

Abstract
Acid sphingomyelinase deficiency in type B Niemann-Pick disease leads to lysosomal sphingomyelin storage, principally affecting lungs, liver, and spleen. Infused recombinant enzyme is beneficial, yet its delivery to the lungs is limited and requires higher dosing than liver and spleen, leading to potentially adverse reactions. Previous studies showed increased enzyme pulmonary uptake by nanocarriers targeted to ICAM-1, a protein overexpressed during inflammation. Here, using polystyrene and poly(lactic-co-glycolic acid) nanocarriers, we optimized lung delivery by varying enzyme dose and nanocarrier concentration, verified endocytosis and lysosomal trafficking in vivo, and evaluated delivered activity and effects. Raising the enzyme load of nanocarriers progressively increased absolute enzyme delivery to all lung, liver, and spleen, over the naked enzyme. Varying nanocarrier concentration inversely impacted lung versus liver and spleen uptake. Mouse intravital and postmortem examination verified endocytosis, transcytosis, and lysosomal trafficking using nanocarriers. Compared to naked enzyme, nanocarriers increased enzyme activity in organs and reduced lung sphingomyelin storage and macrophage infiltration. Although old mice with advanced disease showed reactivity (pulmonary leukocyte infiltration) to injections, including buffer without carriers, antibody, or enzyme, younger mice with mild disease did not. We conclude that anti-ICAM nanocarriers may result in effective lung enzyme therapy using low enzyme doses.
Keywords: type B Niemann-Pick disease, acid sphingomyelinase deficiency, enzyme replacement therapy, pulmonary effects, ICAM-1 targeting, polymer nanocarriers
Graphical Abstract
Type B Niemann-Pick disease is a pulmonary/hepatosplenic disorder caused by acid sphingomyelinase (ASM) deficiency. Intravenously infused, recombinant ASM is promising, but suboptimal lung versus hepatosplenic uptake requires high dosing, leading to adverse reactions. Muro and colleagues show enhanced mouse lung delivery and effects of ASM using ICAM-1-targeted polymer nanocarriers.
Introduction
Type B Niemann-Pick disease (NPD-B) (OMIM 607616) is an inherited lysosomal storage disorder (LSD) caused by deficiency of lysosomal acid sphingomyelinase (ASM) (EC 3.1.4.12).1, 2 Its prevalence is thought to be 0.5–1 in every 100,000 live births,1 although this may be underestimated due to under- or mis-diagnosis.1, 3 ASM is a 57- to 75-kDa enzyme (depending on post-translational modifications and proteolytic maturation),1, 3 which hydrolyzes sphingomyelin into ceramide and phosphocholine.3 Hence, its deficiency leads to excessive sphingomyelin storage within lysosomes.1, 3 However, as this compartment plays a key role in the recycling of the cell membrane, substrate accumulation also affects the plasmalemma.3 Sphingomyelin is particularly abundant in lipid raft domains and its product, ceramide, plays a central role in cell signaling.4 As such, ASM deficiency causes cellular alterations due to storage of undegraded substrates, and also structural and functional aberrancies that alter the normal cell physiology.3, 4
NPD-B affects numerous tissues, including visceral, skeletal, vascular, immune, and other systems.3, 5, 6, 7 The lungs, liver, and spleen are primary targets for intervention, as compromised pulmonary function, liver failure, and splenic rupture are causes of death in NPD-B patients.2, 3, 6, 7 Sphingomyelin accumulates in cells of the reticuloendothelial system, vascular component, and tissue-specific cells in these organs, including alveolar macrophages, Kupffer cells, epithelial and endothelial cells, vascular smooth muscle cells, hepatocytes, fibroblasts, etc.2, 3, 6, 7 Due to the main role that sphingomyelin-ceramide signaling pathways play in endothelial and immune cells, inflammation is also a hallmark in NPD-B.3 This causes increased lung permeability with cellular infiltration, accumulation of lipid-laden macrophages in alveoli and septa, and alterations in surfactant composition.3 This results in lung infections and compromised respiratory function, leading to NPD-B morbidity and mortality.5, 6
Neurological symptoms are either absent or minor in NPD-B, leading to slow, mild disease progression.2, 3, 6 This contributes to the late-onset and chronic form of NPD-B, which associates with residual ASM activity in patients.3 This is in contrast to type A NPD (NPD-A), an early-onset, severe phenotypic manifestation of ASM deficiency, characterized by rapid neurological decline and fatality within the first years of life.3, 6 Main peripheral (versus brain) effects, broadly distributed (versus specific) body targets, easily accessible compartments (the endothelium, liver, spleen, lysosomes), and slow disease progression make NPD-B a plausible target for therapeutic interventions.
Experimental studies and clinical trials support this, i.e., enzyme replacement therapy (ERT) is well suited to help manage NPD-B.8, 9, 10 ERT consists of intravenous (i.v.) infusions of recombinant ASM, a 75-kDa protein,8 which is endocytosed and trafficked to lysosomes due to the presence of mannose-6-phosphate on this enzyme and the interaction with respective receptors on cells.1, 11 ASM i.v. injections in ASM knockout (ASMKO) mice attenuate NPD-B phenotype in the liver and spleen and, to a lesser extent, the lungs.1, 8 This differential efficacy may be due to the liver and spleen being highly accessible to circulating compounds. Increasing the enzyme dose improves lung treatment, but this is limited by side effects such as enhanced cytokine levels, hepatic inflammation, and cardiovascular shock.10, 12 In safety and tolerability clinical trials, ERT is also showing great promise, yet dose-associated adverse reactions have also been observed.12, 13 Elevated levels of circulating ceramide resulting from high ASM doses may contribute to this outcome.10 Hence, lowering the administered enzyme dose may increase safety, but also compromises lung delivery, leaving room for improvement.
Enhancing lung delivery while preserving liver and spleen uptake may reduce the ASM dose required for treatment. Our prior studies showed that coupling ASM to nanocarriers (NCs) targeted to intercellular adhesion molecule 1 (ICAM-1), a protein expressed on most cell types during inflammation, may help achieve this goal.14, 15, 16, 17 Experiments in ASMKO mice have showed that anti-ICAM NCs increased ASM accumulation through the body compared to enzyme alone.14, 15, 16 In cell models, NCs shifted the enzyme uptake mechanism from a mannose-6-phosphate-dependent, clathrin-mediated route to ICAM-1-dependent endocytosis via the cell adhesion molecule (CAM) pathway, resulting in enhanced lysosomal delivery.18, 19, 20, 21, 22 Similar outcomes were observed for delivery of lysosomal enzymes deficient in other LSDs.23, 24, 25 Here, we aimed to (1) explore strategies to manipulate the ASM dose delivered to the main NPD-B target organs, (2) verify in vivo endocytosis and lysosomal delivery of anti-ICAM/ASM NCs, and (3) examine lung-specific effects of this strategy. Our findings reveal key points for future pre-clinical development of this therapeutic strategy.
Results
Modulation of the Delivered ASM Dose by Varying the Enzyme Load per Anti-ICAM NC
We first examined the biodistribution of ASM coupled to anti-ICAM NCs in circulation and main NPD-B target organs, i.e., lung, liver, and spleen (Figure 1). For our initial study, we used model polystyrene NCs, because this non-biodegradable material allows us to focus on targeting without confounding effects of polymer degradation. Prior literature14, 26 and subsequent experiments here verify this model against more clinically relevant polymer NCs. By varying the ASM concentration during NC preparation (0.5–5 μM), three anti-ICAM/ASM NC formulations were obtained, which contained an average of 25, 133, and 236 ASM molecules per NC (see Materials and Methods and Table 1). The loading efficiency, expressed as a percent of the total ASM used for NC preparation, ranged between 80% and 95% in all cases. The average number of anti-ICAM molecules was 217, 186, and 109 antibodies per NC, respectively, and that of non-specific IgG NCs in IgG/ASM NCs (at the highest ASM loading) was 110 antibodies per NC. Formulations had hydrodynamic size of 198–212 nm in diameter, polydispersity index (PDI) of 0.10–0.15, and ζ-potential between −38.3 and −39.6 mV (Table 1), in accord with previous reports.14
Figure 1.

Biodistribution of Anti-ICAM/ASM NCs Containing Different ASM Loads
Wild-type (C57BL/6) mice were i.v. injected with anti-ICAM/125I-ASM polystyrene NCs containing 0.06, 0.3, or 0.6 mg ASM/kg body weight (all at 1.8 × 1013 NCs/kg body weight). (A) The 125I content of blood samples taken 1, 15, and 30 min after injection was used to calculate the percent of the injected dose (%ID) in the circulation. (B) Similarly, the 125I content in the lungs, liver, and spleen isolated at sacrifice 30 min after injection, was used to determine the biodistribution (percent of the injected dose) in these organs. (C) The specificity index was calculated by normalizing tissue-over-blood accumulation for the targeted (anti-ICAM/ASM NCs) versus naked (ASM) enzyme, at matching ASM doses (see Materials and Methods). (D) The absolute amount of ASM delivered to these organs was calculated from their 125I-ASM content and specific radioactivity (counts per minute/milligram) of the enzyme. Data are mean ± SEM (n = 3–6 mice). *Compares formulations to the lowest ASM load; #compares highest load formulation to the intermediate ASM load (p ≤ 0.1 by Student’s t test).
Table 1.
Anti-ICAM/ASM NCs Characterization
| Formulation | Size (nm) | PDI | ζ-Potential (mV) | Anti-ICAM Molecules/NC | ASM Molecules/NC |
|---|---|---|---|---|---|
| 0.06 mg ASM/kg BW | 212 ± 6 | 0.157 ± 0.051 | −38.3 ± 0.8 | 217 ± 3 | 25 ± 6 |
| 0.3 mg ASM/kg BW | 204 ± 4 | 0.107 ± 0.008 | −38.7 ± 0.4 | 186 ± 9 | 133 ± 12 |
| 0.6 mg ASM/kg BW | 198 ± 4 | 0.128 ± 0.04 | −39.6 ± 0.8 | 109 ± 3 | 236 ± 17 |
Data are expressed as mean ± SEM. BW, body weight.
Anti-ICAM/ASM NCs were i.v. injected in wild-type mice at a concentration equivalent to 1.8 × 1013 NCs/kg of body weight and either 0.06, 0.3, or 0.6 mg ASM/kg body weight, which is within the safe range previously tested for pre-clinical and clinical studies on naked ASM.8, 10, 12, 13 Anti-ICAM/ASM NCs rapidly disappeared from the circulation: 2.3, 2.7, and 3.2 percent of the injected dose (%ID) by 30 min (Figure 1A), similarly to non-specific IgG/ASM NCs (3.4%ID by this time; not shown), whereas matching doses of naked ASM circulated longer (15.6%ID, 43.7%ID, and 41.1%ID, respectively [Figure S1A]). However, despite these differences in circulation, all anti-ICAM/ASM NCs targeted the lungs specifically (21%ID–23%ID; Figure 1B), an organ with high ICAM-1 levels,14 in contrast to IgG/ASM NCs and naked ASM (between 0.94%ID and 1.25%ID in all cases; not shown). In fact, the localization ratio (LR), which reflects the organ-over-blood ASM level, was markedly higher for anti-ICAM/ASM NCs over naked ASM (Figure S1B). Therefore, the specificity index (the LR ratio between the targeted and naked enzyme), showed between 137- and 325-fold increase in lung targeting of anti-ICAM/ASM NCs (Figure 1C). Anti-ICAM NCs bearing increasing ASM doses resulted in increasing amounts of ASM reaching the lungs (5.0, 12.7, and 27.8 μg of ASM per lung gram of lung; Figure 1D). Lung targeting did not preclude from liver and spleen accumulation, also required for this application, regardless of the enzyme dose, as shown by the percent injected dose of anti-ICAM/ASM NCs in these organs (43%ID–51%ID in liver and 3%ID–5%ID in spleen; Figure 1B) and the comparative LR versus naked ASM (Figure S1B). As a result, NCs enhanced ASM specificity index in the liver by 21- to 40-fold over naked ASM and by 44- to 92-fold in the spleen (Figure 1C), resulting in 0.6–6.4 μg of ASM per gram of liver and 0.7–11.1 μg of ASM per gram of spleen, depending on the dose used (Figure 1D). Hence, ASM loading of anti-ICAM NCs can be varied to modulate the absolute enzyme dose delivered in vivo, with marked lung enhancement offered by ICAM-1 targeting.
Modulation of the Delivered ASM Dose by Varying the Concentration of Anti-ICAM NCs
Next, we tested the effect of varying the concentration of NCs without varying the ASM load per NC (Figure 2). For this purpose, we used anti-ICAM NCs bearing the highest load (265 ASM molecules per NC) and compared 1.8 × 1013 versus 2.7 × 1013 NCs/kg injections (equivalent to 0.6 mg versus 0.9 mg ASM/kg). Although again anti-ICAM/ASM NCs disappeared more rapidly from the circulation than matching doses of naked ASM (Figure S2A), disappearance was somewhat more slow for the higher NC concentration (3.2 versus 5.3%ID by 30 min; Figure 2A). This coincided with lower liver and spleen uptake for high versus low NC concentration, reflected by their lower percent of the injected dose (51% and 32% reductions; Figure 2B), specificity index (74% and 77% reductions; Figure 2C), and absolute ASM delivered to these organs (20% and 40% reductions; Figure 2D). Conversely, lung targeting increased with increasing NC concentration, whether measured as percent of the injected dose or absolute ASM delivered (1.6- and 1.8-fold increases, respectively; Figures 2B and 2D). Once again, ASM tissue-to-blood location ratio showed marked improvement for anti-ICAM NCs versus the naked enzyme (Figure S2B).
Figure 2.

Biodistribution of Anti-ICAM/ASM NCs at Different NC Concentrations
Wild-type (C57BL/6) mice were i.v. injected with anti-ICAM/125I-ASM polystyrene NCs at 1.8 × 1013 or 2.7 × 1013 NCs/kg body weight (0.6 or 0.9 mg/ASM/kg body weight). (A) Blood 125I content was used to calculate the percent of the injected dose (%ID) in the circulation. (B) The lungs, liver, and spleen 125I content were used to determine the biodistribution (percent of the injected dose) in these organs 30 min after injection. (C) The specificity index was calculated by normalizing tissue-over-blood accumulation for the targeted of anti-ICAM/ASM NCs over matching doses of naked ASM. (D) The amount of ASM delivered to organs was calculated from their 125I-ASM content and its specific radioactivity (counts per minute/milligram). Data are mean ± SEM (n = 3–5 mice). *p ≤ 0.1 by Student’s t test.
Endocytosis and Lysosomal Trafficking of Anti-ICAM/ASM NCs In Vivo
We then aimed at examining endocytic uptake of anti-ICAM/ASM NCs in real time in mice. We focused on the endothelium as this lining is in contact with the circulation and is a key NPD-B target.1 The mesentery of a live mouse was exteriorized under anesthesia, followed by i.v. injection of green Fluoresbrite-labeled anti-ICAM/ASM carriers, which were 1 μm in diameter to enable visualization of single particles in vivo. Texas Red dextran was co-injected to provide a label for blood vessels and also because prior cell culture studies have revealed that when anti-ICAM NCs induce endocytosis, Texas Red dextran labels the fluid phase entering the induced endocytic vesicles, as it co-internalizes with carriers.27 Hence, using fluorescence microscopy, endothelial endocytosis should be detected by appearance of dot-like, vesicular structures in the red (dextran) channel, co-localizing with and of similar size as green anti-ICAM/ASM carriers. As shown in a time-lapse video (Movie S1) and illustrated in Figure 3, this was the case: an initial image taken 15 min (15′00″) after injection shows four carriers firmly bound on the endothelium, which did not move with the flow over time. A fifth carrier was loosely bound, as it slowly moved downstream over time (white arrow). At this initial time, two out of the four firmly bound carriers already co-localized with dextran-positive vesicular structures (black arrowheads), suggesting that the corresponding carriers were endocytosed. An image taken 30 s later (15′30″) shows that the two remaining firmly bound carriers began to co-localize with dextran-positive vesicles (new black arrowheads), indicative of their subsequent endocytosis. In the next frame (16′00″), one new carrier bound firmly on the endothelium (white arrowhead) and immediately co-localized with dextran (new black arrowhead). At 17′00″, a new carrier also bound firmly (new white arrowhead) but did not co-localize with dextran until 1 min later (new black arrowhead at 18′00″). The fact that two loosely bound carriers moving over the endothelial surface (two white arrows) never co-localized with punctate dextran, validates that dextran co-localization with green carriers is not unspecific and rather marks carrier endocytosis, as previously seen in cell culture.27 This was also validated by the fact that co-localization with dextran often occurred after previous visualization of firmly bound carriers: e.g., compare carrier and dextran channels (appearance of black arrowheads) at 15′00″ versus 15′30″, and at 17′00″ versus 18′00″. This is the first time that live endothelial endocytosis of ICAM-1-targeted carriers is shown in vivo.
Figure 3.

In Vivo Imaging of Anti-ICAM/ASM Carrier Endocytosis in Mice
Time-lapse fluorescence micrographs of 1-μm, green Fluoresbright-labeled anti-ICAM/ASM polystyrene carriers (dark background panels) co-injected i.v. with the fluid-phase marker Texas Red dextran (light background panels) in wild-type (C57BL/6) mice. Images from exteriorized mesentery were taken every 30 s beginning 15 min after injection. Carriers free flowing in the circulation are not visible. Carriers loosely bound on the endothelial surface are dextran negative and move downstream with time (white arrows). Carriers that firmly bind on the endothelium after beginning imaging are marked by white arrowheads. Presence of punctate dextran co-localizing with carriers indicates carrier endocytosis by the endothelium (black arrowheads). Scale bar, 10 μm.
We then used this technique to image endocytosis of 200 nm anti-ICAM/ASM NCs similar to those used for circulation and biodistribution studies shown in Figures 1 and 2, for which we focused on the lungs (Figure 4). Ten minutes after injection, postmortem examination showed abundant green NC signal covering lung blood vessels (suggesting firm binding), but little to none endocytosis (lack of co-localization with red dextran). However, 30 min after injection, this co-localization was clearly visible (yellow color; right panels), indicating relatively fast uptake by cells (presumably endothelial) in the lungs.
Figure 4.

Endocytosis of Anti-ICAM/ASM NCs in Mouse Lungs
Green Fluoresbright-labeled, 200-nm anti-ICAM/ASM polystyrene NCs co-injected i.v. with Texas Red dextran in wild-type (C57BL/6) mice. At sacrifice (10 min versus 30 min), the lungs were collected and imaged by fluorescence microscopy under the red (dextran) and green (NC) channels (left and middle panels; scale bar, 50 μm). In merged images (right panels, magnified from the areas marked with white boxes in the left and middle panels; scale bar, 10 μm), co-localization of these labels indicates NC endocytosis (yellow).
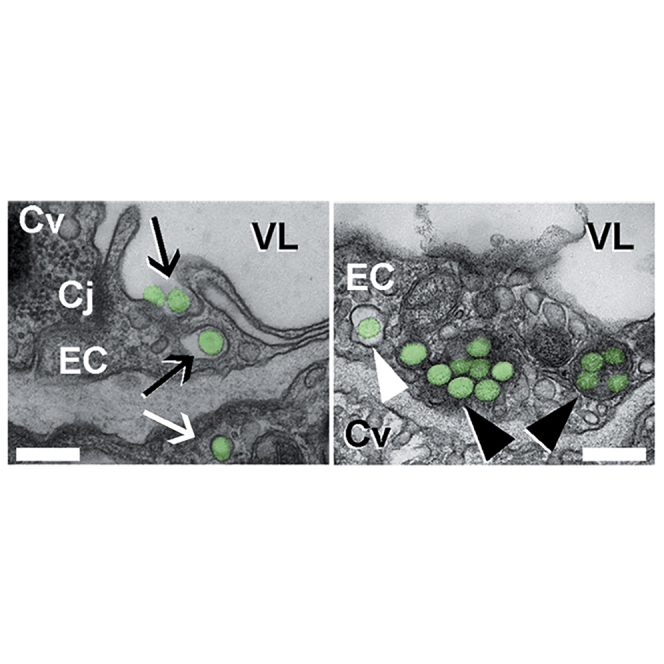
Further proof of endocytic transport was provided by transmission electron microscopy (TEM) of postmortem mouse lung, spleen, and liver specimens collected 3 hr after injection with anti-ICAM/ASM NCs (Figure 5; Figure S3). Images show NCs (green pseudocolor) bound at the luminal surface of the pulmonary endothelium and being engulfed by the endothelial plasmalemma (Figure 5A, left; black arrows), and also within endothelial cells surrounded by membranous vesicles with a clear lumen, consistent with endosomes (Figure 5A, right; white arrowhead). None of these structures had an electron-dense clathrin coat and they were morphologically different from caveolar vesicles (Figure 5A, right; Cv), in agreement with previous reports showing anti-ICAM NC uptake via the non-clathrin, non-caveolar CAM pathway.18, 28, 29 NCs were also visualized within cells in larger vesicles containing an electron-dense lumen, consistent with lysosomes (Figure 5A, right; black arrowheads). Some NCs were observed within epithelial cells underneath the endothelial lining (Figure 5A, left; white arrow), despite endothelial cell junctions appearing intact (Figure 5A, left; Cj). This is consistent with endothelial transcytosis of anti-ICAM NCs, previously reported in cell culture models.30 NCs were additionally visualized in the endothelial lining and subjacent tissue cells in the spleen (Figure 5B) and liver (Figure 5C), where they also resided within electron-dense lysosomal compartments (black arrowheads). Hence, anti-ICAM/ASM NCs not only target these organs but provide transcytosis and lysosomal trafficking in the endothelium, and subjacent cells, all of which is required for efficient ERT of NPD-B.
Figure 5.

Binding, Endocytosis, and Lysosomal Trafficking of Anti-ICAM/ASM NCs In Vivo
Transmission electron microscopy of wild-type mouse lungs (A), spleen (B), and liver (C) showing anti-ICAM/ASM polystyrene NCs (pseudocolored in green) interacting with endothelial cells (ECs), splenic parenchyma cells (SP), and hepatocytes (HP), 3 hr after i.v. injection. NCs can be seen being engulfed by cells (black arrows), within cell endosomes (white arrowheads) and lysosomes (black arrowheads), and transcytosed across the endothelium into subjacent epithelial cells (white arrow). VL, vessel lumen; Cv, caveolar vesicles; Cl, clathrin vesicles; Cj, cell junction. Scale bars, 300 nm.
ASM Activity Delivered by Anti-ICAM NCs in NPD Model Mice
We then tested the functional status of the enzyme delivered by anti-ICAM NCs. First, ASMKO mice were i.v. injected with the formulation containing 0.6 mg of ASM and 1.8 × 1013 NCs/kg body weight, and ASM activity was measured in postmortem tissue homogenates (Figure 6A). Relative comparison with untreated ASMKO mice showed enhanced ASM activity in the lung, liver, and spleen as fast as 30 min after a single injection with anti-ICAM/ASM NCs (8.0-, 5.7-, and 17.8-fold enhancement, respectively), in accord with fast and effective targeting and uptake (Figures 1 and 4). In marked contrast, respective enhancement in enzyme activity in these organs was of 1.1-, 1.7-, and 1.7-fold for animals injected with naked ASM, despite the fact that a longer time (48 h) and higher enzyme dose (3 mg/kg body weight) had been used to compensate for the lower lung uptake of this formulation (Figure 6A). Theoretical normalization to the dose of anti-ICAM/ASM NCs showed even lower effectiveness for naked ASM (Figure S4A).
Figure 6.

Enhanced Enzymatic Function of ASM Delivered by Anti-ICAM/ASM NCs in Mice
(A) ASM activity was measured by monitoring (using HPLC) the generation of fluorescent ceramide from BODIPY-FL-C12-sphingomyelin, in tissue homogenates obtained from 20- to 25-week-old ASMKO mice, 30 min after i.v. injection with anti-ICAM/ASM polystyrene NCs (0.6 mg ASM/kg body weight) or 48 hr after i.v. injection with naked ASM (3 mg ASM/kg body weight). Longer time and higher dose were used for naked ASM given the markedly lower lung accumulation. The enzyme activity was expressed as fold increase compared to ASMKO mice that did not receive enzyme. (B) The sphingomyelin (SM) content of 18- to 20-week-old ASMKO mice was determined in tissue homogenates using an Amplex Red-based enzymatic reaction and fluorescence spectrometry, 24 hr after 12 days of injections (one every other day) with anti-ICAM/ASM PLGA NCs at 0.6 mg ASM/kg body weight, or 28 days of injections (one every week) with naked ASM at 3 mg ASM/kg body weight. Treatment was longer and with higher dose for naked ASM due to its lower lung targeting. SM reduction was calculated by comparison to SM levels in untreated ASMKO mice and wild-type mice (see Materials and Methods). Data are mean ± SEM. (n ≥ 3 mice). *p ≤ 0.1 by Student’s t test.
Then, we examined the ability of the delivered enzyme to reduced sphingomyelin storage in these organs, the main pathological marker in NPD-B.3 Because this type of therapeutic effect would require multiple administrations, we switched to clinically relevant poly(lactic-co-glycolic acid) (PLGA) NCs. PLGA NCs were similarly coated with anti-ICAM antibodies and loaded with ASM, as in the case of polystyrene models (0.6 mg of ASM/kg body weight). Anti-ICAM/ASM PLGA NCs were minimally larger in size and polydispersity than polystyrene counterparts (242 nm, 0.16 PDI). This formulation was i.v. injected every other day in 18- to 20-week-old ASMKO mice, which have an advanced disease state with clear sphingomyelin storage, for a 12-day treatment period. Untreated ASMKO mice and wild-type mice were used as controls. We found an elevated variability in the sphingomyelin levels of all animal types, in agreement with the reported variability underscoring the NPD-B phenotype.3, 6 Nevertheless, the enzyme delivered by NCs seemed to reduce sphingomyelin levels in the lungs (98.3% reduction; see Materials and Methods) and, to a lesser extent, liver and spleen (35.4% and 28.1% reduction, respectively). Hence, anti-ICAM NCs delivered functional ASM to these organs, particularly the main NPD-B target, i.e., the lungs. In contrast, although effective in the liver and spleen, naked ASM showed minimal reduction in lung sphingomyelin (15.7% reduction), even when administered for a longer regime (28 days) at much higher dose (3 mg/kg). Theoretical normalization to the dose of anti-ICAM/ASM NCs showed much lower effectiveness for all organs in the case of naked ASM (Figure S4B).
In Vivo Effects of Anti-ICAM/ASM NCs on NPD-B Pulmonary Inflammation
In addition to examining the effect of anti-ICAM/ASM NCs on the sphingomyelin storage phenotype, we evaluated their effects regarding other NPD-B hallmarks, i.e., elevated macrophage counts in the alveoli and pulmonary infiltration of other white blood cells (WBCs).2, 3, 7 This was done by determining the total and differential cell counts in the bronchoalveolar lavage fluid (BALF) of 20-week-old ASMKO mice, after a similar regime of i.v. injections with anti-ICAM/ASM NCs (six injections, one every other day; Figure 7A). At this age, advanced pulmonary inflammation and cellular infiltrate have been reported.8, 31, 32 After treatment, the number of alveolar macrophages was significantly reduced compared to untreated ASMKO mice (36% reduction), and also the lymphocyte and total WBC counts seemed diminished (20% and 29% reduction, respectively). This suggests that anti-ICAM/ASM NCs can achieve attenuation of the NPD-B inflammatory phenotype. However, the number of infiltrated neutrophils seemed enhanced (51% increase).
Figure 7.

Effects of Anti-ICAM/ASM NCs of NPD-B-Associated Lung Inflammation in Mice
Total white blood cell (WBC) and differential (macrophages, lymphocytes, neutrophils) counts in the bronchoalveolar lavage fluid (BALF) after the following treatments: (A) six total i.v. injections (one every other day) with anti-ICAM/ASM PLGA NCs in 20-week-old ASMKO mice (represented as percentage of untreated ASMKO mice); (B) one i.v. injection with anti-ICAM/ASM PLGA NCs or “vehicle” (buffer without carriers, antibody, or enzyme) in 20-week-old ASMKO mice versus wild-type mice; (C) one i.v. injection with anti-ICAM/ASM PLGA NCs or “vehicle” (see B) in wild-type (20-week-old) or ASMKO mice (20 versus 4 weeks old). The horizontal dashed line in (B) and (C) corresponds to untreated ASMKO mice. Data are mean ± SEM (n = 3–5 mice). *Compares NC-treated to untreated ASMKO mice in (A), and ASMKO to wild-type mice in (B) and (C); #compares 20-week-old to 4-week-old ASMKO mice (p ≤ 0.1 by Student’s t test).
To examine the cause for this response, we obtained BALF cell counts from 20-week-old ASMKO mice that had not received any injection versus those that received one injection of either anti-ICAM/ASM NCs and compared these results to similar injections in age-matching wild-type mice. ASMKO mice injected with NCs presented elevated neutrophils counts over untreated ASMKO mice (18.5-fold increase; Figure 7B) and also enhanced total WBC counts (3.8-fold increase; Figure 7C). However, injection of NCs in wild-type mice did not result in high neutrophil counts in the BALF (43-fold below ASMKO levels; Figure 7B), and the same was found for total WBC counts (6.3-fold below ASMKO; Figure 7C), indicating lack of inherent toxicity of the NC formulation. Unexpectedly, i.v. injection of “vehicle” alone (the buffer without carriers, antibody, or enzyme) in ASMKO mice rendered similar increase in neutrophils as NCs (11-fold increase; Figure 7B), and the same outcome was observed for total WBC counts (3.5-fold increase; Figure 7C). These results suggest that the injected materials are safe, and the observed reactivity rather associates with the ASMKO phenotype.
Given this, we then examined whether this outcome relates to the state of disease progression. For this purpose, similar injections were performed in 4-week-old ASMKO mice, which present a less advanced disease state.8, 31, 32 As postulated, younger animals showed a much lower level of WBC infiltration as a response to NCs (4.0-fold below older animals; Figure 7C), similar to untreated mice (1.0-fold compared to 20-week-old mice; Figure 7C). Hence, therapeutic intervention may be more amenable when administered in early disease states due to inherent reactivity of older mice.
Discussion
Efficient NPD-B treatment by ERT requires enough recombinant ASM enzyme to reach the main target organs.3, 11 In particular, primary storage of aberrant sphingomyelin levels in the lungs, liver, and spleen and secondary consequences, such as subsequent pulmonary inflammation, represent key clinical phenotypes for therapeutic intervention.3, 5, 7, 10 Although current ERT by i.v. injection of naked ASM has established unquestionable clinical value, these studies have also revealed the need to improve lung targeting in order to minimize the administered enzyme dose while retaining therapeutic benefit.8, 9, 10, 12, 13 Our previous studies had demonstrated that ICAM-1-targeted polymer NCs represent an interesting strategy to achieve this goal. Broad ICAM-1 expression throughout the body, and its enhancement under inflammation, has been shown to provide a markedly increased delivery of ASM in all organs of mouse models when using ICAM-1-targeted/ASM NCs.14, 15, 16 Moreover, in cell culture systems, uptake via the CAM-mediated pathway has also been shown to enhance lysosomal delivery and sphingomyelin degradation.18, 19, 20, 21, 22 These findings suggest that anti-ICAM/ASM NCs may help reduce the enzyme dose required to obtain a therapeutic benefit in vivo. Advancing this seminal work, here we have (1) demonstrated methods to manipulate the administered NC-based enzyme dose, (2) verified in vivo NC uptake with transcytosis and lysosomal trafficking in cells of the main target organs, (3) provided proof of enzyme activity delivered by anti-ICAM NCs in vivo, and (4) demonstrated attenuation of sphingomyelin storage and cellular infiltration in the lungs.
First, we examined two approaches to manipulate the enzyme dose administered in mice; by varying the enzyme load per NC while keeping the NC concentration constant (Figure 1), versus varying the concentration of the injected NCs while maintaining constant the enzyme-per-NC dose (Figure 2). All formulations provided specific ICAM-1 targeting compared to control IgG/ASM NCs (e.g., ≥20-fold increase in the lungs), and markedly enhanced tissue-to-blood accumulation over naked ASM (e.g., lung specificity indexes were ≥137). However, interestingly, each approach rendered different advantages and disadvantages. Increasing the NC concentration of the injection (Figure 2) was a simple way to increase the total ASM injected dose without the need to manipulate the NC formulation. This resulted in increasing ASM levels in the lungs, but it also caused a reduction in the enzyme levels delivered to the liver and spleen. Although this may be beneficial for other applications, it is likely detrimental for these important NPD-B targets. This effect has been previously observed with formulations voided of a therapeutic cargo.33 Hence, it appears that liver-spleen bulk saturation with NCs causes the observed decay in uptake by these organs, leaving more NCs available for lung targeting. Supporting this, increasing the NC concentration slightly increased their circulation. As a consequence, the tissue-to-blood specificity index for anti-ICAM/ASM NCs over naked ASM decreased when increasing the NC concentration. As increasing NC concentration would also signify an elevation in the amount of polymer injected, this strategy must be used with caution. Instead, increasing the enzyme load per NC while keeping the NC concentration constant (Figure 1) would avoid this effect, although this approach requires preparation of independent formulations. Nonetheless, this allowed us to manipulate the total ASM dose in the injection without compromising targeting to any organs: increasing ASM load per NC increased the final level of enzyme in all three—lungs, liver and spleen—while keeping the relative lung targeting (ASM per gram of tissue) above the liver and spleen levels. Therefore, this seems a more amenable approach for this particular application.
Second, we imaged intravital endocytosis by the endothelium in vivo using time-lapse microscopy, which was then verified in postmortem samples. Our results suggest that the kinetics for this event in vivo is fast and within the range of previous cell culture assays: i.e., we had observed internalization of 50% of all pre-bound NCs between 10 and 20 min for cell culture assay,18, 28 whereas here we observed ∼30%–50% of the NCs to be internalized in vivo by 15–30 min (Figures 3 and 4). Endocytosis was visualized by co-localization of dextran only with firmly bound anti-ICAM/ASM NCs (not loosely bound, moving counterparts), and with a gap of time between firm binding and co-localization. Hence, this method provides an avenue to examine more precise mechanistic aspects in vivo and in real time.
In some cases, endocytosis of single carriers was observed within the range of seconds to very few minutes after their binding, even for the case of micrometer size counterparts (Figure 3). This is in agreement with prior observations of the CAM pathway being efficient with regard to uptake of a wide range of carrier sizes.19, 29 Interestingly, our previous studies have demonstrated that this property is mediated by generation of cell-surface ceramide at sites of ICAM-1 engagement, a key signaling mechanism of the CAM pathway.29, 34 Because the carriers used in this study were precisely loaded with recombinant ASM, an enzyme that generates ceramide from sphingomyelin, it is possible that this further contributes to the efficacy of internalization apart from therapeutic effects of degrading sphingomyelin. Therefore, ASM delivery benefits from anti-ICAM NCs, as NCs provide targeting and endocytosis via the CAM pathway and, paradoxically, anti-ICAM NCs also benefit from ASM cargo, because the product of this enzyme contributes to CAM-mediated uptake, a dual benefit of this strategy (the carrier improves the benefit of the cargo and the cargo improves the benefit of the carrier).
Furthermore, TEM showed both transcytosis and lysosomal trafficking of anti-ICAM/ASM NCs in the pulmonary endothelium (Figure 5). This is in agreement with previous publications in endothelial cell cultures showing this dual destination,18, 30 a finding that remains to be mechanistically deciphered and represents a key focus for our future studies. However, this is beneficial for this particular application because sphingomyelin accumulates in lysosomes of both the endothelium and subendothelial tissue, and hence, this dual transport fits well with the disease requirement. This was not unique to the lungs, as we also viewed NCs in lysosomes within hepatic and splenic cells beyond the endothelium, although the endothelial lining in these organs is discontinuous and passive extravasation (instead of transcytosis) may suffice here.
Importantly, in agreement with effective targeting and lysosomal (endothelial plus subendothelial) delivery, anti-ICAM NCs rendered a significant increase in the ASM activity measured in organs of ASMKO mice after a single i.v. injection with a dose of 0.6 mg/kg body weight, as compared to naked ASM injected at 3 mg/kg (Figure 6A). Injection of anti-ICAM/ASM NCs at this dose also markedly attenuated the sphingomyelin storage in all organs after 12-day treatment, with much higher attenuation in the lungs versus a naked ASM injected for 28 days at a higher dose (Figure 6B). This is in agreement with previous studies that have shown that i.v. injections of the naked enzyme at doses between 0.3 and 3 mg/kg did not reduce sphingomyelin storage in the lungs (although liver and spleen sphingomyelin was reduced), and a 10 mg ASM/kg dose was needed to render 20% sphingomyelin reduction in this organ.8 Noteworthy, doses of ≥10 mg/kg resulted in toxicity in ASMKO mice,12 highlighting the balance that must be reached for a safe and effective treatment. Therefore, despite the variability found in our study, the 52%–98% sphingomyelin reduction we observed in the lungs using a 0.6 mg ASM/kg dose is remarkable and well within the safe range (up to 3 mg ASM/kg) tested for clinical use.10, 13
In addition to sphingomyelin reduction, this dose and regime of anti-ICAM/ASM NCs also decreased pulmonary WBC infiltration, including that of macrophages (Figure 7), a hallmark of NPD-B.1, 31 This further supports the therapeutic value of this strategy. However, neutrophil counts in the BALF were enhanced, which appeared to be a reaction of ASMKO mice to the injection received. The mechanism for this reaction will have to be investigated in detail in future studies, because understanding potential side effects is paramount for translation. Nevertheless, the fact that injection buffer alone induced a similar neutrophil infiltration in these mice, and infiltration was not observed in wild-type mice receiving a similar dose of anti-ICAM/ASM NCs, suggests that the formulation is relatively safe. It is possible that the advanced disease stage associated with the age of the ASMKO mice used (20 weeks old) led to this secondary reaction, not viewed in healthy controls. In fact, in younger (4-week-old) ASMKO mice with a less advanced disease state, anti-ICAM/ASM NCs did not increase the infiltration level compared to untreated mice. Interestingly, younger ASMKO mice that received buffer alone had a higher infiltration level than those that received anti-ICAM/ASM NCs, suggesting that NCs bearing the therapeutic enzyme were able to reduce the infiltration that may originate from the injection. Future studies will focus on examining other phenotypic changes pertaining to lung function, to further establish the potential for this treatment strategy.
In conclusion, anti-ICAM/ASM NCs hold great potential to improve lung delivery of ASM over the naked enzyme (while maintaining liver and spleen uptake needed for this disease). This renders enhanced attenuation of the key NPD-B markers at enzyme doses established to be safe and within the clinically intended range. Moreover, we suggest that beginning treatment at a younger age, when the disease has not yet advanced, may provide optimal benefits to patients.
Materials and Methods
Antibodies and Reagents
Rat monoclonal antibody to mouse ICAM-1, clone YN1, was from ATCC and non-specific rat IgG was from Jackson Immunoresearch. Fluoresbrite-labeled polystyrene particles (100 nm and 1 μm in diameter) were from Polysciences. Thirty-eight-kilodalton PLGA (50:50 copolymer ratio) was from Lakeshore Biomaterials. Na125I was from PerkinElmer Life and Analytical Sciences. Human recombinant ASM was expressed and purified as described.35 Texas-Red dextran (10,000 kDa), BODIPY-FL-C12-sphingomyelin, and Amplex Red Sphingomyelinase Assay Kit were from Thermo Fisher Scientific. Unless otherwise noticed, all other reagents were from Sigma-Aldrich.
Preparation of Anti-ICAM/ASM NCs
Fluoresbrite-labeled polystyrene particles (100 nm or 1 μm in diameter, where indicated) were used as models for studies focusing on biodistribution and endocytosis, as in previous publications.18, 21 For functional assays, we used PLGA NCs prepared by nanoprecipitation, whereby an organic phase consisting of PLGA in acetone was added into the aqueous phase under agitation, followed by overnight solvent evaporation also under agitation, as described.14, 26 The resulting nanoparticle suspension was filtered, dialyzed, and concentrated using a rotary evaporator.
In all cases, particles were coated by absorption with anti-ICAM (3–6 μM) and recombinant ASM (0.5–5 μM), as previously described.14, 21 For experiments quantifying loading and for in vivo biodistribution, ASM was labeled with 125I prior to coating. In parallel, to determine the number of targeting antibodies on the NC surface, preparations contained 125I-anti-ICAM instead. In either case, coating with 125I-ASM or 125I-anti-ICAM was assessed by measuring the 125I content in a gamma counter (2470 Wizard2; Perkin-Elmer) and calculating the number of the corresponding molecules per particle, based on the particle concentration and the 125I-specific activity (counts per minute/mass) of each molecule, as described.23, 36 Uncoated enzyme or antibody were removed by centrifugation and the coated-NC pellet was resuspended in PBS containing 0.3% bovine serum albumin, and sonicated. The final size, PDI, and ζ-potential of the resulting preparations were estimated by dynamic light scattering (DLS) and electrophoretic mobility (Zetasizer Nano-S90; Malvern Instruments), as reported.14, 21 These formulations are relatively stable, showing lack of aggregation (no precipitation and DLS-measured diameter remains same upon incubation with saline or serum), insignificant detachment of coated proteins (≤10% in cell media or serum), and low coating with serum albumin (≤0.5% of total coat molecules).14, 23, 24, 26, 36
In Vivo Biodistribution of Anti-ICAM/ASM Nanocarriers
C57BL/6 (wild-type) mice were anesthetized and i.v. injected with anti-ICAM/125I-ASM polystyrene NCs at 1.8 × 1013 NCs/kg body weight (containing 0.06, 0.3, or 0.6 mg ASM/kg body weight) or 2.7 × 1013 NCs/kg body weight (equivalent to 0.9 mg ASM/kg body weight). In parallel, mice were injected with respective matching doses of naked ASM as controls. Blood samples were collected from the retro-orbital sinus at 1, 15, and 30 min after injection, and key NPD-B target organs (liver, lung, and spleen) were collected at sacrifice (30 min). The radioactive content and weight of the samples was determined to calculate biodistribution parameters, including the following: (1) the percent of the injected dose (counts per minute in the blood or an organ divided by total counts per minute injected), (2) the LR (the percent of the injected dose per gram of an organ divided by the percent of the injected dose per gram of blood), (3) the specificity index (the LR of anti-ICAM/ASM NCs divided by the LR of naked ASM), and (4) the absolute amount of ASM delivered per gram of organ (counts per minute/gram of organ divided by counts per minute/milligram of ASM), as reported.14
Microscopy Visualization of Anti-ICAM/ASM NCs in Mice
C57BL/6 mice were anesthetized, followed by i.v. co-injection with 1-μm-diameter, green Fluoresbrite-labeled anti-ICAM/ASM polystyrene carriers (0.6 mg ASM/kg body weight) and 16 mg/kg of the fluid-phase marker TexasRed dextran. The mesentery was exteriorized, kept wet with warm PBS, and beginning 15 min after injection, intravital images were taken every 30 s by fluorescence microscopy using a 60× PlanApo objective and Eclipse TE2000-U microscope (Nikon), and ORCA-1CCD camera (Hamamatsu Corporation). Image analysis was done using Image-Pro 3.0 software (Media Cybernetics) to visualize the presence of punctate red dextran signal co-localizing with green carrier signal, indicative of carrier endocytosis.27 Similar experiments were performed to examine endocytosis of anti-ICAM/ASM NCs in the lungs, using 200-nm NCs and conducting imaging immediately postmortem, either 10 or 30 min after injection.
Additionally, in vivo endocytosis and lysosomal localization of anti-ICAM/ASM NCs were examined by TEM, as before.14, 29 For this, mice were injected with NCs at 0.6 mg ASM/kg body weight and sacrificed 3 hr after injection. Animals were perfused under anesthesia through the right ventricle, first with PBS and then with fixative (2.5% glutaraldehyde, 4% paraformaldehyde in 0.1 M sodium cacodylate buffer), while maintained under ventilation. The lungs, liver, and spleen were collected and processed for TEM, as reported.29, 37
ASM Activity and Sphingomyelin Levels in Mouse Tissues
For ASM activity assays, anesthetized, 20- to 25-week-old ASMKO mice received one i.v. injection of anti-ICAM/ASM NCs or naked ASM. As biodistribution data had shown much lower ASM delivery of naked enzyme versus anti-ICAM NC counterparts (Figures S1B and S2B), injections contained 3 mg/kg versus 0.6 mg/kg of enzyme, respectively, and activity measurements were performed 48 hr versus 30 min post-injection. The lungs, liver, and spleen were collected and ≈50-mg tissue samples were homogenized in 0.2% Triton X-100. Aliquots were then used to measure protein concentration and, in parallel, incubated for 3 hr at 37°C with BODIPY-FL-C12-sphingomyelin in citrate buffer at pH 5.2. This rendered fluorescent ceramide as a product of ASM activity, which was measured by HPLC. Enzyme activity was calculated as femtomoles of generated ceramide per reaction volume and time units, and normalized per mass of protein in the homogenate.8, 38 The ASM activity found in treated ASMKO mice was expressed as fold increased over that found in animals that did not receive enzyme injection.
For measurements on the level of sphingomyelin in the lungs, liver, and spleen, 18- to 20-week-old ASMKO mice were i.v. injected with anti-ICAM/ASM NCs (0.6 mg ASM/kg body weight), every other day for a total of 12 days. Other mice were similarly injected with naked ASM (3 mg/kg body weight), once a week for a total of 28 days. Untreated ASMKO mice and wild-type mice served as controls. At sacrifice, 24 hr after the last injection, tissue samples were removed, weighed, and homogenized in 1:2 chloroform:methanol. Tissue homogenates were then centrifuged to collect the organic fraction, followed by solvent evaporation and reconstitution in reaction buffer, as per the Amplex Red Sphingomyelinase Assay Kit protocol.8, 38 Briefly, this consists of an enzymatic chain reaction that uses bacterial sphingomyelinase to hydrolyze sphingomyelin to phosphocholine and ceramide, alkaline phosphatase to generate choline, choline oxidase to produce hydrogen peroxide, and horseradish peroxidase to render Amplex Red oxidation into fluorescent resorufin, which is finally measured in a fluorescence spectrophotometer.8, 38 Results were calculated as micrograms of sphingomyelin per milligrams of tissue. Sphingomyelin reduction in the organs of ASMKO mice injected with anti-ICAM/ASM NCs or naked ASM was calculated as follows: 100 × [(sphingomyelin in untreated ASMKO mice – sphingomyelin in treated ASMKO mice): (sphingomyelin in untreated ASMKO mice – sphingomyelin in wild-type mice)].
Total and Differential Cell Counts in Mouse BALF
Wild-type C57BL/6 (20-week-old) and ASMKO (20- or 4-week-old) mice were euthanized 24 hr after one i.v. injection or six injections (one every other day), as indicated in particular experiments, with either anti-ICAM/ASM NCs (0.6 mg ASM and 0.3 mg anti-ICAM/kg body weight), naked anti-ICAM (0.3 mg ASM/kg body weight), or vehicle. A 25G blunt needle was inserted through the trachea, and the airways were washed three times with PBS. For each mouse, pooled samples were centrifuged at 1,000 × g for 15 min, and the cell pellet was subjected to differential cytospins to first lyse erythrocytes and then separate WBCs. WBCs were then stained with hematoxylin-eosin stain and counted using a hemacytometer, as described.9
Animal Protocols
All animal studies were performed under protocols approved by Institutional Animal Care and Use Committee and University regulations.
Statistics
Except for non-quantitative fluorescence and TEM imaging results, all other assays were performed using n = 3–6 (in accord with previous publications of the effects of the naked enzyme) and expressed as the mean ± SEM, where statistical significance was determined by Student’s t test.
Author Contributions
C.G., R.D., and M.S. performed experiments, prepared figures, and helped to write and edit the manuscript. E.H.S. and S.M. conceived the study, guided experiments and interpretation of results, and helped to write and edit the manuscript.
Conflicts of Interest
E.H.S. is a consultant and received research support from Genzyme/Sanofi, a company developing enzyme replacement therapy for NPD-B. E.H.S. is also an inventor on patents licensed to Genzyme/Sanofi and has received royalty income.
Acknowledgments
This work was supported by National Institutes of Health awards R01-HL98416 (to S.M.) and R37-HD28607 (to E.H.S.).
Footnotes
Supplemental Information includes four figures and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2017.05.014.
Contributor Information
Edward H. Schuchman, Email: edward.schuchman@mssm.edu.
Silvia Muro, Email: muro@umd.edu.
Supplemental Information
Time lapse fluorescence microscopy videos of 1 μm, green Fluoresbright®-labeled anti-ICAM/ASM polystyrene carriers (a, dark background panels) co-injected i.v. with the fluid-phase marker Texas Red dextran (b, light background panels) in wild-type (C57Bl/6) mice. Images from exteriorized mesentery were taken every 30 seconds beginning 15 min after injection. Carriers free flowing in the circulation are not visible. Carriers loosely bound on the endothelial surface are dextran negative and move downstream with time. Carriers that firmly bind on the endothelium after beginning imaging do not move. Presence of punctate dextran co-localizing with carriers indicates carrier endocytosis by the endothelium. Scale bar = 10 μm.
{kind=link}
References
- 1.Schuchman E.H. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 2007;30:654–663. doi: 10.1007/s10545-007-0632-9. [DOI] [PubMed] [Google Scholar]
- 2.von Ranke F.M., Pereira Freitas H.M., Mançano A.D., Rodrigues R.S., Hochhegger B., Escuissato D., Araujo Neto C.A., da Silva T.K., Marchiori E. Pulmonary involvement in Niemann-Pick disease: a state-of-the-art review. Lung. 2016;194:511–518. doi: 10.1007/s00408-016-9893-0. [DOI] [PubMed] [Google Scholar]
- 3.Schuchman E.H., Wasserstein M.P. Types A and B Niemann-Pick disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015;29:237–247. doi: 10.1016/j.beem.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Futerman A.H., Hannun Y.A. The complex life of simple sphingolipids. EMBO Rep. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurberg B.L., Wasserstein M.P., Schiano T., O’Brien F., Richards S., Cox G.F., McGovern M.M. Liver and skin histopathology in adults with acid sphingomyelinase deficiency (Niemann-Pick disease type B) Am. J. Surg. Pathol. 2012;36:1234–1246. doi: 10.1097/PAS.0b013e31825793ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassiman D., Packman S., Bembi B., Turkia H.B., Al-Sayed M., Schiff M., Imrie J., Mabe P., Takahashi T., Mengel K.E. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): literature review and report of new cases. Mol. Genet. Metab. 2016;118:206–213. doi: 10.1016/j.ymgme.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 7.McGovern M.M., Lippa N., Bagiella E., Schuchman E.H., Desnick R.J., Wasserstein M.P. Morbidity and mortality in type B Niemann-Pick disease. Genet. Med. 2013;15:618–623. doi: 10.1038/gim.2013.4. [DOI] [PubMed] [Google Scholar]
- 8.Miranda S.R., He X., Simonaro C.M., Gatt S., Dagan A., Desnick R.J., Schuchman E.H. Infusion of recombinant human acid sphingomyelinase into Niemann-Pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J. 2000;14:1988–1995. doi: 10.1096/fj.00-0014com. [DOI] [PubMed] [Google Scholar]
- 9.Ziegler R.J., Brown C., Barbon C.M., D’Angona A.M., Schuchman E.H., Andrews L., Thurberg B.L., McPherson J.M., Karey K.P., Cheng S.H. Pulmonary delivery of recombinant acid sphingomyelinase improves clearance of lysosomal sphingomyelin from the lungs of a murine model of Niemann-Pick disease. Mol. Genet. Metab. 2009;97:35–42. doi: 10.1016/j.ymgme.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 10.McGovern M.M., Wasserstein M.P., Kirmse B., Duvall W.L., Schiano T., Thurberg B.L., Richards S., Cox G.F. Novel first-dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann-Pick disease type B (acid sphingomyelinase deficiency) Genet. Med. 2016;18:34–40. doi: 10.1038/gim.2015.24. [DOI] [PubMed] [Google Scholar]
- 11.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010;2:189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray J.M., Thompson A.M., Vitsky A., Hawes M., Chuang W.L., Pacheco J., Wilson S., McPherson J.M., Thurberg B.L., Karey K.P., Andrews L. Nonclinical safety assessment of recombinant human acid sphingomyelinase (rhASM) for the treatment of acid sphingomyelinase deficiency: the utility of animal models of disease in the toxicological evaluation of potential therapeutics. Mol. Genet. Metab. 2015;114:217–225. doi: 10.1016/j.ymgme.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Wasserstein M.P., Jones S.A., Soran H., Diaz G.A., Lippa N., Thurberg B.L., Culm-Merdek K., Shamiyeh E., Inguilizian H., Cox G.F., Puga A.C. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol. Genet. Metab. 2015;116:88–97. doi: 10.1016/j.ymgme.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garnacho C., Dhami R., Simone E., Dziubla T., Leferovich J., Schuchman E.H., Muzykantov V., Muro S. Delivery of acid sphingomyelinase in normal and Niemann-Pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J. Pharmacol. Exp. Ther. 2008;325:400–408. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 15.Papademetriou J., Garnacho C., Serrano D., Bhowmick T., Schuchman E.H., Muro S. Comparative binding, endocytosis, and biodistribution of antibodies and antibody-coated carriers for targeted delivery of lysosomal enzymes to ICAM-1 versus transferrin receptor. J. Inherit. Metab. Dis. 2013;36:467–477. doi: 10.1007/s10545-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papademetriou I.T., Garnacho C., Schuchman E.H., Muro S. In vivo performance of polymer nanocarriers dually-targeted to epitopes of the same or different receptors. Biomaterials. 2013;34:3459–3466. doi: 10.1016/j.biomaterials.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muro S. VCAM-1 and ICAM-1. In: Aird W.C., editor. Endothelial Biomedicine. Cambridge University Press; 2007. pp. 1058–1070. [Google Scholar]
- 18.Muro S., Schuchman E.H., Muzykantov V.R. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol. Ther. 2006;13:135–141. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 19.Muro S., Garnacho C., Champion J.A., Leferovich J., Gajewski C., Schuchman E.H., Mitragotri S., Muzykantov V.R. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol. Ther. 2008;16:1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappaport J., Garnacho C., Muro S. Clathrin-mediated endocytosis is impaired in type A-B Niemann-Pick disease model cells and can be restored by ICAM-1-mediated enzyme replacement. Mol. Pharm. 2014;11:2887–2895. doi: 10.1021/mp500241y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rappaport J., Manthe R.L., Garnacho C., Muro S. Altered clathrin-independent endocytosis in type A Niemann-Pick disease cells and rescue by ICAM-1-targeted enzyme delivery. Mol. Pharm. 2015;12.5:1366–1376. doi: 10.1021/mp5005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rappaport J., Manthe R.L., Solomon M., Garnacho C., Muro S. A comparative study on the alterations of endocytic pathways in multiple lysosomal storage disorders. Mol. Pharm. 2016;13:357–368. doi: 10.1021/acs.molpharmaceut.5b00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu J., Serrano D., Bhowmick T., Kumar K., Shen Y., Kuo Y.C., Garnacho C., Muro S. Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J. Control. Release. 2011;149:323–331. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu J., Northrup L., Bhowmick T., Muro S. Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine (Lond.) 2012;8:731–739. doi: 10.1016/j.nano.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu J., Bhowmick T., Burks S.R., Kao J.P., Muro S. Enhancing biodistribution of therapeutic enzymes in vivo by modulating surface coating and concentration of ICAM-1-targeted nanocarriers. J. Biomed. Nanotechnol. 2014;10:345–354. doi: 10.1166/jbn.2014.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muro S., Dziubla T., Qiu W., Leferovich J., Cui X., Berk E., Muzykantov V.R. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J. Pharmacol. Exp. Ther. 2006;317:1161–1169. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 27.Muro S., Gajewski C., Koval M., Muzykantov V.R. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105:650–658. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 28.Muro S., Wiewrodt R., Thomas A., Koniaris L., Albelda S.M., Muzykantov V.R., Koval M. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J. Cell Sci. 2003;116:1599–1609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 29.Serrano D., Bhowmick T., Chadha R., Garnacho C., Muro S. Intercellular adhesion molecule 1 engagement modulates sphingomyelinase and ceramide, supporting uptake of drug carriers by the vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 2012;32:1178–1185. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu J., Rappaport J., Muro S. Specific binding, uptake, and transport of ICAM-1-targeted nanocarriers across endothelial and subendothelial cell components of the blood-brain barrier. Pharm. Res. 2014;31:1855–1866. doi: 10.1007/s11095-013-1289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikegami M., Dhami R., Schuchman E.H. Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L518–L525. doi: 10.1152/ajplung.00258.2002. [DOI] [PubMed] [Google Scholar]
- 32.Horinouchi K., Erlich S., Perl D.P., Ferlinz K., Bisgaier C.L., Sandhoff K., Desnick R.J., Stewart C.L., Schuchman E.H. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat. Genet. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 33.Calderon A.J., Bhowmick T., Leferovich J., Burman B., Pichette B., Muzykantov V., Eckmann D.M., Muro S. Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J. Control. Release. 2011;150:37–44. doi: 10.1016/j.jconrel.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ansar M., Serrano D., Papademetriou I., Bhowmick T.K., Muro S. Biological functionalization of drug delivery carriers to bypass size restrictions of receptor-mediated endocytosis independently from receptor targeting. ACS Nano. 2013;7:10597–10611. doi: 10.1021/nn404719c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He X., Miranda S.R., Xiong X., Dagan A., Gatt S., Schuchman E.H. Characterization of human acid sphingomyelinase purified from the media of overexpressing Chinese hamster ovary cells. Biochim. Biophys. Acta. 1999;1432:251–264. doi: 10.1016/s0167-4838(99)00069-2. [DOI] [PubMed] [Google Scholar]
- 36.Serrano D., Manthe R.L., Paul E., Chadha R., Muro S. How carrier size and valency modulate receptor-mediated signaling: understanding the link between binding and endocytosis of ICAM-1-targeted carriers. Biomacromolecules. 2016;17:3127–3137. doi: 10.1021/acs.biomac.6b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garnacho C., Shuvaev V., Thomas A., McKenna L., Sun J., Koval M., Albelda S., Muzykantov V., Muro S. RhoA activation and actin reorganization involved in endothelial CAM-mediated endocytosis of anti-PECAM carriers: critical role for tyrosine 686 in the cytoplasmic tail of PECAM-1. Blood. 2008;111:3024–3033. doi: 10.1182/blood-2007-06-098657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He X., Chen F., McGovern M.M., Schuchman E.H. A fluorescence-based, high-throughput sphingomyelin assay for the analysis of Niemann-Pick disease and other disorders of sphingomyelin metabolism. Anal. Biochem. 2002;306:115–123. doi: 10.1006/abio.2002.5686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time lapse fluorescence microscopy videos of 1 μm, green Fluoresbright®-labeled anti-ICAM/ASM polystyrene carriers (a, dark background panels) co-injected i.v. with the fluid-phase marker Texas Red dextran (b, light background panels) in wild-type (C57Bl/6) mice. Images from exteriorized mesentery were taken every 30 seconds beginning 15 min after injection. Carriers free flowing in the circulation are not visible. Carriers loosely bound on the endothelial surface are dextran negative and move downstream with time. Carriers that firmly bind on the endothelium after beginning imaging do not move. Presence of punctate dextran co-localizing with carriers indicates carrier endocytosis by the endothelium. Scale bar = 10 μm.