Kir channel
inwardly rectifying potassium channel. These ion channels have greater conductance for potassium at negative potentials than at positive potentials. Kir channels do not show voltage-gated behavior, i.e. the current is only a function of the instantaneous voltage as opposed to depending on past voltages too. In neurons and cardiomyocytes Kir channels are critical to stabilize a negative resting membrane potential, close to the potassium reversal potential
- Nav channels
voltage gated sodium channels. When activated by a depolarizing shift in membrane voltage, these ion channels open and conduct sodium ions into the cell. This inward current leads to further depolarization, creating a positive feedback that produces a spike in the membrane voltage. Most NaV channels spontaneously close after a short time, so NaV activation only produces a brief spike in membrane voltage. NaV channels produce the upstroke of the action potential in neurons and cardiomyocytes
- QuasAr
a near infrared fluorescent protein reporter of membrane voltage. This protein responds to changes in membrane voltage quickly and sensitively, but is very dim
- RCaMP
a class of protein-based calcium indicators that emit red fluorescence. The red-shifted spectrum of these reporters facilitates pairing with blue-activated channelrhodopsins
- Reversal potential
the value of the membrane potential at which an ion will neither flow into or out of a cell. The reversal potential for each ion depends on its concentrations inside and outside the cell, via the Nernst equation
Footnotes
Conflict of Interest Statement: AEC is a founder of Q-State Biosciences. HZ is an employee of Q-State Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schroeder KS, Neagle BD. FLIPR: a new instrument for accurate, high throughput optical screening. Journal of Biomolecular Screening. 1996;1(2):75–80. [Google Scholar]
- 2.Epi4K Consortium. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epi4K consortium. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. The Lancet Neurology. 2017;16(2):135–143. doi: 10.1016/S1474-4422(16)30359-3. [DOI] [PubMed] [Google Scholar]
- 4.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purcell SM, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hempel CM, et al. A system for performing high throughput assays of synaptic function. PloS One. 2011;6(10):e25999. doi: 10.1371/journal.pone.0025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng W, et al. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18(21):1067–1073. doi: 10.1016/j.drudis.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eijkelkamp N, et al. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135(Pt 9):2585–2612. doi: 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paz JT, Huguenard JR. Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci. 2015;18(3):351–359. doi: 10.1038/nn.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deisseroth K. Optogenetics : 10 years of microbial opsins in neuroscience. 2015;18:1213–1225. doi: 10.1038/nn.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin JY. A user's guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol. 2011;96(1):19–25. doi: 10.1113/expphysiol.2009.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin MZ, Schnitzer MJ. Genetically encoded indicators of neuronal activity. Nat Neurosci. 2016;19(9):1142–1153. doi: 10.1038/nn.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta S, Zhang J. Reporting from the field: genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems. Annu Rev Biochem. 2011;80:375–401. doi: 10.1146/annurev-biochem-060409-093259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolbat A, Schultz C. Recent developments of genetically encoded optical sensors for cell biology. Biology of the Cell. 2016 doi: 10.1111/boc.201600040. [DOI] [PubMed] [Google Scholar]
- 15.McManus OB. HTS assays for developing the molecular pharmacology of ion channels. Current Opinion in Pharmacology. 2014;15:91–96. doi: 10.1016/j.coph.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Du Y, et al. Development and validation of a thallium flux-based functional assay for the sodium channel NaV1. 7 and its utility for lead discovery and compound profiling. ACS Chemical Neuroscience. 2015;6(6):871–878. doi: 10.1021/acschemneuro.5b00004. [DOI] [PubMed] [Google Scholar]
- 17.Weaver CD, et al. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. Journal of Biomolecular Screening. 2004;9(8):671–677. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 18.Sui J, et al. Optimization of a yellow fluorescent protein-based iodide influx high-throughput screening assay for cystic fibrosis transmembrane conductance regulator (CFTR) modulators. Assay and Drug Development Technologies. 2010;8(6):656–668. doi: 10.1089/adt.2010.0312. [DOI] [PubMed] [Google Scholar]
- 19.Sawada K, Yoshinaga T. Automated patch clamping. Patch Clamp Techniques: From Beginning to Advanced Protocols. 2012:323–332. [Google Scholar]
- 20.Farre C, et al. Ion channel screening–automated patch clamp on the rise. Drug Discovery Today: Technologies. 2008;5(1):e23–e28. doi: 10.1016/j.ddtec.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Chambers C, et al. High-Throughput Screening of Na(V)1.7 Modulators Using a Giga-Seal Automated Patch Clamp Instrument. Assay Drug Dev Technol. 2016;14(2):93–108. doi: 10.1089/adt.2016.700. [DOI] [PubMed] [Google Scholar]
- 22.Zhang ZN, et al. The voltage-gated Na+ channel Nav1.8 contains an ER-retention/retrieval signal antagonized by the beta3 subunit. J Cell Sci. 2008;121(Pt 19):3243–3252. doi: 10.1242/jcs.026856. [DOI] [PubMed] [Google Scholar]
- 23.Vanoye CG, et al. Mechanism of sodium channel NaV1.9 potentiation by G-protein signaling. J Gen Physiol. 2013;141(2):193–202. doi: 10.1085/jgp.201210919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheel O, et al. Action potential characterization of human induced pluripotent stem cell–derived cardiomyocytes using automated patch-clamp technology. Assay and Drug Development Technologies. 2014;12(8):457–469. doi: 10.1089/adt.2014.601. [DOI] [PubMed] [Google Scholar]
- 25.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100(24):13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyden ES, et al. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8(9):1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 27.Lin JY, et al. ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat Neurosci. 2013;16(10):1499–1508. [Google Scholar]
- 28.Klapoetke NC, et al. Independent optical excitation of distinct neural populations. Nat Meth. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erbguth K, et al. Bimodal activation of different neuron classes with the spectrally red-shifted channelrhodopsin chimera C1V1 in Caenorhabditis elegans. PLoS One. 2012;7:e46827. doi: 10.1371/journal.pone.0046827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govorunova EG, et al. Characterization of a highly efficient blue-shifted channelrhodopsin from the marine alga Platymonas subcordiformis. J Biol Chem. 2013;288(41):29911–29922. doi: 10.1074/jbc.M113.505495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang F, et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11(6):631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin JY, et al. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96(5):1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berndt A, et al. Bi-stable neural state switches. Nat Neurosci. 2009;12(2):229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 34.Kleinlogel S, et al. Ultra light-sensitive and fast neuronal activation with the Ca2+-permeable channelrhodopsin CatCh. Nat Neurosci. 2011;14:513–518. doi: 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- 35.Inoue K, et al. A light-driven sodium ion pump in marine bacteria. Nat Commun. 2013;4:1678. doi: 10.1038/ncomms2689. [DOI] [PubMed] [Google Scholar]
- 36.Govorunova EG, et al. Natural light-gated anion channels: A family of microbial rhodopsins for advanced optogenetics. Science. 2015;349(6248):647–650. doi: 10.1126/science.aaa7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gradinaru V, et al. eNpHR: a Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008;36(1):129–139. doi: 10.1007/s11068-008-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chow BY, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berndt A, et al. Structure-guided transformation of channelrhodopsin into a light-activated chloride channel. Science. 2014;344(6182):420–424. doi: 10.1126/science.1252367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wietek J, et al. Conversion of channelrhodopsin into a light-gated chloride channel. Science. 2014;344(6182):409–412. doi: 10.1126/science.1249375. [DOI] [PubMed] [Google Scholar]
- 41.Hochbaum DR, et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Methods. 2014;11:825–833. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang YL, et al. A photostable silicon rhodamine platform for optical voltage sensing. J Am Chem Soc. 2015;137(33):10767–10776. doi: 10.1021/jacs.5b06644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collot M, et al. CaRuby-Nano: a novel high affinity calcium probe for dual color imaging. Elife. 2015:4. doi: 10.7554/eLife.05808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoue M, et al. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nature Methods. 2015;12(1):64–70. doi: 10.1038/nmeth.3185. [DOI] [PubMed] [Google Scholar]
- 45.Dana H, et al. Sensitive red protein calcium indicators for imaging neural activity. Elife. 2016:5. doi: 10.7554/eLife.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagal SK, et al. Voltage gated sodium channels as drug discovery targets. Channels. 2015;9(6):360–366. doi: 10.1080/19336950.2015.1079674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park J, et al. Screening fluorescent voltage indicators with spontaneously spiking HEK cells. PLoS One. 2013;8(12):e85221. doi: 10.1371/journal.pone.0085221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McNamara HM, et al. Optically Controlled Oscillators in an Engineered Bioelectric Tissue. Physical Review X. 2016;6(3):031001. [Google Scholar]
- 49.Zhang H, et al. Optical electrophysiology for probing function and pharmacology of voltage-gated ion channels. ELife. 2016;5:e15202. doi: 10.7554/eLife.15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agus V, et al. Bringing the light to high throughput screening: use of optogenetic tools for the development of recombinant cellular assays. Proc SPIE. 2015;9305 doi: 10.1117/12.2077579. [DOI] [Google Scholar]
- 51.Kang S, et al. CaV1. 3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nature Communications. 2012;3:1146. doi: 10.1038/ncomms2149. [DOI] [PubMed] [Google Scholar]
- 52.Maher MP, et al. HCN channels as targets for drug discovery. Comb Chem High Throughput Screen. 2009;12(1):64–72. doi: 10.2174/138620709787048028. [DOI] [PubMed] [Google Scholar]
- 53.Stierl M, et al. Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J Biol Chem. 2011;286(2):1181–1188. doi: 10.1074/jbc.M110.185496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inglés-Prieto Á, et al. Light-assisted small-molecule screening against protein kinases. Nature Chemical Biology. 2015 doi: 10.1038/nchembio.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mylvaganam S, et al. Roles of gap junctions, connexins, and pannexins in epilepsy. Frontiers in Physiology. 2014;5:172. doi: 10.3389/fphys.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirkton RD, Bursac N. Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat Commun. 2011;2:300. doi: 10.1038/ncomms1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9(4):323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 58.Cohen AE, Venkatachalam V. Bringing bioelectricity to light. Annu Rev Biophys. 2014;43:11.1–11.22. doi: 10.1146/annurev-biophys-051013-022717. [DOI] [PubMed] [Google Scholar]
- 59.Mahaut-Smith MP, et al. A role for membrane potential in regulating GPCRs? Trends Pharmacol Sci. 2008;29(8):421–429. doi: 10.1016/j.tips.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 60.Vickery ON, et al. Membrane potentials regulating GPCRs: insights from experiments and molecular dynamics simulations. Current Opinion in Pharmacology. 2016;30:44–50. doi: 10.1016/j.coph.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kucka M, et al. Dependence of multidrug resistance protein-mediated cyclic nucleotide efflux on the background sodium conductance. Mol Pharmacol. 2010;77(2):270–279. doi: 10.1124/mol.109.059386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luker GD, et al. MDR1 P-glycoprotein reduces influx of substrates without affecting membrane potential. J Biol Chem. 2001;276(52):49053–49060. doi: 10.1074/jbc.M105192200. [DOI] [PubMed] [Google Scholar]
- 63.Howard EM, Roepe PD. Purified human MDR 1 modulates membrane potential in reconstituted proteoliposomes. Biochemistry (N Y) 2003;42(12):3544–3555. doi: 10.1021/bi026706i. [DOI] [PubMed] [Google Scholar]
- 64.Zhou Y, et al. SIGNAL TRANSDUCTION. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science. 2015;349(6250):873–876. doi: 10.1126/science.aaa5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang M, Brackenbury WJ. Membrane potential and cancer progression. Frontiers in Physiology. 2013;4:185. doi: 10.3389/fphys.2013.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bailes HJ, et al. Reproducible and sustained regulation of Gαs signalling using a metazoan opsin as an optogenetic tool. PloS One. 2012;7(1):e30774. doi: 10.1371/journal.pone.0030774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oh E, et al. Substitution of 5-HT1A receptor signaling by a light-activated G protein-coupled receptor. J Biol Chem. 2010;285(40):30825–30836. doi: 10.1074/jbc.M110.147298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Airan RD, et al. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458(7241):1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- 69.Zhang K, Cui B. Optogenetic control of intracellular signaling pathways. Trends Biotechnol. 2015;33(2):92–100. doi: 10.1016/j.tibtech.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Polstein LR, Gersbach CA. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nature Chemical Biology. 2015;11(3):198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lim ST, et al. A novel targeting signal for proximal clustering of the Kv2. 1 K channel in hippocampal neurons. Neuron. 2000;25(2):385–397. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- 72.Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465(7301):1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tilley DC, et al. Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc Natl Acad Sci U S A. 2014;111(44):E4789–96. doi: 10.1073/pnas.1406876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmidt-Dannert C, Arnold FH. Directed evolution of industrial enzymes. Trends Biotechnol. 1999;17(4):135–136. doi: 10.1016/s0167-7799(98)01283-9. [DOI] [PubMed] [Google Scholar]
- 75.Huang C, et al. Characterization of voltage-gated sodium-channel blockers by electrical stimulation and fluorescence detection of membrane potential. Nat Biotechnol. 2006;24(4):439–446. doi: 10.1038/nbt1194. [DOI] [PubMed] [Google Scholar]
- 76.Sills GJ. The mechanisms of action of gabapentin and pregabalin. Current Opinion in Pharmacology. 2006;6(1):108–113. doi: 10.1016/j.coph.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 77.Lynch BA, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101(26):9861–9866. doi: 10.1073/pnas.0308208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karila P, Blom C. The development of cell-based assays for pain drug discovery. Drug Discovery World. 2015:40–44. [Google Scholar]
- 79.DiPilato LM, Zhang J. The role of membrane microdomains in shaping β2-adrenergic receptor-mediated cAMP dynamics. Molecular Biosystems. 2009;5(8):832–837. doi: 10.1039/b823243a. [DOI] [PubMed] [Google Scholar]
- 80.Tantama M, et al. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4:e2550. doi: 10.1038/ncomms3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lou S, et al. Genetically targeted all-optical electrophysiology with a transgenic Cre-dependent Optopatch mouse. J Neurosci. 2016;36(43):11059–11073. doi: 10.1523/JNEUROSCI.1582-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kiskinis E, et al. Alterations in neuronal excitability in ALS probed through optical electrophysiology studies of human iPSC-derived motor neurons. 2016 Submitted. [Google Scholar]
- 83.Jackson RE, Burrone J. Visualizing Presynaptic Calcium Dynamics and Vesicle Fusion with a Single Genetically Encoded Reporter at Individual Synapses. Frontiers in Synaptic Neuroscience. 2016;8 doi: 10.3389/fnsyn.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miesenbock G, et al. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 85.Marvin JS, et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods. 2013;10(2):162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ohkura M, et al. Optogenetics. Springer; 2015. Optogenetic Manipulation and Probing; pp. 133–147. [Google Scholar]
- 87.Barral J, Reyes AD. Synaptic scaling rule preserves excitatory-inhibitory balance and salient neuronal network dynamics. Nat Neurosci. 2016;19(12):1690–1696. doi: 10.1038/nn.4415. [DOI] [PubMed] [Google Scholar]
- 88.Millet LJ, et al. Microfluidic devices for culturing primary mammalian neurons at low densities. Lab on a Chip. 2007;7(8):987–994. doi: 10.1039/b705266a. [DOI] [PubMed] [Google Scholar]
- 89.Sofroniew NJ, et al. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. BioRxiv. 2016;055947 doi: 10.7554/eLife.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tsai PS, et al. Ultra-large field-of-view two-photon microscopy. Optics Express. 2015;23(11):13833–13847. doi: 10.1364/OE.23.013833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Magee JC, Cook EP. Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci. 2000;3(9):895–903. doi: 10.1038/78800. [DOI] [PubMed] [Google Scholar]
- 92.Bernstein JG, Boyden ES. Optogenetic tools for analyzing the neural circuits of behavior. Trends Cogn Sci. 2011;15:592–600. doi: 10.1016/j.tics.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tye KM, Deisseroth K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat Rev Neurosci. 2012;13(4):251–266. doi: 10.1038/nrn3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Johansen JP, et al. Controlling the elements: an optogenetic approach to understanding the neural circuits of fear. Biol Psychiatry. 2012;71(12):1053–1060. doi: 10.1016/j.biopsych.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin D, et al. Functional identification of an aggression locus in the mouse hypothalamus. Nature. 2011;470(7333):221–226. doi: 10.1038/nature09736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aponte Y, et al. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14(3):351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Daou I, et al. Remote optogenetic activation and sensitization of pain pathways in freely moving mice. J Neurosci. 2013;33(47):18631–18640. doi: 10.1523/JNEUROSCI.2424-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iyer SM, et al. Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat Biotechnol. 2014;32(3):274–278. doi: 10.1038/nbt.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsunematsu T, et al. Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J Neurosci. 2011;31(29):10529–10539. doi: 10.1523/JNEUROSCI.0784-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hamel EJ, et al. Cellular Level Brain Imaging in Behaving Mammals: An Engineering Approach. Neuron. 2015;86(1):140–159. doi: 10.1016/j.neuron.2015.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim TH, et al. Long-Term Optical Access to an Estimated One Million Neurons in the Live Mouse Cortex. Cell Reports. 2016;17(12):3385–3394. doi: 10.1016/j.celrep.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jeong J, et al. Wireless optofluidic systems for programmable in vivo pharmacology and optogenetics. Cell. 2015;162(3):662–674. doi: 10.1016/j.cell.2015.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chi KR. Revolution dawning in cardiotoxicity testing. Nature Reviews Drug Discovery. 2013;12(8):565–567. doi: 10.1038/nrd4083. [DOI] [PubMed] [Google Scholar]
- 104.Colatsky T, et al. The comprehensive in vitro proarrhythmia assay (CiPA) initiative—Update on progress. J Pharmacol Toxicol Methods. 2016;81:15–20. doi: 10.1016/j.vascn.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 105.Klimas A, et al. OptoDyCE as an automated system for high-throughput all-optical dynamic cardiac electrophysiology. Nature Communications. 2016;7 doi: 10.1038/ncomms11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cerignoli F, et al. High throughput measurement of Ca2+ dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. J Pharmacol Toxicol Methods. 2012;66:246–256. doi: 10.1016/j.vascn.2012.08.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dempsey GT, et al. Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. J Pharmacol Toxicol Methods. 2016 doi: 10.1016/j.vascn.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 108.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hou JH, et al. Temporal dynamics of microbial rhodopsin fluorescence reports absolute membrane voltage. Biophys J. 2014;106(3):639–648. doi: 10.1016/j.bpj.2013.11.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brinks D, et al. Two-photon fluorescence lifetime imaging microscopy (2P-FLIM) of genetically encoded voltage indicators as a probe of absolute membrane voltage. Biophys J. 2015;109:914–921. doi: 10.1016/j.bpj.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thestrup T, et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nature Methods. 2014;11(2):175–182. doi: 10.1038/nmeth.2773. [DOI] [PubMed] [Google Scholar]
- 112.Jensen TP, et al. Monitoring single-synapse glutamate release and presynaptic calcium concentration in organised brain tissue. Cell Calcium. 2017 doi: 10.1016/j.ceca.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 113.Clements IP, et al. Optogenetic stimulation of multiwell MEA plates for neural and cardiac applications. 2016:96902C–96902C-10. [Google Scholar]