

Summary
Objective
To search for new therapies aimed at ameliorating the neurologic symptoms and epilepsy developing in patients with Lafora disease.
Methods
Lafora disease is caused by loss-of-function mutations in either the EPM2A or EPM2B genes. Epm2a−/− and Epm2b−/− mice display neurologic and behavioral abnormalities similar to those found in patients. Selenium is a potent antioxidant and its deficiency has been related to the development of certain diseases, including epilepsy. In this study, we investigated whether sodium selenate treatment improved the neurologic alterations and the hyperexcitability present in the Epm2b−/− mouse model.
Results
Sodium selenate ameliorates some of the motor and memory deficits and the sensitivity observed with pentylenetetrazol (PTZ) treatments in Epm2b−/− mice. Neuronal degeneration and gliosis were also diminished after sodium selenate treatment.
Significance
Sodium selenate could be beneficial for ameliorating some symptoms that present in patients with Lafora disease.
Keywords: Lafora bodies, Epilepsy, Oxidative stress, Epm2b−/− mouse, Sodium selenate
Lafora disease (OMIM 254780; ORPHA501) (LD) is a rare autosomal recessive form of progressive myoclonus epilepsy that presents in adolescence with absence, visual, myoclonic, and tonic–clonic seizures or cognitive decline. Myoclonus and seizures respond temporarily to treatment, although they gradually become untreatable. Rapid neurologic deterioration including ataxia, dementia, dysarthria, amaurosis, and respiratory failure leads to death within 5–10 years of disease onset.1,2 The principal pathologic feature of LD is the presence of periodic acid–Schiff (PAS)-positive intracellular inclusions of polyglucosans, known as Lafora bodies (LBs), which accumulate in brain, liver, heart, and other tissues.1,3–5 LD is caused by recessive mutations either in the EPM2A gene encoding a dual-specificity phosphatase known as laforin (OMIM 607566)6–9 or in the EPM2B gene encoding malin (OMIM 608072), an E3 ubiquitin ligase.10,11 At present, no therapy exists for this disease.
Different mouse models of LD have been generated by disrupting either the Epm2a12 or the Epm2b gene.13–15 Both Epm2a−/− and Epm2b−/− mice display many of the neurologic and behavioral abnormalities found in LD patients, including neuronal degeneration and the development of LBs in different organs.12,15 Absence of laforin and malin in LD models also produces reactive astrogliosis.14,16 Moreover, these models show altered motor activity, impaired motor coordination, and episodic memory deficits.17 LD mice models present different degrees of spontaneous epileptic activity such as spontaneous single spikes, polyspikes, and spike-wave and poly spike-wave complexes correlating with myoclonic jerks. Occasionally Epm2a−/− mice, but not Epm2b−/− mice, also show spontaneous tonic–clonic seizures as recorded using electroencephalography (EEG) analysis.17 Both, laforin and malin-deficient mice present an increased sensitivity to the chemoconvulsant pentylenetetrazol,18 an antagonist of the type A γ-aminobutyric acid (GABAA) receptor.19 The presence of hyperphosphorylated tau aggregates has also been reported in the brain of Epm2a−/− mice.20,21 At the cellular level, Emp2a−/− and Epm2b−/− mice present impaired autophagy and defects in the ubiquitin–proteasome system resulting in alterations in the machinery responsible for protein clearance. 15,22,23 An increase in oxidative stress and an impaired antioxidant response in the brain has been described in both mouse models.24
Selenium is an essential trace element with antioxidant and antiinflammatory effects, and it has been implicated in the production of active thyroid hormone and in psychological functioning.25,26 It regulates the expression and activity of selenoenzymes, and thus provides protection from oxidative stress–induced cell damaged, which otherwise would lead to neuropsychiatric diseases and disorders like cerebrovascular disease, Alzheimer’s disease (AD), Parkinson’s disease, obsessive compulsive disorders, stroke, and epilepsy. 27 Low selenium status has been associated with cognitive decline in patients with AD, and with an increased propensity to seizures in humans and laboratory animals. 27,28 Sodium selenate regulates the phosphorylation of some key proteins involved in oxidative stress, energy metabolism, and protein degradation in mouse models of different pathologies.29 Moreover, sodium selenate reduces tau hyperphosphorylation by activating PP2A, halts the formation of neurofibrillary tangles, and prevents neurodegeneration, thereby improving memory and motor performance in mouse models of tauopathies.30,31 By dephosphorylating tau, sodium selenate also suppresses the epileptic seizures induced by various epileptogenic substances in rodent models, 32,33 and attenuates brain damage, improving behavioral outcomes in rat models of traumatic brain injury.34 Another oxidized form of selenium, sodium selenite, induces an anticonvulsant effect in PTZ-induced seizures in mice.27
In order to find a new treatment that could improve the devastating neurologic alterations present in patients with LD, we analyzed the effects of sodium selenate in Epm2b−/− mice on motor behavior, memory function, and seizure susceptibility.
Methods
Animals
Malin-deficient mice were used for our study. Epm2b−/− mutant mice were generated by deletion of the single exon encoding malin, as described in Criado et al.15 Four groups of 12–25 animals were used per condition: wild-type mice; Epm2b−/− mice; Epm2b−/− mice with selenate treatment for 4 weeks; and Epm2b−/− mice with selenate treatment for 10 weeks.
The mouse colonies were bred at the IIS-Jiménez Díaz Foundation Animal Facility, and were maintained in separate cages, on a 12:12-h light/dark cycle under constant temperature (23°C), and with access to food and water ad libitum. The experiments were conducted in accordance with the Declaration of Helsinki principles and the guidelines of the Institutional Animal Care and Use Committee, and were approved by the IIS-Fundación Jiménez Díaz Ethical Review Board.
Sodium selenate treatment
Treatment with sodium selenate was performed in Epm2b−/− mice at 9 months of age. Sodium selenate (Sigma Chemicals, St. Louis, MO, U.S.A.) was dispensed at a dose of 1.2 mg/100 ml in water ad libitum31 during 4 or 10 weeks, and animals were then subjected to the tests detailed below. After 1 month of analysis, five animals from each group of treatment were anesthetized and transcardially perfused with 4% phosphate-buffered paraformaldehyde for histochemical analysis. Brains were recovered from mice at 11 months of age for the group with 4 weeks of treatment, and at 12 months of age for the group with 10 weeks of treatment, and processed for histologic analysis.
Motor coordination and balance
An accelerating rotarod (Panlab/Harvard) was used to test neuromuscular abnormalities and resistance to fatigue. The ability of the mice to remain on the rod was measured according to the time elapsed until they fell (latency time) after 2 days of training.17
Tail suspension test (TST)
The TST was used to evaluate dyskinesia and abnormal hindlimb clasping response of mice when subjected to tests of vertical suspension from the tail. Each mouse was vertically suspended from the middle of the tail for 30 s and their responses were scored using a behavioral scale ranging from “0,” when the hindlimbs were completely extended (normal wild-type posture); “1,” when one or both hindlimbs were intermittently extended and bent; and “2,” when both hindlimbs were completely bent and folded into the abdomen. The number of animals falling under each behavioral-scale value was represented as a percentage of the total.
Spontaneous activity
A computerized actimeter (PanLab/Harvard) was used to study the spontaneous motor activity of the mice. The number of times that the animal crossed the infrared light beams located at the actimeter cage was counted for spontaneous surface displacement, and for rearing and stereotyped movements with the Sedacom 1.4 sofware (Panlab/Harvard) at 15-, 30-, 45-, and 60-min intervals.17 The arena of the actimeter was divided into two zones—the central and the surrounding area—and the percentage of time spent in the central zone was also quantified (Actitrack 2.7.13 Panlab/Harvard).
Object recognition task (ORT)
We used the ORT to measure episodic memory retention. Briefly, two objects of similar texture, color, and size (Lego toys) were placed in the center of a black wood chamber.17 The time employed in exploring the two equivalent objects (tA and tB, objects A and B) was measured for each mouse in the sample familiarization phase. The test session was performed 2 h later, and the times (tA and tC) that animal spent exploring two objects, a familiar object (A), and a novel object (C), were recorded. A discrimination index (DI) was calculated as the ratio of the difference between the exploration times of the new object (tC) and the familiar object (tA) with respect to the total (tT) exploration time, and was expressed as the following equation: DI = tC−tA/tT.
PTZ treatment
PTZ (Sigma Chemicals) was administered intraperitoneally as a single injection at 30 and 50 mg/kg.35 The percentage of mice showing PTZ-induced myoclonic jerks and generalized seizures was monitored over a period of 45 min. In addition to measuring epileptic activity, PTZ-induced lethality was also analyzed for each drug dose.
Histology and Immunohistochemistry
The animals were anesthetized and perfused transcardially with 4% phosphate-buffered paraformaldehyde. Brains were removed and embedded in paraffin. Brain blocks were then sectioned in serial arrays of 3-μm-thick sections and processed for PAS staining, as described previously. 36 For immunohistochemistry, slices were rehydrated in graded alcohols, incubated in boiling 0.1 M sodium citrate buffer pH 6.0, and subjected to two cycles of microwave irradiation for 2 min each for antigen retrieval. Sections were incubated with glial fibrillary acidic protein (GFAP) antibody (Millipore, Temecula, CA, U.S.A.), a neuronal specific marker (NeuN; Millipore) and AT8, an antibody directed against phospho-PHF-tau pSer202/Thr205 (Thermo Fisher Scientific Inc., Waltham, MA, U.S.A.) and stained using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, U.S.A.). Immunoreactivity was developed with diaminobenzidine and H2O2 (DakoCytomation, CA, U.S.A.).
Statistical analysis
Values are given as means − standard error of means (SEMs) or percentages. Differences between groups were analyzed by one-way analysis of variance (ANOVA) or chi-square test. Statistical significance was considered to be reached at *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.05; ##p < 0.01; ###p < 0.001 (Graph-PadPrism2.0) (n = 12–25).
Results
Motor coordination and abnormal gait improvement after treatment with sodium selenate in malin-deficient mice
Malin-deficient mice showed alterations in motor coordination when they were analyzed using a rotarod, displaying a significantly lower mean latency than age-matched controls before falling from the rod15 (Fig. 1A). Here, in malin mutant mice, we studied the effects of the administration of sodium selenate on their motor coordination. After 4 weeks of treatment, sodium selenate slightly increased the time that Epm2b−/−mice stood on the rod, causing a significant increase in this time after 10 weeks of treatment, such that the differences with control mice disappeared (Fig. 1A).
Figure 1.

Analysis of motor coordination and abnormal postures in Epm2b−/− mutant mice after treatment with sodium selenate. (A) Rotarod-based analysis of motor coordination in controls (n = 23) and Epm2b−/− mice (n = 24), in Epm2b−/− mice with selenate treatment for 4 weeks (n = 22), and in Epm2b−/− mice with selenate treatment over 10 weeks (n = 15) (trials 1–4). The mean latencies (time to fall from the rotarod) were significantly lower for Epm2b−/− mice than for controls. Sodium selenate treatment for 4 weeks in Epm2b−/− mice slightly increased the latency period, whereas a longer treatment significantly improved performance. Student’s t-test was performed for statistical evaluation. (B) Percentage of animals showing normal posture (0), partially altered (1) or high abnormal (2) stereotypical clasping of the hind limbs upon tail suspension. The frequent hind-limb clasping of Epm2b−/− mice improved progressively with sodium selenate treatment (n = 23 for wild-type, n = 24 for ML, n = 20 for ML + Sel [4 weeks], and n = 15 for ML + Sel [10 weeks] mouse groups). A chi-square test was performed for statistical analysis. *Indicates p < 0.05, **indicates p < 0.01, and ***indicates p < 0.001 when control mice were compared to Epm2b−/− mice; #indicates p < 0.05 and ###indicates p < 0.001 when Epm2b−/− mice were compared to Epm2b−/− mice with selenate treatment for 10 weeks. ML, malin-deficient mouse; Sel, sodium selenate.
Lack of malin in mice also produced hind-limb clasping and abnormal gait response in the TST15 (Fig. 1B). Although the 4-week treatment with sodium selenate significantly improved the abnormal postures of mice, a longer treatment with this drug further ameliorated these alterations (Fig. 1B).
Spontaneous locomotor activity alterations and anxiety-related behavior of malin-deficient mice after treatment with sodium selenate
Analysis of accumulated activity, rearing, and stereotyped movements in a computerized actimeter showed the presence of motor alterations in malin-deficient mice15 (Fig. 2A–C). Here, we also measured the permanence of Epm2b−/− mice in the central zone of the arena to analyze exploration behavior (Fig. 2D). We observed that Epm2b−/− mice spent a lower percentage of time in the central zone of the arena than in the surrounding areas, indicating anxiety-like behavior (Fig. 2D). After a short, 4-week treatment with sodium selenate, alterations of spontaneous accumulated (Fig. 2A), rearing (Fig. 2B), and stereotyped (Fig. 2C) movements, as well as anxiety-like behavior (Fig. 2D) significantly improved. Unexpectedly, sodium selenate treatment over 10 weeks reversed this effect, and Epm2b−/− mice presented similar altered spontaneous activity than untreated malin-deficient mice (Fig. 2A–C). This longer administration of sodium selenate improved exploration and anxiety in malin-deficient mice but to a lesser extent than the shorter treatment (Fig. 2D).
Figure 2.

Motor and mood abnormalities in Epm2b−/− mice after sodium selenate treatments. Patterns of accumulated motor activity in controls, Epm2b−/− mice, and Epm2b−/− mice with selenate treatment for 4 and 10 weeks, measured as (A) accumulated, (B) rearing, and (C) stereotyped movements (n = 25 for wild type, n = 25 for ML, n = 25 for ML + Sel [4 weeks], and n = 15 for ML + Sel [10 weeks] mouse groups). Quantitative data represent mean + SEM. Student’s t-test was performed for statistical evaluation. (D) Percentage of time spent by all the four groups in the central zone of arena as a measure of anxiety-like behavior (n = 12 for wild type, n = 12 for ML, n = 12 for ML + Sel [4 weeks], and n = 12 for ML + Sel [10 weeks] mouse groups). Statistical analysis was performed with one-way ANOVA or the chi-square test. *p < 0.05; **p < 0.01; ***p < 0.001. Note that after 4 weeks of treatment with selenate, motor performance and anxiety improved, whereas a longer treatment eliminated this advance in motor performance and reduced mood improvement. ML, malin-deficient mouse; Sel, sodium selenate.
Treatments with sodium selenate improved the episodic memory performance of malin-deficient mice
The effect of sodium selenate on episodic memory in Epm2b−/− mice was studied by measuring object recognition learning in the ORT. We previously reported that deletion of the malin gene in Epm2b−/− mice produced a deficit in episodic memory15 (Fig. 3). Herein we show that treatment with selenate for 4 weeks produced a significant improvement in memory performance in malin-deficient mice, while a longer treatment completely abrogated these memory deficits (Fig. 3).
Figure 3.
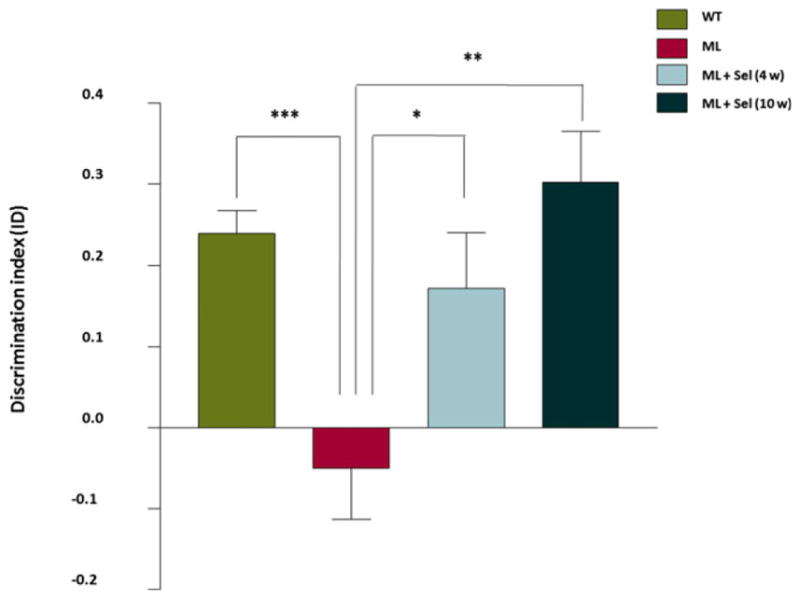
Assessment of memory performance with the object recognition task. The ORT was performed in control mice (n = 20), Epm2b−/− mice (n = 19), and Epm2b−/− mice with sodium selenate treatment for 4 (n = 20) and 10 (n = 12) weeks. The discrimination index (DI) was calculated as the ratio of the difference between the exploration time of the new (tC) and the familiar object (tA), and the total (tT = tA + tC) exploration time (DI = tC−tA/tT). The DI of Epm2b−/− mice was poor, indicating deficits in the retention of episodic memory. Treatments with sodium selenate significantly and gradually improved the memory performance of malin-deficient mice. Values are expressed as mean + SEM. One-way ANOVA was performed.*p < 0.05; **p < 0.01. ML, malin-deficient mouse; Sel, sodium selenate.
PTZ-induced myoclonus, generalized tonic–clonic seizures, and lethality in mice lacking malin are reduced after treatment with sodium selenate
Following injection of a convulsive dose of PTZ (50 mg/kg), mice displayed intervals of immobility and convulsive activity, which included hyperactivity, twitching, and hyperextension of the limbs that at times progressed to generalized tonic–clonic seizures, and occasionally to death. We previously recorded the percentages of PTZ-injected Epm2b−/− mice that presented muscular jerks, generalized tonic–clonic seizures, and lethality, and reported that lack of malin in Epm2b−/− mice increased the sensitivity to PTZ.18 We show here that sodium selenate treatments decreased the hypersensitivity of malin-deficient mice to PTZ (Fig. 4). Thus, the percentages of Epm2b−/− mice showing myoclonic jerks induced by PTZ at subconvulsive (Fig. 4A) and convulsive doses (Fig. 4B) (30 and 50 mg/kg, respectively) were slightly reduced after treatment with sodium selenate. Moreover, treatment with selenate for 4 weeks also decreased the percentage of mice showing PTZ-induced generalized seizures and lethality even under the control levels (Fig. 4C,D), whereas a longer treatment with selenate for 10 weeks completely eliminated generalized seizures and lethality in malin-deficient mice (Fig. 4C, D).
Figure 4.

Sensitivity of Epm2b−/− mice to the chemoconvulsant agent PTZ after treatment with sodium selenate. Percentage of Epm2b−/− mice with myoclonic jerks after intraperitoneal injection of PTZ at doses of (A) 30 mg/kg and (B) 50 mg/kg significantly decreased after treatment with selenate for 10 weeks. Sodium selenate administration also reduced PTZ-induced (C) generalized seizures, and (D) lethality after 4 week of treatment, whereas at 10 weeks, generalized seizures and lethality disappeared (n = 25 for wild-type, n = 25 for ML, n = 15 for ML + Sel [4 weeks], and n = 12 for ML + Sel [10 weeks] mouse groups). Chi-square was performed for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001. ML, malin-deficient mouse; Sel, sodium selenate.
Effects of treatment with sodium selenate on gliosis, neuronal degeneration, and accumulation of PAS-positive LBs
Absence of laforin and malin in LD models also produces gliosis.14,16 We estimated the amount of GFAP-positive cells in the dentate gyrus of malin-deficient mice after 4 and 10 weeks of sodium selenate treatment. After 4 weeks of treatment, the number of reactive astrocytes was slightly decreased, whereas selenate treatment for 10 weeks significantly reduced the gliosis produced by the lack of malin (Fig. 5A,D). In addition, we analyzed the number of neuronal nuclei (NeuN) staining and the presence of PAS-positive LBs in equivalent sections of the hippocampus of malin-deficient mice after 4 and 10 weeks of treatment with sodium selenate. Treatment with sodium selenate reduced the neuronal loss observed in Epm2b−/− mice, as evaluated by quantifying NeuN staining (Fig. 5B,E). The number of LBs in CA1 of malin null mice did not change significantly after sodium selenate treatments (Fig. 5C,F).
Figure 5.

Neuronal loss, gliosis and Lafora bodies in the hippocampus of Epm2b−/− mice after treatment with sodium selenate. Images of representative immunostained sections from dentate gyrus with GFAP (A) and from CA1 with NeuN (B) and PAS staining (C) are shown from wild-type, Epm2b−/−, and Epm2b−/− after selenate treatment for 4 and 10 weeks (scale bars 25 μm). Graphs show the quantification of GFAP-positive cells (D), NeuN-positive cells (E), and PAS+ inclusions (F) in wild-type, Epm2b−/−, and Epm2b−/− after sodium selenate treatment for 4 or 10 weeks. Visual blinded quantification was performed in the dentate gyrus and CA1 regions of the hippocampus. Both hemispheres of the brain were quantified in three mice of each group. One-way ANOVA was performed for statistical analysis. *p < 0.05; **p < 0.01; ***p < 0.001. ML, malin-deficient mouse; Sel, sodium selenate.
Discussion
In this study, we used the Epm2b−/− malin-deficient mouse model of LD to search for new therapies aimed at ameliorating the neurologic symptoms and epilepsy developing in patients with LD. We report here the effects of sodium selenate, an oxidized form of selenium that presents antioxidant and anticonvulsant properties,27,29 on the neurologic alterations produced by the deficiency of the malin protein expression in the Epm2b−/− model at 11 months of age. In carrying out this experiment, we found that sodium selenate ameliorates some of these characteristic symptoms. Thus, motor coordination in the rotarod gradually improves after selenate treatment, as was reported previously for certain models of tauopathies,30,31 rendering Epm2b−/− mice indistinguishable from controls after 10 weeks of treatment. Sodium selenate also gradually improved the episodic memory performance of malin null mice, such that after 10 weeks of treatment these deficits disappeared and mutant mice reached the control levels. An improvement in memory performance was also shown previously in mouse models of AD after treatment with sodium selenate.31 The abnormal postures and dyskinesia that characterize malin-deficient mice, are also reduced after selenate treatment. Moreover, sodium selenate reduces the sensitivity of Epm2b−/− mice to PTZ, diminishing PTZ-induced myoclonus, generalized tonic–clonic seizures, and lethality. This amelioration of the hyperexcitability after treatment with sodium selenate was also described previously for other mouse models of epilepsy.32 A faint improvement in spontaneous motor activity of malin null mice was shown after a short treatment with sodium selenate, whereas a longer treatment gradually worsened this behavior. This effect was also observed when the time spent in the central zone of the arena was measured. Thus, a short selenate treatment improved the anxiety-like behavior of Epm2b−/− mice, although a longer treatment lessened this favorable effect. Astrogliosis and neuronal cell degeneration was also diminished after sodium selenate treatment. The positive effects of sodium selenate on these neurologic alterations of malin-deficient mice may be attributed to the reduction of oxidative stress and to the amelioration of neuronal cell death and gliosis rather than to an action on abnormal glycogen aggregation, since no effects on the amount of LBs were observed after treatment. As noted earlier, sodium selenate was shown to improve neurologic symptoms and epilepsy in different rodent models throughout the activation of PP2A and dephosphorylation of tau, thus preventing the appearance of neurodegenerative processes and seizures.30–32 Previous reports on laforin-deficient mice have shown the presence of hyperphosphorylated tau aggregates in different regions of the brain.20,21 However, we did not observe hyperphosphorylated tau aggregates in the brain of the malin null mice (data not shown). The lack of tau neuropathology in our LD model indicates that the positive effects of sodium selenate are not due to the activation of PP2A and prevention of tau hyperphosphorylation. Further experiments must be performed to assess the glycogen response to sodium selenate and the mechanisms of action involved in the neurological improvement of our malin-deficient model.
Several treatment-related adverse events have been reported in sodium selenate clinical trials, for example, nausea, diarrhea, fatigue, muscle spasms, alopecia, and nail disorders. 37 Long-term exposure to selenium also produced fatigue and other symptoms.38 No treatment-related events were observed during sodium selenate administration to malin-deficient mice, although the gradual decline of the initial improvement in motor and anxiety-related behavior during treatment may be due to side effects of selenium. A novel sodium selenate clinical trial has reported to be safe and well tolerated in patients with AD, at doses up to 30 mg per day for 24 weeks.39
A previous report from Berthier et al. and our group40 showed that administration of certain chemicals ameliorated some neurologic alterations found in our Epm2b−/− malin-deficient mouse model. Thus, both the chaperone 4-phenylbutyric acid (4-PBA) and the neuroprotector metformin produced beneficial effects on the performance of malin-deficient mice in different functional assays. 4-PBA and metformin are already approved for clinical use in different neurologic pathologies, and selenium is a common ingredient over-the-counter. Therefore, assays with sodium selenate in combination with other substances should be performed in LD animal models, and further tested in clinical trials for their ability to ameliorate some symptoms that present in patients with Lafora disease.
Key Points.
The Epm2b−/− mouse model of Lafora disease displays neuronal degeneration, Lafora bodies, neurologic alterations, and increased seizure susceptibility
Sodium selenate, a potent antioxidant trace element, diminishes gliosis and neuronal degeneration in malin-deficient mice
Motor, memory, and anxiety-related alterations improved after sodium selenate treatment
Sodium selenate reduces seizure sensitivity of Epm2b−/− mice to the convulsant agent PTZ
Treatment with sodium selenate may be beneficial for ameliorating some symptoms of Lafora disease
Acknowledgments
We thank Nuria Cabrero and Maximilià Bautista for technical support, Oliver Shaw for editing assistance, Drs. Alberto Rábano and José Ramón Fortes, Department of Anatomical Pathology and the Animal Facility of the IIS-Jiménez Díaz Foundation. This work was supported by grants from the Fondo de Investigacion Sanitaria/Carlos III Institute of Health (PI13/00865) from the Spanish Ministry of Health (FEDER), and from the Spanish Ministry of Economy (SAF2014-59594-R to JMS). GSE is supported by a fellowship from the Fundación Conchita Rábago.
Biography
Gentzane Sánchez-Elexpuru is a doctoral student at the IIS-Jiménez Díaz Foundation in Madrid, Spain.
Footnotes
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Berkovic SF, Andermann F, Carpenter S, et al. Progressive myoclonus epilepsies: specific causes and diagnosis. N Engl J Med. 1986;315:296–305. doi: 10.1056/NEJM198607313150506. [DOI] [PubMed] [Google Scholar]
- 2.Van Heycop Ten Ham MW, De Jager H. Progressive myoclonus epilepsy with Lafora bodies. Clinical-pathological features. Epilepsia. 1963;4:95–119. doi: 10.1111/j.1528-1157.1963.tb05214.x. [DOI] [PubMed] [Google Scholar]
- 3.Lafora GR. The presence of amyloid bodies in the protoplasm of the ganglion cells: a contribution to the study of the amyloid substance in the nervous system. Bull Gov Hosp Insane. 1911;3:83–92. [Google Scholar]
- 4.Harriman DG, Millar JH, Stevenson AC. Progressive familial myoclonic epilepsy in three families: its clinical features and pathological basis. Brain. 1955;78:325–349. doi: 10.1093/brain/78.3.325. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter S, Karpati G. Ultrastructural findings in Lafora disease. Ann Neurol. 1981;10:63–64. doi: 10.1002/ana.410100116. [DOI] [PubMed] [Google Scholar]
- 6.Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 7.Serratosa JM, Gomez-Garre P, Gallardo ME, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum Mol Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. [DOI] [PubMed] [Google Scholar]
- 8.Ganesh S, Agarwala KL, Ueda K, et al. Laforin, defective in the progressive myoclonus epilepsy of Lafora type, is a dual-specificity phosphatase associated with polyribosomes. Hum Mol Genet. 2000;9:2251–2261. doi: 10.1093/oxfordjournals.hmg.a018916. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Stuckey JA, Wishart MJ, et al. A unique carbohydrate binding domain targets the Lafora disease phosphatase to glycogen. J Biol Chem. 2002;277:2377–2380. doi: 10.1074/jbc.C100686200. [DOI] [PubMed] [Google Scholar]
- 10.Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 11.Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ganesh S, Delgado-Escueta AV, Sakamoto T, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11:1251–1262. doi: 10.1093/hmg/11.11.1251. [DOI] [PubMed] [Google Scholar]
- 13.DePaoli-Roach AA, Tagliabracci VS, Segvich DM, et al. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to Lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285:25372–25381. doi: 10.1074/jbc.M110.148668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valles-Ortega J, Duran J, Garcia-Rocha M, et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3:667–681. doi: 10.1002/emmm.201100174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Criado O, Aguado C, Gayarre J, et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet. 2012;21:1521–1533. doi: 10.1093/hmg/ddr590. [DOI] [PubMed] [Google Scholar]
- 16.Turnbull J, DePaoli-Roach AA, Zhao X, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 2011;7:e1002037. doi: 10.1371/journal.pgen.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Cabrero AM, Marinas A, Guerrero R, et al. Laforin and malin deletions in mice produce similar neurologic impairments. J Neuropathol Exp Neurol. 2012;71:413–421. doi: 10.1097/NEN.0b013e318253350f. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Cabrero AM, Sanchez-Elexpuru G, Serratosa JM, et al. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front Neurosci. 2014;8:291. doi: 10.3389/fnins.2014.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone WE. Convulsant actions of tetrazole derivatives. Pharmacology. 1970;3:367–370. doi: 10.1159/000136093. [DOI] [PubMed] [Google Scholar]
- 20.Machado-Salas J, Avila-Costa MR, Guevara P, et al. Ontogeny of Lafora bodies and neurocytoskeleton changes in Laforin-deficient mice. Exp Neurol. 2012;236:131–140. doi: 10.1016/j.expneurol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puri R, Suzuki T, Yamakawa K, et al. Hyperphosphorylation and aggregation of Tau in laforin-deficient mice, an animal model for Lafora disease. J Biol Chem. 2009;284:22657–22663. doi: 10.1074/jbc.M109.009688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puri R, Suzuki T, Yamakawa K, et al. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum Mol Genet. 2012;21:175–184. doi: 10.1093/hmg/ddr452. [DOI] [PubMed] [Google Scholar]
- 23.Aguado C, Sarkar S, Korolchuk VI, et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 2010;19:2867–2876. doi: 10.1093/hmg/ddq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roma-Mateo C, Aguado C, Garcia-Gimenez JL, et al. Increased oxidative stress and impaired antioxidant response in Lafora disease. Mol Neurobiol. 2015;51:932–946. doi: 10.1007/s12035-014-8747-0. [DOI] [PubMed] [Google Scholar]
- 25.Benton D. Selenium intake, mood and other aspects of psychological functioning. Nutr Neurosci. 2002;5:363–374. doi: 10.1080/1028415021000055925. [DOI] [PubMed] [Google Scholar]
- 26.Brenneisen P, Steinbrenner H, Sies H. Selenium, oxidative stress, and health aspects. Mol Aspects Med. 2005;26:256–267. doi: 10.1016/j.mam.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Rehni AK, Singh TG. Selenium induced anticonvulsant effect: a potential role of prostaglandin E(1) receptor activation linked mechanism. J Trace Elem Med Biol. 2013;27:31–39. doi: 10.1016/j.jtemb.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 28.Cardoso BR, Ong TP, Jacob-Filho W, et al. Nutritional status of selenium in Alzheimer’s disease patients. Br J Nutr. 2010;103:803–806. doi: 10.1017/S0007114509992832. [DOI] [PubMed] [Google Scholar]
- 29.Chen P, Wang L, Wang Y, et al. Phosphoproteomic profiling of selenate-treated Alzheimer’s disease model cells. PLoS One. 2014;9:e113307. doi: 10.1371/journal.pone.0113307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corcoran NM, Martin D, Hutter-Paier B, et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J Clin Neurosci. 2010;17:1025–1033. doi: 10.1016/j.jocn.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 31.van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci USA. 2010;107:13888–13893. doi: 10.1073/pnas.1009038107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones NC, Nguyen T, Corcoran NM, et al. Targeting hyperphosphorylated tau with sodium selenate suppresses seizures in rodent models. Neurobiol Dis. 2012;45:897–901. doi: 10.1016/j.nbd.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Liu SJ, Zheng P, Wright DK, et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain. 2016;139:1919–1938. doi: 10.1093/brain/aww116. [DOI] [PubMed] [Google Scholar]
- 34.Shultz SR, Wright DK, Zheng P, et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138:1297–1313. doi: 10.1093/brain/awv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shitak R, Sahai AK, Hota D, et al. Anti-seizure efficacy of nimodipine in pentylenetetrazole and kainic acid combined seizure models in mice. Indian J Physiol Pharmacol. 2006;50:265–272. [PubMed] [Google Scholar]
- 36.Mitsuno S, Takahashi M, Gondo T, et al. Immunohistochemical, conventional and immunoelectron microscopical characteristics of periodic acid-Schiff-positive granules in the mouse brain. Acta Neuropathol. 1999;98:31–38. doi: 10.1007/s004010051048. [DOI] [PubMed] [Google Scholar]
- 37.Corcoran NM, Hovens CM, Michael M, et al. Open-label, phase I dose-escalation study of sodium selenate, a novel activator of PP2A, in patients with castration-resistant prostate cancer. Br J Cancer. 2010;103:462–468. doi: 10.1038/sj.bjc.6605798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holness DL, Taraschuk IG, Nethercott JR. Health status of copper refinery workers with specific reference to selenium exposure. Arch Environ Health. 1989;44:291–297. doi: 10.1080/00039896.1989.9935896. [DOI] [PubMed] [Google Scholar]
- 39.Malpas CB, Vivash L, Genc S, et al. A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. J Alzheimers Dis. 2016;54:223–232. doi: 10.3233/JAD-160544. [DOI] [PubMed] [Google Scholar]
- 40.Berthier A, Paya M, Garcia-Cabrero AM, et al. Pharmacological interventions to ameliorate neuropathological symptoms in a mouse model of Lafora disease. Mol Neurobiol. 2016;53:1296–1309. doi: 10.1007/s12035-015-9091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]