

Abstract
Background
The cytosolic arrestin proteins mediate desensitization of activated G protein-coupled receptors (GPCRs) via competition with G proteins for the active phosphorylated receptors. Arrestins in active, including receptor-bound, conformation are also transducers of signaling. Therefore, this protein family is an attractive therapeutic target. The signaling outcome is believed to be a result of structural and sequence-dependent interactions of arrestins with GPCRs and other protein partners. Here we elucidated the detailed evolution of arrestins in deuterostomes.
Results
Identity and number of arrestin paralogs were determined searching deuterostome genomes and gene expression data. In contrast to standard gene prediction methods, our strategy first detects exons situated on different scaffolds and then solves the problem of assigning them to the correct gene. This increases both the completeness and the accuracy of the annotation in comparison to conventional database search strategies applied by the community. The employed strategy enabled us to map in detail the duplication- and deletion history of arrestin paralogs including tandem duplications, pseudogenizations and the formation of retrogenes. The two rounds of whole genome duplications in the vertebrate stem lineage gave rise to four arrestin paralogs. Surprisingly, visual arrestin ARR3 was lost in the mammalian clades Afrotheria and Xenarthra. Duplications in specific clades, on the other hand, must have given rise to new paralogs that show signatures of diversification in functional elements important for receptor binding and phosphate sensing.
Conclusion
The current study traces the functional evolution of deuterostome arrestins in unprecedented detail. Based on a precise re-annotation of the exon-intron structure at nucleotide resolution, we infer the gain and loss of paralogs and patterns of conservation, co-variation and selection.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-1001-4) contains supplementary material, which is available to authorized users.
Keywords: Arrestin, Signaling, Gene duplication, Evolution, Receptor specificity, Retrogene
Background
Arrestins are cytosolic proteins with a molecular weight of about 40-45 kDa involved in the regulation of cell signaling. The binding of arrestins to activated and phosphorylated G protein-coupled receptors (GPCRs) blocks the inter-helical cavity of active GPCR, thereby precluding its coupling to cognate G proteins [1, 2]. Thus, arrestins contribute to the fast and precise shut-off of GPCR signaling via G proteins. In particular, arrestin binding is indispensable for a high temporal resolution in vision [3, 4]. Beyond their “arresting”-function that gave the protein family its name, diverse other biological functions of arrestins have been described in the last two decades. Among them are the scaffolding, subcellular localization, and regulation of kinases, phosphatases and ubiquitin ligases, G protein independent signaling and GPCR trafficking (for review see [5, 6]). In recent years, considerable efforts were made towards the design of arrestins that modulate GPCR signaling and facilitate biased signaling [7].
Arrestin proteins consist of two domains each with the β-sandwich at its core, the arrestin_N and arrestin_C domain. The domains are connected by a highly flexible linker region. The N domain contains the only α-helix (Fig. 1 a). Arrestin proteins belong to the arrestin clan and were named β-arrestins by [8] or true arrestins by [2, 9]. Below, we will refer to this group of proteins as arrestins although there are additional members in the clan that share the anti-parallel β-sandwich fold and are involved in cellular trafficking. These are the arrestin-domain containing proteins and a set of families that are rather distantly related to arrestins with maximal 10% sequence identity [8]. These distant relatives encompass the VPS26 family (including DSCR3) and RGP1 that are represented in human (Homo sapiens), as well as fungal arrestin-related trafficking adapters, amoebal arrestin domain-containing proteins and the Spo0M family in bacteria and archaea [8, 10].
Fig. 1.
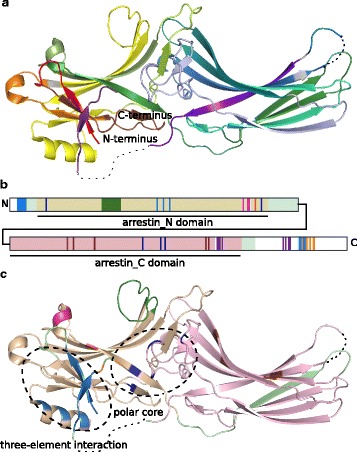
Functional elements of arrestins. a – Crystal structure of bovine ARRB1 colored according to the conserved exon-borders in vertebrates (rainbow coloring from exon 2 - red to exon 16 - dark violet). Exons 1, 15 as well as parts of exons 13, 14 and 16 are missing in the crystal structure (shown as dotted lines if not situated on the N- or C-terminus). Amino acids whose codons are split among two exons are shown in grey [112]. b – Schematic, linear representation of bovine ARRB1 with important functional elements shown in bright colors (orange - AP-2 binding site, light blue - three-element interaction, dark blue - polar core, green - finger loop, brown - high affinity IP6 binding site, pink - low affinity IP6 binding site, red - phosphate sensor, purple - clathrin binding sites). Arrestins encode two key domains, the arrestin_N domain (wheat) and the arrestin_C domain (light pink). Other regions that are present in the crystal structure are shown in light green, while sequence parts missing therein are shown in white. c – Functional elements depicted in B are mapped to the crystal structure of bovine ARRB1. The clathrin binding sites are missing in the crystal structure as they are situated on exons 13 and 15. PDB: 1G4R [55]. Crystal structure images were created with Pymol 1.8.4.0 Open-Source [113]
Arrestins have been found in Choanoflagellata, Filasterea and Metazoa, which all belong to Holozoa [11]. Within Metazoa, arrestins are found in both deuterostomes and protostomes [8, 9, 11]. In contrast to the rest of the arrestin clan, the sequences of arrestins are highly conserved [5]. Mammalian arrestins were studied extensively in the past [12–14]. There are four paralogs, functionally divided into the visual and non-visual group, each composed of two members. The visual arrestins, arrestin-1 (formerly known as rod arrestin) and arrestin-4 (formerly known as cone arrestin or X-arrestin) are encoded by the genes SAG and ARR3, respectively. The non-visual arrestins, arrestin-2 and arrestin-3 (also known as b-arrestin1 and b-arrestin2), are encoded in humans by the genes ARRB1 and ARRB2. Both functional groups are clearly monophyletic. Visual arrestins exhibit a much higher evolutionary rate than non-visual arrestins [15, 16].
GPCRs engage the concave side of arrestins [17–19]. Receptor binding leads to the reorganization of arrestin’s polar core and three-element interaction inducing a conformational change and resulting in the release of arrestin’s C-terminus [17, 20, 21]. The C-terminus of non-visual arrestins harbors binding sites for AP-2 and clathrin (Fig. 1 b, c) [1].
Arrestin-1 is the prevalent form in mouse cones, suggesting that it can bind to cone pigments [22]. Arrestin-1 is well known for binding to rhodopsin with high specificity, preferring it over other GPCRs [19, 23]. In contrast, binding specificity of arrestin-4 is ensured by its co-expression with cone opsins in cone photoreceptors, as in vitro arrestin-4 binds non-visual GPCRs fairly well [24]. In contrast, non-visual arrestins are expressed in all cell types and have a broad receptor specificity recognizing several hundred different GPCRs.
Individual arrestins from non-mammalian vertebrates have been cloned for functional studies. Among them are visual arrestins from frogs [25–27], salamander [13] and gecko [28]. Phylogenetic analyses support 1:1 orthology with their human counterparts. [29] reported co-expression of two distinct arrestin-1 genes, termed SAGa and SAGb, in rods of medaka (Oryzias latipes) and [3] identified two zebrafish paralogs (Danio rerio) for each visual arrestin ortholog in human, as well as two zebrafish paralogs for arrestin-3. They concluded that three additional arrestin genes originated from the teleost-specific whole genome duplication event (3R-WGD). [15] reported the expression of a visual and a non-visual arrestin in arctic lamprey’s pineal organ (Lethenteron camtschaticum). [30] showed that the vase tunicate (Ciona intestinalis), has only a single arrestin with functional features of both visual and non-visual subtypes. This suggests that the divergence of visual and non-visual arrestins is indeed associated with the vertebrate-specific whole genome duplications (2R-WGD). A comprehensive phylogenetic analysis to test this hypothesis, however, still has been missing.
While the cloning of individual arrestins led to the discovery of unexpected duplications and subfunctionalizations, the evolutionary history of arrestins has not been studied systematically. The information on arrestin homologs presently available covers only a very limited range of species [8] and an incomplete set of paralogs for most species investigated [9]. The objective of this study was to systematically investigate the duplication and deletion history of arrestins in deuterostomes. Sequence and exon-intron structure conservation are evaluated to gain insight into possible functional changes of the less studied members of the protein family and to elucidate nature’s repertoire of signaling interfaces relating to arrestins.
Results
We were working with two data sets that resolve arrestin phylogeny on two levels. In a database analysis, we placed arrestins in a wider evolutionary context (first subsection), while in a second analysis we focused on a narrower set of sequences covering only deuterostome arrestins (all other subsections). The interest in the detection of positive selection and co-variation requires a complete collection of paralogs per genome, a highly accurate annotation of the exon-intron structure on nucleotide level and transfer of the functional annotation between homologs. For this purpose, we needed carefully reconstructed coding sequences of the individual family members even when situated on genome fragments (see Methods). This level of accuracy is currently not provided by databases for non-model organisms. This has been a limitation to previous studies on arrestin evolution. We applied the ExonMatchSolver (EMS) pipeline and manually curated the annotation of deuterostome arrestins. We demonstrate that in comparison to a coarse database analysis, the exon-intron structure focused homology search is in fact a more successful strategy to trace the details of arrestin evolution. For example, considering paralog counts, OrthoDB under- and overpredicted the number of paralogs in 20 and five of 57 species, respectively. In general, we found paralogs that are missing from OrthoDB (Fig. 2). OrthoDB overpredicted sequences due to mis-assembly (in pig, Sus scrofa), inclusion of a pseudogene (in opossum, Monodelphis domestica), a naming mistake (in human), and included two additional sequences without any further reference (in lancelet, Branchiostoma floridae and acorn worm, Saccoglossus kowalevskii). We added five species critical to resolve the arrestin genealogy that were not included in OrthoDB (Lytechinus variegatus, Patiria miniata, Leucoraja erinacea, arctic lamprey and Orycteropus afer afer). The updated annotation is in general more complete than the respective database entries and represent a fundamental improvement in regard to the annotation of splice sites, short and terminal exons. We argue that our approach demonstrates how detailed curation can change and improve the detailed duplication and deletion history of an individual gene. The updated arrestin annotation represents one of the very rare instances of a highly curated set of paralogous genes and thus is ideal for evaluation of gene annotation tools and orthology prediction tools.
Fig. 2.
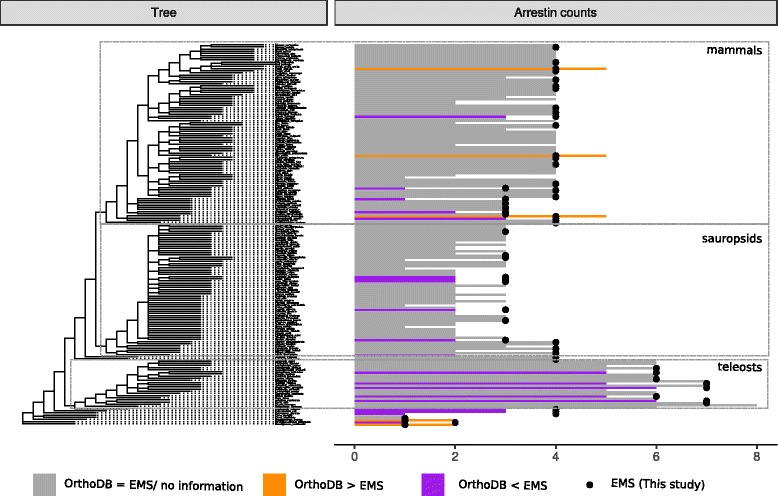
Number of deuterostome arrestin paralogs resulting from the application of the ExonMatchSolver (EMS) pipeline and manual curation in comparison with the OrthoDB database. Higher and lower paralog counts were obtained by genome mining in combination with manual curation for 20 species (purple) and five species (orange), respectively, as compared to the OrthoDB. The paralog counts and annotations obtained with the EMS approach and that are based on an expert opinion, are assumed to be correct. OrthoDB overpredicted sequences due to mis-assembly (Sus scrofa), inclusion of a pseudogene (Monodelphis domestica), a naming mistake (Homo sapiens), included two additional sequences without any further reference (Branchiostoma floridae, Saccoglossus kowalevskii). Arrestins correspond to the OrthoDB group EOG091G05M2
Placing deuterostome arrestins in a wider evolutionary context
To obtain an updated overview of the evolution of proteins that harbor an arrestin_N and arrestin_C domain, we queried UniProtKB and OrthoDB in a jackhmmer search with profile Hidden Markov Models (pHMMs) built from the four human arrestin full-length sequences. We found very remote homology to the scaffolding proteins DSCR3 and VPS26B, that contain a Vps26 domain, as reported previously in [8, 10]. The following domains are members of the Arrestin N-like clan (CL0135) in Pfam 31.0, which corresponds to the arrestin clan described in the literature: arrestin_C, arrestin_N, Spo0M, Vps26 and transcends this classification by inclusion of the domains LDB19 and Bul1_N (both restricted to Fungi). Restricting our search to a homology level, where the arrestin_N domain and arrestin_C domain can be detected with their respective Pfam HMMs (PF00339, PF02752), results in a set of ten members in human in accordance with [8] (ARRDC1-5, TXNIP, SAG, ARRB1, ARRB2, ARR3). We refer to this group as the arrestin fold family. These homologs form four orthologous groups supported by phylogenetic inference with both, full-length and single domain sequences (arrestins, ARRDC1, ARRDC2/ARRDC3/ARRDC4/TXNIP, ARRDC5, Additional file 1: Figure S1, Additional file 2). The monophyly of each group, arrestins and the ARRDC2-4/TXNIP, is further supported by the strict conservation of their exon-intron structure within the respective groups in humans (arrestins: 13-16 exons, see Fig. 10, ARRDC2-4/TXNIP: 8 exons). ARRDC1 shares three exon-intron boundaries with the ARRDC2-4/TXNIP group supporting ARRDC1 as the closest outgroup to ARRDC2-4/TXNIP, while ARRDC5 shares the two existing exon-intron boundaries with both, ARRDC1 and ARRDC2-4/TXNIP. The arrestin and ARRDC2-4/TXNIP groups expanded at the base of vertebrates with generally lower paralog numbers in non-vertebrate deuterostomes and protostomes (Additional file 1: Figures S2 and S4). While the majority of vertebrate arrestin fold family members belongs to one of these four orthology groups (OrthoDB-IDs: EOG091G0B0Y, EOG091G07XG, EOG091G0CVZ, EOG091G05M2), more diversity is seen in protostomes with numerous lineage- or clade-specific extensions (Additional file 1: Figure S2 A). Striking lineage-specific extensions occurred e.g. in Caenorhabditis (nematodes) and Polypedilum (flies), that posses up to 30 arrestin homologs as described by [8, 11]. The emergence of the arrestin, ARRDC1 and ARRDC2-4/TXNIP groups predates the split of protostomes and deuterostomes, while the ARRDC5 group is amniota-specific (Additional file 1: Figure S2 A). At least two of the four surveyed metazoan species outside of Bilateria additionally possess three orthology groups that do not have representatives in human (Additional file 1: Figure S2 A). To determine the existence of arrestin homologs in even deeper branching clades, we considered the results of the scan of Pfam arrestin domain models (PF00339, PF02752) and the full-length human arrestins against UniProtKB, which covers more species than OrthoDB (Additional file 1: Figure S3). 79% of all our hits against the UniProtKB database with the full-length query contain at least one arrestin_N and one arrestin_C domain covering the clades of Metazoa, Fungi, Amoebozoa, Alveolata and Stramenophiles with at least three species representatives of each of these clades (Additional file 1: Figures S3 and S5).
Fig. 10.

Evolutionary changes in exon-intron structure of arrestins. a - Exon-intron structure of the bovine ARRB1 gene. Exon and intron numbering is imposed onto arrestin homologs by sequence alignment. Positions of introns refer to their position on the amino acid sequence of cow arrestin-2 with a-c indicating their position after the first, second or third base of the codon, respectively. b - Exon-intron structure of arrestins (right hand side) is associated with a simplified gene tree (left hand side). Exons are shown as grey and colorful boxes, whereby homologous regions are “aligned” below each other. Colored exons highlight differences in exon-intron structure (intron gain, intron loss, exon loss). Changes in intron positions in comparison to the reference amino acid sequence of cow arrestin-2 are given whenever deviating except for the positions surrounding exons 13 and 15, which occasionally deviated by few nucleotides in our annotation (see Additional file 1: Appendix 7). Information about the corresponding exons was not available in the genomes if boxes are surrounded by a dotted line, but are assumed to be the same as in the 1:1 ortholog of the closest relative. If an unequivocal scenario of intron loss or gain is in accordance with the maximum parsimony principle, these events are indicated in the phylogenetic tree. Paralogs of species that share the exon-intron structure are summarized to phylogenetic clades, e.g. ARRB1 vertebrates. Structural differences in comparison to the family are shown right below associated with the corresponding species or phylogenetic clade. Losses of coding sequence (exons) are indicated by black pentagons with respective exons given as a number in the pentagon. The phylogenetic tree was created using Treegraph 2.0.54 [114]. c - Exon-intron structure of lamprey arrestins. Note that the length of the exon boxes is drawn to scale
We additionally detected hits in the following clades with one representative each: bacteria (Sorangium cellulosum), virus (Canarypox virus) and Chlorophyta (Chlorella variabilis). Our results confirm the absence of arrestin fold proteins in Embryophyta and their low abundance in Chlorophyta described by [11]. Our phylogenetic inference also confirms that the arrestin fold protein in Canarypox virus probably originated from horizontal gene transfer of a vertebrate member of the ARRDC2-4/TXNIP group (Additional file 1: Figure S1) [10]. Arrestins clearly form a monophyletic group within the group of arrestin fold proteins, which expanded in deuterostomes to give rise to the four paralogs seen in humans (Fig. 2).
Emergence of the four vertebrate arrestin paralogs by whole genome duplications (2R-WGD)
The arrestin sequences retrieved from the genomes of jawed vertebrates (the updated annotation) fall into four well separated orthology groups, each of which contains one of the four human arrestins (Additional files 3 and 4). Phylogenetic trees of the gene family, furthermore, show that the visual arrestins, SAG and ARR3, form a well supported monophyletic group. Disregarding ARRB2 of lampreys, the same applies to the non-visual arrestins, ARRB1 and ARRB2. The split of non-visual arrestins and ARR0 is well supported in the Bayesian tree and the Maximum likelihood tree with selected sequences (see ML tree with basal arrestins in Additional file 1: Figure S7 and Bayesian tree in Additional file 4), while this split is not well supported in the ML tree including all curated arrestin sequences. In order to check that this tree topology is not the result of convergent evolution of visual arrestins, we removed the parts of the sequence that are known to mediate receptor binding [2, 17, 19, 23, 31–33]. The truncated alignment still produces the same tree topology (Additional file 5).
The scenario best supported by the data is the existence of one visual and one non-visual proto-arrestin derived from a single arrestin, referred to hereafter as ARR0 (Fig. 3). ARR0 subsequently gave rise to two arrestins each (Fig. 3 b). All investigated non-vertebrate arrestins cluster together in a well-supported subtree. ARR0 is most similar to the non-visual vertebrate arrestins, especially to ARRB1 (average identity of all ARR0 to human ARRB1 61.9%).
Fig. 3.
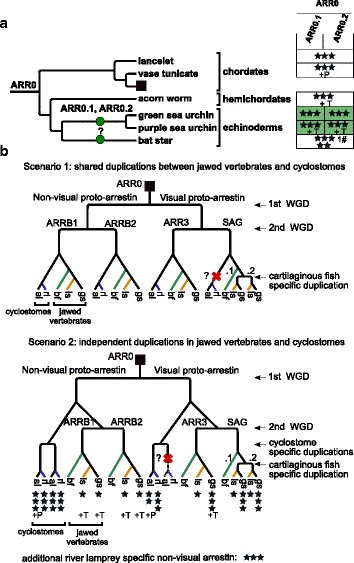
Duplication and deletion of arrestin paralogs within basal deuterostomes. a - Species tree of basal deuterostomes with mapped duplication events of arrestins (dots). b - Schematic arrestin gene tree for vertebrates (square in A). A cross indicates a gene loss. New gene names are given above the dot or branch. The gene loss/ duplication pattern was simplified for bony fish (bf), see Fig. 6, Fig. 8 and Additional file 1: Figure S13 for details. The completeness of arrestin annotations in the respective genomes is depicted with three stars indicating 0-3 missing exons, two stars 4-8 missing exons, one star more than 8 missing exons and dash (-) that no gene fragments were detected. Additional support from other omics-data for cartilaginous fishes and jawless fishes and from experimentally validated genbank entries is indicated by the following abbreviations: T - transcriptome evidence, P - protein evidence. The hash (#) indicates the number of frame shift mutations contained in the exon annotation. Note that the order of cyclostome-specific and cartilaginous fish-specific duplications in relation to each other was chosen arbitrarily. An additional non-visual arrestin detected in the germline genome of river lamprey was not included in the scenario (see Additional file 1: Appendix 1). Phylogenetic trees were created with Treegraph 2.0.54 [114]
In order to pinpoint the exact timing of the divergence of the four vertebrate arrestins, we focused on arrestins in available genomes of river lamprey (Petromyzon marinus) and arctic lamprey. Cyclostomes, including lampreys, are the sister clade of the jawed vertebrates (Gnathostomes). The pattern of arrestin distribution in lampreys is heterogeneous with different numbers of paralogs retrieved from the germline and somatic genome of river lamprey. However, the germline genomes of both lamprey species harbor at least one visual and two non-visual arrestins that are clearly 1:1 orthologs (Fig. 4, Additional file 3). A third, complete non-visual arrestin is encoded in the germline genome of river lamprey (see Additional file 1: Appendix 1 for details about arrestins in lampreys). One group of lamprey non-visual arrestins (ARRB2 lampreys) clusters with ARR0 with high support, while the other forms a monophyletic group with vertebrate ARRB1, albeit with low support (Fig. 4, Additional file 4). The visual arrestin from arctic lamprey clusters together with vertebrate ARR3 with high support. The position of the putative lamprey ARRB1 and ARR3 within the tree is in agreement with a shared 2R-WGD. However, the exact timing of the emergence of the four arrestin paralogs and thus the exact timing of the first and second round of the 2R-WGD cannot be resolved unambiguously with the available data, see Fig. 3 b for two possible tree scenarios. We return to this issue in the discussion.
Fig. 4.
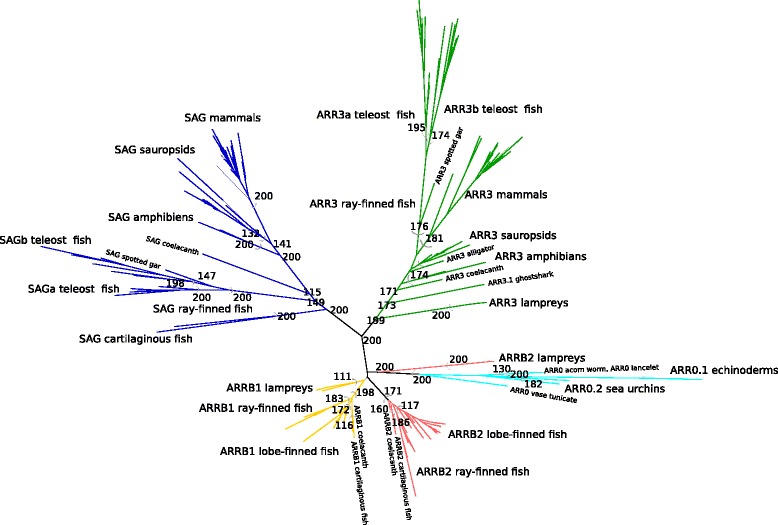
Maximum likelihood tree of arrestins. The tree was constructed from an amino acid alignment of deuterostome arrestins using PhyML (model JTT+I+G with α 1.04, 0.05% of invariable sites and 200 bootstraps). The different monophyletic and well-supported orthology groups are highlighted in different colors. Bootstrap support values from 50...100% are shown for the labeled monophyletic groups. The phylogenetic tree was visualized with Dendroscope 3.5.7 [115]
Tandem duplication of ARR0 in sea urchins
Most non-vertebrate deuterostome genomes encode a single ARR0 gene (Fig. 3 a). The most notable exceptions are three echinoderms. The sea urchins Strongylocentrotus purpuratus and Lytechinus variegatus possess two paralogous ARR0 genes with a mean sequence identity of 61%. They are located about 110 kb apart indicating that they are the result of a tandem duplication. The ARR0.1 genes show an accelerated substitution rate in comparison to ARR0.2 as indicated by long branch lengths within the phylogenetic tree. ARR0.1 of both sea urchins carries specificity determining positions (SDP) that are distinct from homologous positions in all other investigated ARR0s. These differences include charge reversing substitutions at positions known to be important for phosphate sensing [19], inositol-6-phosphate (IP6) binding [34] and AP-2 binding [35] (Fig. 5 a, c, d, see Additional file 6). Furthermore, receptor binding residues are different (Fig. 5 b). After the duplication and before speciation of green and purple sea urchin, different fractions of sites evolved under positive selection in ARR0.1 and ARR0.2, 15% and 5%, respectively (see Additional file 6). Before speciation of green and purple sea urchin, positions involved in or neighboring to receptor binding sites as well as to IP6 binding residues are positively selected in the ARR0.1 branch (Table 1). Furthermore, we find sequences in the bat star Patiria miniata suggesting the presence of two ARR0 genes, despite their near identity (exonic nucleotide sequences are 98.7% identical, intronic nucleotide sequences are 89% identical). Clearly, these two copies are the result of a very recent duplication independent of the duplication event that generated the much older paralogs in the sea urchins.
Fig. 5.

Specificity determining positions discriminating between sea urchin ARR0.1 and ARR0s including ARR0.2 from sea urchins. Amino acid frequency logos are shown for ARR0 and ARR0.1 of sea urchins ordered by functionality of motifs known from studies in vertebrate arrestins (a–d). Positions that are known to directly confer the respective functionality are marked by arrows. Some mutations change the charge of the respective residue (marked with *). Positions identified by SDP analysis are highlighted by black boxes. As receptor specificity is mediated by a rather big interface, only the SDPs are shown that are known to be involved in receptor binding and their direct neighbors. An additional position that shows differences in both groups (manually identified) and is associated with the respective function is highlighted by a dotted box. The numbering of the positions refers to bovine ARRB1. See the following references: [2] (pos. 14), [32] (pos. 67, 78, 80, 82 in finger loop region), [17] (pos. 154, 233), [19] (pos. 242), [23] (pos. 245, 247, 248, 249) for receptor binding residues, [34] (pos. 157, 160, 161, 165, 232, 236, 250, 324, 326) for IP6 binding residues, [19] (pos. 165, 169) for phosphate sensing and [35] (pos. 385, 388, 391, 393, 395) for AP-2 binding residues. Results are also summarized in Additional file 6. The figure was created with Weblogo [116]
Table 1.
Positively selected residues detected with the BEB method
| Foreground branch | BEB sites | Homologous position in cow paralog | Function known from homologs | SDP? |
|---|---|---|---|---|
| ARR0.1 sea urchins | S76 | N83 | Second neighboring to receptor binding residue | x |
| E95 | E102 | - | x | |
| K116 | K157 | Low affinity IP6 binding site | x | |
| N121 | N162 | Neighboring to low affinity IP6 binding site | x | |
| N184 | N225 | Second neighboring to receptor binding residue | - | |
| C201 | C242 | Receptor binding | x | |
| N296 | N382 | Second neighboring to clathrin binding site | - | |
| ARR0.2 sea urchins | K82 | P89 | Neighboring to PxxP motif | - |
| SAG.1 ghost shark | K2 | K2 | - | na |
| V106 | P134 | Neighboring to receptor binding residue | na | |
| N114 | R171 | Phosphate sensor | na | |
| T128 | G185 | Neighboring to PxxP motif | na | |
| N160 | G217 | - | na | |
| H205 | E262 | Receptor binding | na | |
| Q248 | N305 | Second neighboring to polar core | na | |
| V277 | T334 | Second neighboring to high affinity IP6 binding site | na | |
| S27 | G27 | Second neighboring to receptor binding residue | na | |
| SAGb teleost | V14 | V35 | Second neighboring to polar core | x |
| R126 | W194 | Receptor binding | - | |
| SAGb Acanthopterygii | P72 | P93 | Neighboring to PxxP motif | na |
| A112 | A180 | Neighboring to PxxP motif | na | |
| D142 | S210 | - | na | |
| ARR3b Euteleosteomorpha | Y42 | M55 | Neighboring to mu2 adaptin binding site | x |
| C150 | F254 | Neighboring to receptor binding residue | x |
The branch-site model of the PAML package was used to identify sites under positive selection in the specified foreground branch. The position in column two refers to the position within the group alignment, while the homologous position in cow serves as a reference. The position in ARR0 is given in respect to ARRB1 in cow. The function assignment is based on literature review. See Additional file 6 for further details. Positions that were also identified as specificity determining position (SDP), are marked by a cross. SDP were not determined for all subgroups as indicated by “na”
Tandem duplication of SAG in cartilaginous fishes
The jawed vertebrates are divided into two major subgroups, the bony fish (including reptiles, birds, and mammals) and the cartilaginous fish comprising the chimaeras, sharks, and rays. SAG is tandem-duplicated in the ghost shark genome, the only available chimaera genome (Callorhinchus milii). The two copies, SAG.1 and SAG.2, are located about 8 kb apart on opposite strands. With the help of the EMS pipeline and additional manual curation, we also found support for a second SAG gene in the draft assembly of the genome of the little skate, Leucoraja erinacea (see Additional file 1: Appendix 2 for details on annotation of arrestins in cartilaginous fish). Therefore, the tandem duplication of SAG occurred before the split of chimaeras and sharks between 413-473 mya (Fig. 3 b). The protein sequences of arrestin-1.1 and arrestin-1.2 of ghost shark have an identity of 51% and 55%, respectively, to the single arrestin-1 of spotted gar. About 13% of sites are under positive selection in ghost shark SAG.1 (Table 1, see Additional file 6). Among these are two residues involved or directly neighboring to a receptor binding residue. The basic residue R171 is conserved among SAG, with only a few exceptions. However, it is replaced by an acidic asparagine in ghost shark’s arrestin-1.1., probably impairing its function as a phosphate sensor.
Increase of arrestin number in ray-finned fish as a consequence of 3R-WGD
The bony fish are formed by the class of lobe-finned and ray-finned fish, with the majority of living representatives of the latter falling into the infraclass of teleosts (Fig. 6). The genome of spotted gar (Lepisosteus oculatus), a ray-finned fish outside the teleosts, encodes four arrestin paralogs, while all investigated teleosts have six or seven arrestin genes (Fig. 6), confirming and extending the results of [29] and [3]. The increased number of paralogs is explained by the teleost-specific round of genome duplications (3R-WGD) that happened between 230-315 mya (Additional file 1: Figure S7). 3R-WGD potentially resulted in eight arrestin paralogs [3]. We hypothesize that one copy of ARRB1 was lost already before the divergence of Otomorpha and Euteleosteomorpha during the initial 85 million years (max.) after the 3R-WGD (Fig. 6, Additional file 1: Figure S7) [36]. The other three pairs of copies are retained in the ancestor of the eight investigated teleosts. The ancestral ARRB2b evolved under neutral evolution and was lost independently along two different branches of Euteleosteomorpha (Fig. 6), while the majority (80%) of ARRB2a sites evolved under purifying selection directly after duplication. ARRB2a and ARRB2b of Otomorpha are overall very similar (average of 90% identity), while ARRB2b of stickleback (Gasterosteus aculeatus) and pufferfish (Takifugu rubripes) are a lot more divergent from ARRB2a of the same species (average 79.3% identity). Supervised multiple correspondence analysis (MCA) shows each of these two sequences as a separate cluster that is also clearly distinct from the group of all other ARRB2 in teleosts. This observation strongly suggests a change in function. Differences identified by manual inspection concern residues binding to IP6 (K161Q) [34], the phosphate sensor R166 (mutated to Q) [19], AP-2 binding residues (R395G) [35] as well as residues that mediate receptor specificity (e.g. P253Q/S) [19] (see Additional file 6).
Fig. 6.
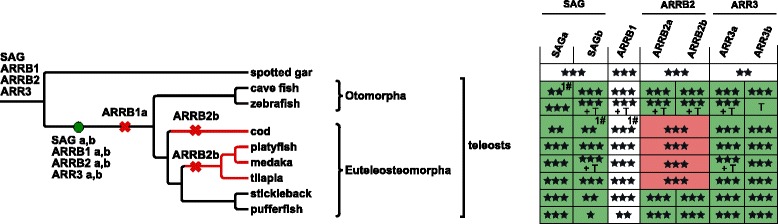
Duplication and deletion of arrestin paralogs within ray-finned fish. The teleost whole genome duplication (WGD) increases the number of arrestin paralogs within the two clades Otomorpha and Euteleosteomorpha. The two resulting copies of each paralog (SAG, ARRB1, ARRB2, ARR3) are depicted as a and b. Zebrafish ARR3b was annotated in GRCz10 as the gene was missing in the originally investigated genome version Zv9. The species tree was created based on [117] using Treegraph 2.0.54 [114]. Crosses depict gene losses. See caption of Fig. 3 for additional description of symbols
The paralogous pairs of SAGa and SAGb as well as ARR3a and ARR3b persisted in all investigated teleost species and evolved with similar rates since their emergence. SAGas/SAGbs and ARR3as/ARR3bs are recognized as separate groups in unsupervised MCA applied to alignments of SAG and ARR3 in fish, respectively, emphasizing their sequence divergence. SDPs of both paralogous groups are involved in phosphate sensing and receptor binding (Fig. 7 a–d). About 17% and 13% of residues evolved under positive selection in the ancestral branches of SAGa and SAGb, respectively (Table 1, Additional file 6). See Additional file 1: Appendix 3 for a detailed description of the evolution of SAGb and ARR3b in different orders of teleosts.
Fig. 7.

Specificity determining positions discriminating each pair of duplicated visual arrestins in teleosts. Amino acid frequency logos are shown for SAGa vs. SAGb (a, b) and for ARR3a vs. ARR3b (c, d) in teleosts. Positions that are known to directly confer a specific functionality in mammalian arrestins are marked by arrows. Of these, some mutations change the charge of the respective residue (marked with *). Positions identified by SDP analysis are highlighted by black boxes. As receptor specificity is mediated by a rather big interface, only the SDPs are shown that are known to be involved in receptor binding and their first and second order neighbors. Additional positions that show differences in both groups (manually identified) and might be associated with the respective function are highlighted with a dotted box. See [2] (pos. 10, 77, 81/76, 82, 319/313), [33] (pos. 195, 254/248), [23] (pos. 52, 54/49, 265), [17] (pos. 157, 273), [19] (pos. 90/85, 244, 267, 246/240, 261/255) for references of receptor binding residues, [19] (pos. 171/165, 175/169) for phosphate binding and [118] (pos. 163/157, 166/160, 167/161) for IP6 binding residues. The numbering refers to the position numbers in bovine SAG and ARR3, respectively. Results are also summarized in Additional file 6. The figure was created with Weblogo [116]. Ins - Insertion in comparison to reference
Loss or pseudogenization of ARR3 in Afrotheria, Xenarthra, and the common shrew
Within the second clade of bony fish, the lobe-finned fish, a single gene for each of the four paralogs is retained with a few exceptions: (1) Loss or pseudogenization of ARR3 in Afrotheria, Xenarthra and common shrew (Sorex araneus); (2) Retrogene formation and pseudogenization of ARRB1 and ARRB2 in marsupials; (3) likely loss of ARRB2 in birds (Fig. 8).
Fig. 8.
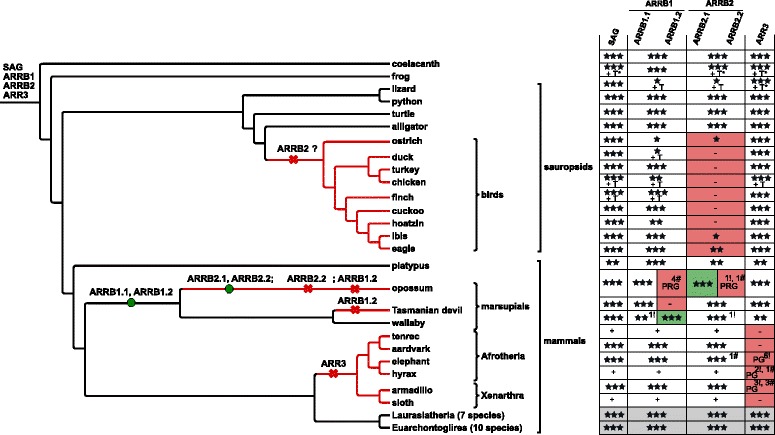
Duplication and deletion of arrestin paralogs in lobe-finned fish. ARRB2 could not be detected in the genomes and transcriptomes of birds (see Additional file 1: Table S1 for other 41 investigated bird species). Additional omics-data was investigated for sauropsids. The gene loss/duplication pattern was simplified for the monophyletic groups highlighted in light grey (see Additional file 1: Appendix 4). See caption of Fig. 3/6 for description of symbols. The exclamation mark (!) indicates the number of stop codons contained in the exon annotation, while plus (+) indicates that gene (parts) are encoded within the respective genome, but were not annotated in detail. Note that the order of the ARRB2.2 and ARRB1.2 losses is arbitrary. The phylogenetic tree was created using Treegraph 2.0.54 [114]. PG - pseudogene; PRG - pseudo-retrogene
In the genomes of African elephant, Loxodonta africana, and rock hyrax, Procavia capensis, ARR3 orthologs are degraded to pseudogenes to different extents (see Additional file 1: Appendix 4 for details about the investigation of the ARR3 locus in the respective species). Both species belong to the superorder Afrotheria. In contrast, ARR3 is completely lost in the genome of armadillo, Dasypus novemcinctus, which belongs to the taxonomic group of Xenarthra. An independent degradation of ARR3 to a pseudogene was observed in the genome of common shrew (see Additional file 1: Appendix 4).
Retrogene formation and pseudogenization of ARRB1/ARRB2 in marsupials
Another peculiarity within mammals is the identification of an additional ARRB1 and ARRB2 gene in the marsupial genomes of opossum and wallaby (Macropus eugenii) (Fig. 8). Both genes are encoded by a single exon, the main characteristic of a retrogene. While the ARRB1.2 retrogene seems functional in wallaby (Additional file 1: Figure S6), it has turned into a pseudo-retrogene in opossum indicated by four frame shift mutations within the potentially protein-coding region. Applying the parsimonious principle, we assume that a processed ARRB1-mRNA was inserted into the nuclear genome of the common ancestor of both species between 82-177 mya before split of Didelphimorphia and Australidelphia [37] (Fig. 8, Additional file 1: Figure S7). Remarkably, both ARRB1.2 retrogenes share high conservation within the putative 5’ untranslated region as annotated by Ensembl for wallaby multi-exon ARRB1.1 (Additional file 1: Figure S6). In the third investigated marsupialian genome, the Tasmanian devil (Sarcophilus harisii), the ARRB1 retrogene is completely lost.
Additionally, an ARRB2 retrogene was inserted within a cluster of zinc-finger transcription factors on chromosome 3 in the lineage leading to opossum. However, the retrogene turned into a pseudogene containing a premature stop codon and an insertion resulting in a frame-shift mutation (Fig. 8).
Possible loss of ARRB2 in birds
To our surprise, hardly any fragments of ARRB2 were detected in bird genomes or lizard (Anolis carolinensis), while the respective ortholog was easily detectable in the genomes of other Sauropsids, e.g. alligator (Alligator mississippiensis), turtle (Pelodiscus sinensis) and python (Python molurus). This raised the possibility of a loss of the ARRB2 gene within these species. Extensive homology search in 50 bird genomes retrieved only five species that harbor two or more complete exons of this 15 exon gene ARRB2 (Additional file 6). All detected exons have a high sequence identity to orthologous exons in turtle (on average 91.3%, at least 83.9%). The potential loss was further tested by investigating genomic synteny of ARRB2 and expression of ARRB2 in transcriptome data (see Additional file 1: Appendix 5 for details). Neither strategy provided evidence to reject the hypothesis that ARRB2 has been lost in birds. In contrast, a query of the NCBI EST database retrieved both non-visual arrestin transcripts in lizard confirming the integrity of the ARRB2 gene in reptiles.
Loss and gain of functional elements
Scanning the Pfam28.0 database using hmmscan confirmed that more than 95% of all annotated deuterostome arrestins possess an arrestin_C and an arrestin_N domain (see Additional file 1: Appendix 6 for details about other domains). As expected, known key functional motifs such as the phosphate sensing residues [6], the polar core residues [20], the residues involved in the three element interaction, and the sequence of the receptor-binding finger loop [32] are conserved in all ARR0 and vertebrate arrestins (Fig. 9). The great majority of residues of all arrestins evolved under strong purifying selection and are highly conserved. However, recently duplicated paralogs can behave differently in respect to conservation and selection (Additional file 6).
Fig. 9.
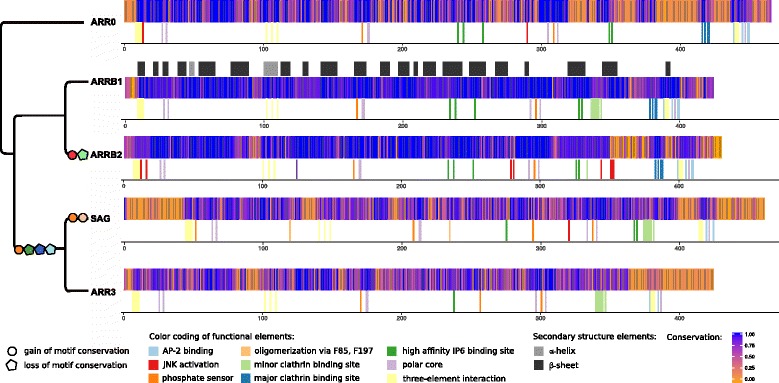
Changes in conservation patterns and functional motifs of arrestins. Conservation of alignment positions of the individual orthology groups is shown. The conservation was calculated according to the Method of Karlin [110] using AACon [111] for each orthology group separately. Sequences with a coverage < 90% as well as all lamprey sequences were excluded. Functional motifs characterized in one or several paralogs were projected onto the individual alignments solely based on sequence homology. Putative loss (pentagon) and gain (circle) events based on conservation of the respective motifs were projected onto a simplified arrestin gene tree. The order of motif gain and loss on the respective branch was chosen arbitrarily. Positions were not marked if they did not conserve the amino acid known to be part of the motif in that orthology group in a majority of representatives. Some positions are marked although their conservation is restricted to a phylogenetic group as indicated by their lower conservation score (e.g. oligomerization is specific for Sarcopterygii SAG). The secondary structure based on the crystal structure of 1G4R (Fig. 1) is mapped onto the alignment of ARRB1. Note that only a selection of known motifs are shown
We propose that the duplication of ARR0 led to the emergence of new functionalities that are commonly conserved in the respective orthology group in vertebrates. For example, arrestin-3 binds and activates JNK3, while arrestin-2 does not [38–40]. The residues S13 and C17 previously identified to mediate JNK3 binding and activation are strictly conserved in all ARRB2 except for lamprey and ARRB2b pufferfish [41] (Fig. 9). ARR0 not only shows residues conserved among non-visual arrestins, but also paralog-specific positions with ARRB1 and ARRB2 in the N-terminal 25 residues. The conservation of most other positions known to mediate JNK activation is restricted to a phylogenetic group of ARRB2 such as conservation of H350D351H352 in mammals and of L278xS280 in lobe-finned fish, respectively [40]. An exception is position V343 in the C domain of arrestin, which is conserved in all ARRB2 except Otomorpha ARRB2a. Interestingly, all sea urchin ARR0.1 sequences carry a conserved valine here, while all other ARR0 carry threonine at the homologous position, which is characteristic for arrestin-2.
In both visual arrestins, the high affinity IP6 binding site, the AP-2 binding site and the major clathrin binding site are not conserved or loosely conserved, in contrast to non-visual arrestins (Fig. 9). Other key mutations in comparison to ARR0 involve A253D, which was hypothesized to weaken the hydrogen bond network of the pre-activated state in comparison to non-visual arrestins [42]. An additional phosphate-binding residue, R18, is conserved in all SAG sequences [24]. The residues F85 and F197, which are known to be involved in oligomerization of SAG [43] are strictly conserved in SAG of the lobe-finned fish. The C-terminus of teleost ARR3 is shorter than in ARR3 of other vertebrates. For example, the C-terminus of ARR3a and ARR3b in zebrafish is 31 and 24 amino acids, respectively, shorter than the C-terminus of ARR3 in spotted gar. The residues missing in zebrafish are known to be responsible for the three-element interaction, AP-2 binding and contribute an arginine to the polar core [10] (Figs. 9 and 10).
Fine-tuning of receptor endocytosis is regulated by various post-translational modifications at positions conserved within but not across orthology groups (not shown). Phosphorylation of S412 of ARRB1 regulates clathrin binding and endocytosis [44]; phosphorylation of S/T360 in ARRB2 regulates clathrin-mediated internalization [45]; nitrolysation of C409 in ARRB2 promotes binding to clathrin and AP-2 [46]. Other positions known to be phosphorylated and involved in the regulation of internalization, binding of clathrin (T382 in ARRB2) and interaction with mu2-adaptin (Y54 in ARRB1) are mammalian-specific and, thus, represent recent evolutionary innovations. Additional information on conservation of possible isoforms can be found in Additional file 1: Appendix 7 and Additional file 6.
Hotspot of exon gain/loss at positions determining receptor specificity
The exon-intron structure of the vertebrate arrestin paralogs is highly conserved, preserving the majority of exon-intron boundaries of their last common ancestor, ARR0 (Fig. 10). Nevertheless, changes in gene structure including loss of coding sequence, intron gain or loss are much more frequent in the arrestin gene family than in other vertebrate gene families [47]. In accordance with the propensity for these events in paralogous gene families as discussed by [48, 49], these gene structure changes mainly occurred within arrestin genes that underwent a tandem duplication (exemplified by loss of exon 16 in SAG of ghost shark) or WGDs (loss of exon 16 in ARR3 of teleosts, gain of intron 85c in ARRB2a of Euteleosteomorpha, loss of intron 138c in ARRB2b and of intron 333b in ARR3b of Otomorpha). This can be further illustrated by to the emergence of the four arrestin paralogs by 2R-WGD from ARR0 accompanied by at least one intron loss event (intron 7b) in SAG and a loss of coding sequence in the ancestor of SAG and ARR3, as well as in ARRB2 (exons 15 and 13, respectively) (Fig. 10 b, Additional file 1: Figure S7). Interestingly, we observed the gain of intron 85c between 148-230 mya in the ancestor of Euteleosteomorpha, a branch of teleosts, for which frequent intron gains were described previously for several GPCRs and the serpin gene family [47, 50, 51].
A parsimony reconstruction of intron loss and gain points out a hotspot of intron gain at position 85c (Fig. 10 b/c, see Additional file 1: Appendix 8 for details). Introns were gained five times independently at position 85c of deuterostome arrestins. Four of these events occurred at the exact same position, while the exact position of intron gain in the river lamprey-specific non-visual arrestin cannot be resolved with the available data. This paralog is excluded from the following conclusions. Two intron gains occurred within vertebrates, a very rare event for this clade [47, 52].
Introns are known to preferentially insert into sequences that carry an upstream AG and a downstream G in respect to the insertion site. This site, “AG |intron|GY”, has been termed protosplice site in literature [53], whereby | denotes a splice site. Alignment of the intron-containing paralogs with their intron-deficient orthologs of closely related species revealed a prevalence of intron gain at this position caused by the existence of a protosplice site in all intron-containing paralogs (Fig. 11). Newly gained intron sequences of the respective arrestin paralogs did not have any apparent sequence homology. This architecture suggests intron-insertion mediated by an endonuclease, which cuts downstream of AGGY in the exon thereby producing sticky ends. A transposon than inserts into this locus [54]. Missing nucleotides are probably filled up by a DNA polymerase resulting in two identical motifs at the 5’- and 3’-end of the inserted sequence establishing the canonical splice site AG |GT-intron-AG |GY. There is no codon that spans exons 5.1 and 5.2, the first and the second part of exon 5, respectively. The last codon of exon 5.1, CAG, is translated into glutamine, which is conserved in all but two inspected arrestins (Fig. 10). The first codon of exon 5.2 is much less conserved translating into different non-polar, aliphatic amino acids (L, M, I, A, V) in visual arrestins (V90 in SAG, V85 in ARR3) and into small amino-acids (A, S) in non-visual arrestins with three exceptions (S86 in ARRB1, A87 in ARRB2).
Fig. 11.

Alignment of exon-intron borders after insertion of intron 85c into exon 5. Intron 85c is found in ARR0 of bat star (Pmi) and vase tunicate (Cin), but not in acorn worm (Sko) or lancelet (Bfl) (highlighted in grey). Exon 5 of one of the non-visual arrestins in lampreys (shown: Lca) as well as in ARRB2 in all Euteleosteomorpha (Gmo, Gac, Ola, Oni, Tru, Xma) is split into two parts, denoted as 5.1 and 5.2. In contrast, exon 5 of ARRB2 is not split in Otomorpha (Dre, Ame) and spotted gar (Lco) (grey). Only the 5’- and 3’-parts of the intron sequences are shown (green box), while the larger inner region is left out being non-informative (black lines). The proto-splice site motif ‘AGGY’ is conserved for all species genes shown except for Otomorpha (‘AAGC’). The alignment was visualized with Jalview 2.8.1 [93]
Interestingly, V90 in bovine arrestin-1 is not surface-exposed. It is located between the two β-sheets of the N domain, making contacts with several other hydrophobic residues [55]. Its substitution with a small side chain residue characteristic for non-visual arrestins (A or S) enables arrestin-1 binding to non-cognate M2 muscarinic receptor [55]. Therefore, large hydrophobic residue in this position likely makes the N domain more rigid, predisposing an arrestin to be more GPCR subtype-specific [19, 56].
Discussion
We uncovered the complex duplication and deletion history of the arrestin family in deuterostomes based on a careful evaluation of genomic information. Our approach enhanced by manual curation outperforms conventional strategies that rely on uncurated databases to infer orthology relationships (OrthoDB).
We show that the paralog counts differ for 25 species (44%) between the updated annotation and OrthoDB, a frequently used database that is considered to be fairly complete (Fig. 2). The majority of deviations is caused by an underestimation of paralog counts in OrthoDB exemplifying the incompleteness of this database. Although patterns of absence and presence of paralogs are conveniently presented in OrthoDB, it remains an open problem to distinguish paralog losses from missing information for biological and technical reasons in automated procedures. Biological reasons are high degree of divergence, duplications, and pseudogenizations. Over- and especially underprediction of paralogs is mostly caused by technical issues, e.g. due to sequencing techniques, sequencing coverage, annotation and assembly strategies. Particular care should be taken when inferring the expected number of paralogs from the maximum number of paralogs in the database, proposing an exaggerated rate of losses. A successful strategy to perform gene family annotation uses a whole genome homology search, a priori information about genome duplications and exon-intron structure. Consideration of lowly sampled taxonomic groups phylogenetically close to gene loss and gain events is critical to resolve the genealogy. Information extracted from current protein databases can thus just deliver a preliminary overview on paralog counts and orthology relationships. Here, we establish a high-quality standard of a small curated data set that can be used as a benchmark for annotation and orthology prediction tools. As homology search methods propagate erroneous annotations, the improvement of existing annotations and methodology for annotation and orthology predictions is a necessity in computational biology [57, 58].
The majority of deuterostome arrestin paralogs arose by WGD, either at the root of vertebrates or at the root of teleosts. These events promoted the acquisition of new functions and changes in exon-intron structure of arrestins.
The 2R-WGD led to the emergence of the four arrestin paralogs from a prototypical arrestin presumably similar to ARR0 in vase tunicate in accordance to [30, 59]. Arrestins are in line with several other gene families of the phototransduction cascade, e.g. opsins, G protein-coupled receptor kinases and transducins, all of which expanded by the basal vertebrate tetraploidizations and thus paved the way for the development of a sophisticated visual system in the vertebrate clade [59]. Some studies place the 2R-WGD after the split of jawless fish and jawed vertebrates suggesting independent duplications in the lamprey-lineage [60, 61], other studies argue that both 2R-WGDs took place at the root of jawed vertebrates, followed by an immediate split of both groups [62, 63]. The current study of one gene family, also cannot resolve this controversy. It remains unclear, therefore, whether the lamprey arrestins represent 1:1 orthologs to the vertebrate arrestins or arose from independent duplications after a shared first WGD.
A third WGD resulted in further increase in the number of arrestin paralogs in teleosts. Visual arrestins were more readily retained after duplication than non-visual arrestins. [3] made the first attempt to functionally characterize ARR3a and ARR3b in zebrafish, which they found specifically expressed in the outer layer of either M- and L-cones (ARR3a) or of S- and UV-sensitive cones (ARR3b).
In addition to spatial subfunctionalization, we propose that expansion and diversification of opsins is paralleled by functional diversification of ARR3a and ARR3b. This is supported by [3], and our comparative analysis revealing mutations of receptor binding residues. As a second example of functional subfunctionalization, we find SDPs of phosphate and IP6 binding residues affecting binding specificity, in agreement with functional studies showing that SAGa and SAGb have different binding affinities for phosphorylated rhodopsin in carp [64].
The duplicated non-visual arrestins, ARRB2a and ARRB2b, were shown to have similar functions in modulating the distribution of the chemokine ligand Cxcl12a, but different spatial expression patterns in zebrafish primordial germ cells [65]. These paralogs are nearly identical in zebrafish. In contrast, ARRB2 of stickleback and pufferfish carry mutations in key functional motifs presumably impairing their function.
In addition to 3R-WGDs, local duplications such as tandem duplications or retrogene insertions contributed to the emergence of more arrestin paralogs within smaller classes or infraclasses. The sea urchin-specific tandem duplication of ARR0 seems to be in line with the over-representation of arrestin-interaction partners such as the secretin-like GPCR superfamily [66] and the rhodopsin-type GPCRs expressed in sensory appendages and the nervous system in purple sea urchin [67]. Furthermore, the Ras-superfamiliy of G proteins regulated by arrestins, as well as receptor protein tyrosine phosphatases regulating arrestin binding to GPCRs, are expanded in sea urchin hinting at a general expansion of molecules involved in GPCR signaling [68, 69]. ARR0.1 is suggested to have acquired a new function in connection with receptor binding, enhanced phosphate sensing and, possibly, reduced binding to the clathrin adapter protein AP-2.
In conclusion, a common theme for fine tuning or specialization of arrestins after duplication seem to be the following two degrees of freedom: receptor binding and phosphate sensing.
In addition to expansions of the arrestin system we also found some losses, in particular, the well-documented pseudogenization/loss of ARR3 in Afrotheria, Xenarthra, and common shrew. Less obvious is the possible loss of ARRB2 in birds. The study, simply based on comparative genomics, is strongly dependent on genome coverage and the quality of the available assemblies. As these differ widely among vertebrate genomes, we were particularly cautious hypothesizing about exon or gene losses. Whenever possible, multiple data sources and strategies were used. When available, we took into account additional transcriptome data and investigated arrestin genes in additional, closely related species to obtain information on synteny. Nevertheless, we cannot rule out that individual genes, such as bird ARRB2 escaped sequencing or assembly even after having considered 50 bird genomes in this study. The incompleteness of avian genomes, and the difficulty of sequencing certain regions in these genomes is a well known, albeit poorly understood, phenomenon [70–72]. Regions known to cause difficulties in sequencing and assembly are heterochromatin and repeat regions [73], and also bird microchromosomes in general. It remains unclear, therefore, whether the single, detected exons of ARRB2 in birds and of ARR3 in hedgehog are remnants of a pseudogene or of an intact gene. The exons we identified for ARRB2 have a high sequence conservation in comparison to mammalian ARRB2, cover different parts of the gene and are generally situated on short contigs suggesting difficulties in sequencing and/or assembly. On the other hand, we did not detect any transcripts of ARRB2 in various transcriptome data sets representing different tissues and developmental states of several bird species. One has to bear in mind, though, that the expression of the ubiquitous arrestin-3 could be too low to detect its transcripts as arrestin-3’s expression is 10-20-times lower than that of arrestin-2 in most mammalian cells [74].
In contrast, the loss of ARR3 could be shown explicitly for Afrotheria and Xenarthra based on the synteny information. ARR3 is specifically expressed in cones and pinealocytes [75], where it inactivates phosphorylated cone opsin and interacts with additional binding partners e.g. Mdm2, JNK3 [76], Als2Cr4 [77] or MKK4, ASK1 [4] acting as a scaffolding molecule. Whereas it is the only visual arrestin expressed in L- and M-cones in humans and several primates [78], both, SAG and ARR3, are expressed in primate S-cones [78] and all mouse cones [22].
The evolutionary need for ARR3 has already been discussed in the literature emphasizing differences between SAG and ARR3, namely the ability of SAG to self-assemble and the transient binding affinity of ARR3 to receptors [4]. ARR3 null mice have an impaired contrast sensitivity and visual acuity when young, while their cones seem to degenerate slower with increasing age in comparison to wild type (WT) mice as shown recently [79]. However, other studies in mice suggest that the arrestin-4 function can be fulfilled by arrestin-1. The response of S-dominant cones of ARR3 null mice to light stimuli is similar to WT mice, while recovery from flashes is greatly slowed down in SAG/ARR3 double-knock out mice [22]. The authors concluded that at least one visual arrestin is necessary for inactivation of S- or M-opsin in mice. Additionally, [80] showed that arrestin-1 can inactivate S-opsin metaII in transgenic mice expressing S-opsin instead of rhodopsin in rods, although arrestin-1 does not seem to be necessary for dim-flash response in WT cones. Thus, these studies suggest that arrestin-1 could take over the arrestin-4 function if expressed in cones, which is consistent with the loss of ARR3 reported in our study. Alternatively, other adaptions could have evolved in Afrotheria and Xenarthra to compensate for the loss of ARR3.
Although the evolution of e.g. mammalian arrestins has been examined previously [9], the present study uncovered numerous previously unreported gene gain and loss events within arrestins in deuterostomes. Identification of residues that determine specificity and are positively selected after duplication was made possible by a high quality alignment obtained by genome inquiries, dense species sampling and consideration of fragmented loci from poorly assembled genomes. The residues identified during this study as evolutionary “adjusting screws” are candidate positions for construction of biased arrestins that were already approved by nature.
Methods
Database scans
For performing the homology search, we generated a pHMM using jackhmmer with an alignment of the four human arrestins as input querying the UniProtKB database (accessed via https://www.ebi.ac.uk/Tools/hmmer/, 30 June 2017). The level of homology retrieved with different jackhmmer iterations was checked by comparing to the results of a homology search with the arrestin_N domain and arrestin_C domain HMM as downloaded from Pfam 31.0 (PF00339, PF02752, E-value < 0) in order to obtain a good overlap of both strategies. The full-length set of homologs obtained from UniProtKB was filtered according to length (422 > length > 195, μ +- σ), E-value (< 0) and identity of the full-length sequences for each species separately (< 80%). The identity filter cut-off was chosen to balance the removal of isoforms and retention of paralogs. We obtained a set of 2962 sequences, of which 2348 contained at least one arrestin_N and one arrestin_C domain (Additional file 1: Figure S5). 142 sequences did not have either of both domains and were excluded. We proceeded with the full-length sequences of this set under exclusion of hits that were not assigned to one specific species, for phylogenetic inference, and for reporting paralog counts projected on the NCBI phylogeny. In order to exclude effects on phylogenetic inference that can arise from aligning sequences that are not homologous in full-length, we additionally generated individual domain sets for the arrestin_N and arrestin_C domain, respectively, and also proceeded to phylogenetic inference. These sets consist of the respective Pfam model hit in the UniProtKB database restricted to the actual hmmsearch hit length. Both sets were filtered according to identity (see above).
Furthermore, we queried OrthoDB (as of Feb. 2017) with full-length arrestin pHMMs (E-value < 0) obtained with jackhmmer. OrthoDB is considered to be a high quality orthology database, which contains unique orthology group assignments for proteins of interest on a given taxonomic level. We restricted our analysis to the OrthoDB groups that are annotated on Metazoa level and retrieved 3487 hits that belong to 109 orthology groups. For better visibility while plotting, we only distinguish between groups that have more than 29 members. These 9 groups cover 88% of all sequences. Other orthology groups are denoted as “Other” (Additional file 1: Figure S2). The NCBI species tree was retrieved with the ete toolkit [81].
Detailed gene annotation
Automated methods frequently fail to correctly predict multi-exon genes. We therefore used exon- and paralog-specific pHMMs to update the annotation of arrestin genes in different genomes of interest ([82], see next paragraph). Exon models were built from an initial, manually curated protein alignment of mammalian arrestins (see Additional file 1: Appendix 9 for details about reconstruction of the initial alignment). It was then extended by adding the translated exon sequences from arrestins successively annotated in other lobe-finned and ray-finned fishes using HMMER 3.1b1 [83]. These exon- and paralog-specific models were then handed over to the EMS pipeline [82]. Simplified, the EMS performs a two step procedure: (1) a homology search with the provided pHMMs, (2) assignment of exon- and paralog-specific hits to one paralog based on an integer linear programming formulation of the paralog-to-contig assignment problem. In contrast to other methods, the EMS pipeline considers paralogs that are fragmented over several, often short contigs and assembles these paralog fragments to a more complete annotation. The assignment of paralogs to contigs is explicitly solved and provides the starting point for manual curation.
In case of a systematic failure to detect a specific arrestin exon within a monophyletic family with the EMS pipeline, the candidate region was re-investigated with different homology-based methods. These included local blastall 2.2.26 querying a region between two exon hits with the nucleotide sequence of the missing exon(s) applying blastn or, with the protein sequence of the conceptually translated exon, respectively, applying tblastn [84]. To detect exons that differed substantially among homologous groups, we aligned the corresponding regions of at least three close relatives of one group with tba.v12 [85] and applied RNAcode 0.3 to detect conserved regions with protein coding potential [86].
Genome versions used for the current study
Unmasked genomes were extracted from Ensembl, EnsemblPre! or Ensembl Metazoa if available and from the listed sources otherwise (Additional file 1: Table S1). For ghost shark, only a soft-masked version of the genome was available. To clarify the potential loss of ARRB2 in birds, all available 48 bird genomes from the Avian Phylogenomics Project [87], the genomes of kiwi (Apteryx australis mantelli) and gold eagle (Aquila chrysaetos) were investigated additionally. All four arrestin paralogs were annotated in nine birds in total (ostrich, chicken, turkey, duck, finch, ibis, hoatzin, cuckoo, bald eagle). Insertions and stop codons were occasionally observed within exons of arrestin genes in genomes with low coverage and/or poor quality assemblies. We interpreted these as sequencing or assembly errors because the remainder of the protein-coding sequence was usually highly conserved, except in cases which we explicitly identified as pseudogenes in the current study (e.g. elephant ARR3).
Investigation of transcriptome, EST and SRA data
Transcriptome data sets, in particular the NCBI Expressed Sequence Tag (EST) and NCBI Transcriptome Shotgun Assembly data sets, were additionally queried whenever the analysis of the corresponding genome was not conclusive. We used the NCBI webinterface to tblastn with protein sequences of closely related species as queries in these cases (Additional file 1: Table S2). Clades that were queried are “Sauropsida”, “Aves”, “Marsupilia”, “Chondrichthyes” and “Cyclostomata” [88]. NCBI Short Read Archive (SRA) was queried with the known arrestin kiwi exons against SRA data of ostrich (Struthio camelus) and tinamu (Tinamus guttatus) as well as with arrestin exons from bald eagle (Haliaeetus leucocephalus) against SRA data of white-tailed eagle (Haliaeetus albicilla) and gold eagle. As the NCBI blast did not provide a blast-database for EST data of lizard, this was built locally and queried.
Alignment and building of phylogenetic trees
For generating a bootstrapped phylogenetic tree of the arrestin fold family, we aligned all hits obtained after filtering from the OrthoDB with clustalo 1.2.4. Next, we built an approximate maximum likelihood tree with FastTree with the -pseudo option for fragmented/gapped sequences and the following options to increase its accuracy/tree exploration -spr 4 -mlacc 2 -slownni.
For the tree of arrestins, we considered Genbank annotations of arrestins with experimental evidence (NP-entries) whenever available and more complete than the genomic annotations. The same is true for transcript evidence of arrestin paralogs. Coding DNA sequences were aligned according to codons with MACSE 1.01b [89] and further edited in mega 4.0.2 [90]. Maximum likelihood trees were built from protein sequences using PhyML 3.0.1 [91] after testing for optimal model parameters with ProtTest 3.4 allowing for the following substitution models: JTT, Dayhoff, WAG, LG, DCMut, Blosum62, an estimation of amino acid frequencies (-F), the fraction of invariable sites and a gamma-distribution (-all-distributions) [92]. Unknown amino acids were substituted by “?” in the alignment for tree building. The tree that obtained the best information content (BIC and AIC) applying ProtTest was used as starting tree for PhyML. The tree topology was validated by bootstrapping (1000 iterations unless stated otherwise). Manual inspection of the alignment revealed conservation or disruption of functional motifs previously investigated experimentally in mammals and known from literature, that were marked within the Jalview 2.8.2 alignment program ([93], Additional file 7).
Bayesian trees were constructed based on the amino acid alignment with the BEAST2 software [94] under the Birth-Death model with a relaxed molecular clock. We compared different model settings pairwise employing PathSampling [95, 96] to estimate the marginal likelihoods and calculating the Bayes factor (BF). A model was accepted if BF >3 [97]. Otherwise, the simpler model was chosen. The model settings differed in their birth-death priors and regarding estimation or fixation of different priors to specific values, while using the parameters determined with ProtTest as site model parameters.
Here, the best model had the following parameters: Relaxed Clock Log normal, birthRate Uniform, relative Death rate β-distributed (alpha = 1, beta = 10). For every model setting, several chains were combined after confirming that they converged to the same set of parameters with the help of Tracer v1.6 [98] and logcombiner. Trees were analyzed with treeannotator and visualized in FigTree [99].
Identification of SDPs
For identification of SDPs of closely related paralogs that arose from a recent duplication, respective sequences were grouped, aligned and filtered to contain a redundancy < 98% and coverage > 70%. The following groups were investigated: teleost SAGa, b, teleost ARR3a, b, teleost ARRB2a, b, all ARR0 including sea urchin ARR0.1. The filtered alignments were analyzed with four complementary SDP detection tools, the entropy-based sequence harmony approach (SH) [100, 101], the machine-learning approach multi-RELIEF [102, 103], Xdet, which is based on analysis of mutational behavior [104] and S3det based on MCA [105]. The first two approaches were run via the webserver [106], while the latter two are implemented in the program jdet 1.4.5. Positions retrieved with the default values of the respective programs (exception: S3det -m 2) were filtered according to the following, conservative cut-offs: SH z-scores < -6, multi-RELIEF-scores > 0.7 and Xdet-scores < 0.6. Group distinction was computed automatically (unsupervised) in S3det except for teleost ARRB2. Positions were only considered as specificity determining if they were retrieved with at least two of the four methods (see Additional file 6 for detailed results).
Testing for natural selection
To test for natural selection, we constructed alignments of coding DNA sequence restricted to specific sub-branches of interest. Regions encoding frame shift mutations, containing stop codons or gaps were excluded from further analysis. We excluded potential recombinant sequences by testing for recombination in the group alignments with the RDP4 software [107] (SAGa, b zebrafish, ARR3 stickleback). We assume that recombination and gene conversion can only occur within the same species and thus excluded incomplete lineage sorting for the species considered. Positive selection was tested on predefined foreground branches with the branch-site model of codeml inside the PAML program [108] (kappa to be estimated, F3X4 and Codon table tested as Codon frequency models). The significance of difference of the maximum log-likelihoods of the null model (w2=1) and the alternative model (w2≥1) was assessed by comparing the results of the likelihood ratio test with the distribution of p-values (<0.05). In case that the alternative model was significantly better than the null model, specific sites under positive selection were assessed according to the significance levels of the BEB method. Additionally, we performed bootstrapping and assessed the distribution and confidence intervals of the bootstrapped estimates with the codeml_sba [109] method. Some data sets show a slightly bimodal distribution of w2 and/or p1 and thus obtained rather uncertain parameter estimates (reported as μ, σ and upper and lower quartiles in Additional file 6). The fraction of sites under positive selection (p2) was calculated as follows: p2=1−(p0+p1).
Calculation of sequence conservation
Sequence conservation was calculated with the Karlin score [110] implemented in AACon [111] for alignments of individual orthology groups (SAG, ARRB1, ARRB2, ARR3) excluding lamprey sequences. To minimize the effect of missing data on conservation calculations, we filtered the alignments to contain sequences with a coverage > 90%.
The exon and intron numbering used throughout the manuscript is based on homology (refer to Fig. 10 a as reference). Positions of amino acids within the protein always refer to the homologous position in cow (Bos taurus, for ARR0, bovine ARRB2 is considered). Mutations are also reported in this coordinate system. D297Y therefore means that tyrosine (Y) is found in the species of interest at the amino acid position homologous to position 297 of the corresponding bovine arrestin, which is aspartic acid (D). Gene names are used according to recommendations of the HUGO Gene Nomenclature Consortium.
Additional files
This file includes Figures S1–S7, Table S1 (Genomes used) and Table S2 (Transcriptome/EST data used) as well as the following Appendices: Appendix 1 — Arrestin inventories in lampreys. Appendix 2 — Annotation of arrestins in cartilaginous fish. Appendix 3 — Evolution of visual arrestins in different orders of teleosts. Appendix 4 — Investigation of the ARR3 locus in Afrotheria, Xenarthra and common shrew. Appendix 5 — Investigation of loss of ARRB2 in Sauropsids. Appendix 6 — Domains of deuterostome arrestins. Appendix 7 — Isoforms and changes of the conserved exon-intron structure. Appendix 8 — Parsimonious reconstruction of intron gain/loss events. Appendix 9 — Annotation of arrestins in mammals. (PDF 2380 kb)
Approximate ML tree of the arrestin fold family as extracted from UniProtKB (depicted in Additional file 1: Figure S1). Hits were assigned to the arrestin fold family if they contained at least one arrestin_N or arrestin_C domain (see Methods). The tree was generated with the FastTree software and bootstrapping was performed 1000 times with SeqBoot [119]. The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (TREE 78.3 kb)
ML tree of arrestins (depicted in Fig. 4). Starting information for tree reconstruction was an alignment of arrestins investigated in this study, for which sequence information was close to complete. Sequences derived from genomic annotations were substituted by sequences with experimental evidence available if these were more complete. The tree was constructed using PhyML from the amino acid sequences of arrestin paralogs from an alignment of nucleotide sequences aligned with MACSE (model JTT+G+I with α 1.04, p-invariable 0.05, 200x bootstrapping). The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (NW 1340 kb)
Maximum clade credibility tree of arrestins. Starting from the same alignment as used for Additional file 3, we constructed a phylogenetic tree with BEAST2 [94] under the Birth Death model with a relaxed molecular clock (log normal) using the gamma site model (JTT+G+I with α 1.04, p-invariable 0.05). The tree can be visualized with a tree viewer, e.g. Figtree [99]. (TREE 253 kb)
ML tree of arrestins excluding columns known to confer receptor binding. The tree was constructed from the same alignment as Additional file 3 deleting the columns known to confer receptor specificity according to [2, 17, 19, 23, 31–33] (model JTT+G+I with α 1.05, p-invariable 0.05, 200x bootstrapping). The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (NW 7.42 kb)
Chromosomal locations: Chromosomal locations of arrestin genes. Table of chromosomal locations of arrestin genes in species with genomes assembled on chromosome-level. The columns “SAG(a)” contain the location of SAG for non-duplicated species (SAGb does not exist) or of SAGa for teleosts. This applies to all columns. Note that ARR3 is located on the sex chromosome X in all mammals.
SDP ARR0: SDPs distinguishing ARR0.1 from sea urchin ARR0.2 and other ARR0. The classification is based on unsupervised multi-correspondence analysis of all ARR0 with S3det. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARRB1 are listed. SDP SAG: SDPs distinguishing SAGa and SAGb in teleosts. The classification is based on unsupervised MCA of all filtered teleost SAG with S3det. During filtering, SAGb from pufferfish and stickleback were excluded due to sequence coverage < 70%. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine SAG are listed.
SDP ARRB2: SDPs distinguishing ARRB2a and ARRB2b in teleosts. The classification was given in supervised MCA of all filtered teleost ARRB2. Due to redundancy > 98%, ARRB2a of platyfish (Xiphophorus maculatus), pufferfish and medaka were excluded from the analysis. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARRB2 are listed.
SDP ARR3: SDPs distinguishing ARR3a and ARR3b in teleosts. The classification is based on unsupervised MCA of all teleost ARR3 with S3det. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARR3 are listed.
Codeml: Analysis of positive selection after arrestin duplication. Specific branches within the arrestin gene tree were tested for positive selection using the branch-site model of codeml, part of the PAML program. The null-hypothesis assumes purifying or neutral selection on the foreground and background branches, while the alternative model allows for positive selection on the foreground branch. Fractions of sites are given, that are predicted to belong to the respective classes (p) together with their dN/dS ratios (w). If the null hypothesis was rejected, sites that were under positive selection under BEB were mapped onto the respective bovine ortholog. (XLSX 44.5 kb)
Alignment of deuterostome arrestins with functional annotation known from experimental studies of arrestins in mammals. Note that the naming differs from the naming used throughout the manuscript with ARR1, ARR2, ARR3 and ARR4 used instead of SAG, ARRB1, ARRB2 and ARR3, respectively. Please load alignment with annotation file (provided at http://dx.doi.org/10.5281/zenodo.820866) in Jalview alignment viewer. (FA 44.5 kb)
Acknowledgments
We thank Chris Amemiya for sharing the germline genome of river lamprey and two anonymous reviewers for their comments.
Funding
This work was supported by the European Social Fund (ESF) of the European Union (EU) and the Free State of Saxony (grant numbers 100148833/22117017, 100227413 to HI) as well as NIH grants (RO1 DA030103, NS065868 to EVG and RO1 EY011500, R35 GM122491 to VVG).
Availability of data and materials
The data sets supporting the results of this article are available in the zenodo repository, DOI 10.5281/zenodo.820866. [120].
Authors’ contributions
HI annotated arrestin genes and performed subsequent analysis. PFS, SJP and VVG designed the study. VVG and EVG contributed biological knowledge on important sequence motifs and suggestions about regions of interest. All authors contributed in writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- BF
Bayes factor
- EMS
ExonMatchSolver
- EST
Expressed sequence tag
- GPCR
G-protein coupled receptor
- IP6
Inositol-6-phosphate
- MCA
Multiple correspondence analysis
- ML
Maximum likelihood
- mya
Million years ago
- pHMM
Profile Hidden Markov Model
- SDP
Specificity determining position
- SH
Sequence harmony
- SRA
Short read archive
- WGD
Whole genome duplication
- WT
Wild type
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-1001-4) contains supplementary material, which is available to authorized users.
References
- 1.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther. 2006;110(3):465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, Waal PWd, Ke J, Tan MHE, Zhang C, Moeller A, West GM, Pascal BD, van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JCH, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523(7562):561–7. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renninger SL, Gesemann M, Neuhauss StephanCF. Cone arrestin confers cone vision of high temporal resolution in zebrafish larvae. Eur J NeuroSci. 2011;33(4):658–67. doi: 10.1111/j.1460-9568.2010.07574.x. [DOI] [PubMed] [Google Scholar]
- 4.Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV. The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res. 2011;30(6):405–30. doi: 10.1016/j.preteyeres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luttrell LM. Molecular Biology of Arrestins. Progress in molecular biology and translational science, vol. v. 118. Oxford: Elsevier Science & Technology; 2013. [DOI] [PubMed] [Google Scholar]
- 6.Gurevich VV, Song X, Vishnivetskiy SA, Gurevich EV. Enhanced phosphorylation-independent arrestins and gene therapy. Handb Exp Pharmacol. 2014;219:133–52. doi: 10.1007/978-3-642-41199-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Yang Y, Ward R, An S, Guo XX, Li W, Xu TR. Biased signalling: the instinctive skill of the cell in the selection of appropriate signalling pathways. Biochem J. 2015;470(2):155–67. doi: 10.1042/BJ20150358. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol. 2008;8:222. doi: 10.1186/1471-2148-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurevich EV, Gurevich VV. Arrestins: ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7(9):236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aubry L, Klein G. True arrestins and arrestin-fold proteins: a structure-based appraisal. Prog Mol Biol Transl Sci. 2013;118:21–56. doi: 10.1016/B978-0-12-394440-5.00002-4. [DOI] [PubMed] [Google Scholar]
- 11.Mendoza AD, Sebe-Pedros A, Ruiz-Trillo I. The evolution of the GPCR signaling system in eukaryotes: modularity, conservation, and the transition to metazoan multicellularity. Genome Biol Evol. 2014;6(3):606–19. doi: 10.1093/gbe/evu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 1998;391(6670):918–21. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- 13.Smith WC, Gurevich EV, Dugger DR, Vishnivetskiy SA, Shelamer CL, McDowell JH, Gurevich VV. Cloning and functional characterization of salamander rod and cone arrestins. Investig Ophthalmol Vis Sci. 2000;41(9):2445–55. [PubMed] [Google Scholar]
- 14.Maeda T, Ohguro H, Sohma H, Kuroki Y, Wada H, Okisaka S, Murakami A. Purification and characterization of bovine cone arrestin (cArr) FEBS Lett. 2000;470(3):336–40. doi: 10.1016/S0014-5793(00)01334-X. [DOI] [PubMed] [Google Scholar]
- 15.Kawano-Yamashita E, Koyanagi M, Shichida Y, Oishi T, Tamotsu S, Terakita A. beta-arrestin functionally regulates the non-bleaching pigment parapinopsin in lamprey pineal. PloS ONE. 2011;6(1):16402. doi: 10.1371/journal.pone.0016402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hisatomi O, Imanishi Y, Satoh T, Tokunaga F. Arrestins expressed in killifish photoreceptor cells. FEBS Lett. 1997;411(1):12–18. doi: 10.1016/S0014-5793(97)00640-6. [DOI] [PubMed] [Google Scholar]
- 17.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci U S A. 2006;103(13):4900–5. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281(6):3458–62. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem. 2011;286(27):24288–4299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. A Model for Arrestin’s Regulation: The 2.8 Å Crystal Structure of Visual Arrestin. Cell. 1999;97(2):257–69. doi: 10.1016/S0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 21.Kim M, Vishnivetskiy SA, van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, Gurevich VV, Hubbell WL. Conformation of receptor-bound visual arrestin. Proc Natl Acad Sci U S A. 2012;109(45):18407–12. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EN, Craft CM. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59(3):462–74. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface. Structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279(2):1262–8. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 24.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3A: evolution of receptor specificity. J Mol Biol. 2005;354(5):1069–80. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Craft CM, Whitmore DH. The arrestin superfamily: cone arrestins are a fourth family. FEBS Lett. 1995;362(2):247–55. doi: 10.1016/0014-5793(95)00213-S. [DOI] [PubMed] [Google Scholar]
- 26.Mani SS, Besharse JC, Knox BE. Immediate upstream sequence of arrestin directs rod-specific expression in Xenopus. J Biol Chem. 1999;274(22):15590–7. doi: 10.1074/jbc.274.22.15590. [DOI] [PubMed] [Google Scholar]
- 27.Abdulaeva G, Hargrave PA, Smith WC. The sequence of arrestins from rod and cone photoreceptors in the frogs Rana catesbeiana and Rana pipiens. Localization of gene transcripts by reverse-transcription polymerase chain reaction on isolated photoreceptors. Eur J Biochem / FEBS. 1995;234(2):437–42. doi: 10.1111/j.1432-1033.1995.437_b.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Wensel TG, Yuan C. Tokay gecko photoreceptors achieve rod-like physiology with cone-like proteins. Photochem Photobiol. 2006;82(6):1452–60. doi: 10.1111/j.1751-1097.2006.tb09834.x. [DOI] [PubMed] [Google Scholar]
- 29.Imanishi Y, Hisatomi O, Tokunaga F. Two types of arrestins expressed in medaka rod photoreceptors. FEBS Lett. 1999;462(1-2):31–6. doi: 10.1016/S0014-5793(99)01483-0. [DOI] [PubMed] [Google Scholar]
- 30.Nakagawa M, Orii H, Yoshida N, Jojima E, Horie T, Yoshida R, Haga T, Tsuda M. Ascidian arrestin (Ci-arr), the origin of the visual and nonvisual arrestins of vertebrate. Eur J Biochem / FEBS. 2002;269(21):5112–118. doi: 10.1046/j.1432-1033.2002.03240.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J Mol Biol. 2011;406(3):467–78. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szczepek M, Beyrière F, Hofmann KP, Elgeti M, Kazmin R, Rose A, Bartl FJ, Stetten Dv. Heck M, Sommer ME, Hildebrand PW, Scheerer P. Crystal structure of a common GPCR-binding interface for G protein and arrestin. Nat Commun. 2014;5:4801. doi: 10.1038/ncomms5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostermaier MK, Peterhans C, Jaussi R, Deupi X, Standfuss J. Functional map of arrestin-1 at single amino acid resolution. Proc Natl Acad Sci U S A. 2014;111(5):1825–30. doi: 10.1073/pnas.1319402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C. Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem. 2006;281(14):9812–23. doi: 10.1074/jbc.M512703200. [DOI] [PubMed] [Google Scholar]
- 35.Burtey A, Schmid EM, Ford MGJ, Rappoport JZ, Scott MGH, Marullo S, Simon SM, McMahon HT, Benmerah A. The conserved isoleucine-valine-phenylalanine motif couples activation state and endocytic functions of beta-arrestins. Traffic (Copenhagen, Denmark) 2007;8(7):914–31. doi: 10.1111/j.1600-0854.2007.00578.x. [DOI] [PubMed] [Google Scholar]
- 36.Sato Y, Hashiguchi Y, Nishida M. Temporal pattern of loss/persistence of duplicate genes involved in signal transduction and metabolic pathways after teleost-specific genome duplication. BMC Evol Biol. 2009;9:127. doi: 10.1186/1471-2148-9-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nilsson MA, Churakov G, Sommer M, van Tran N, Zemann A, Brosius J, Schmitz J. Tracking marsupial evolution using archaic genomic retroposon insertions. PLoS Biol. 2010;8(7):1000436. doi: 10.1371/journal.pbio.1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science (New York, NY) 2000;290(5496):1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 39.Song X, Coffa S, Fu H, Gurevich VV. How does arrestin assemble MAPKs into a signaling complex? J Biol Chem. 2009;284(1):685–95. doi: 10.1074/jbc.M806124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo J, Tsakem EL, Breitman M, Gurevich VV. Identification of arrestin-3-specific residues necessary for JNK3 kinase activation. J Biol Chem. 2011;286(32):27894–901. doi: 10.1074/jbc.M111.260448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhan X, Stoy H, Kaoud TS, Perry NA, Chen Q, Perez A, Els-Heindl S, Slagis JV, Iverson TM, Beck-Sickinger AG, Gurevich EV, Dalby KN, Gurevich VV. Peptide mini-scaffold facilitates JNK3 activation in cells. Sci Rep. 2016;6:21025. doi: 10.1038/srep21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim YJ, Hofmann KP, Ernst OP, Scheerer P, Choe HW, Sommer ME. Crystal structure of pre-activated arrestin p44. Nature. 2013;497(7447):142–6. doi: 10.1038/nature12133. [DOI] [PubMed] [Google Scholar]
- 43.Hanson SM, Dawson ES, Francis DJ, van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV. A model for the solution structure of the rod arrestin tetramer. Structure. 2008;16(6):924–34. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. Clathrin-mediated Endocytosis of the -Adrenergic Receptor Is Regulated by Phosphorylation/Dephosphorylation of -Arrestin1. J Biol Chem. 1997;272(49):31051–7. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 45.Lin FT, Chen W, Shenoy SK, Cong M, Exum ST, Lefkowitz RJ. Phosphorylation of beta-Arrestin2 Regulates Its Function in Internalization of beta 2 -Adrenergic Receptors †. Biochemistry. 2002;41(34):10692–9. doi: 10.1021/bi025705n. [DOI] [PubMed] [Google Scholar]
- 46.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31(3):395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ragg H, Kumar A, Köster K, Bentele C, Wang Y, Frese MA, Prib N, Krüger O. Multiple gains of spliceosomal introns in a superfamily of vertebrate protease inhibitor genes. BMC Evol Biol. 2009;9:208. doi: 10.1186/1471-2148-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Babenko VN, Rogozin IB, Mekhedov SL, Koonin EV. Prevalence of intron gain over intron loss in the evolution of paralogous gene families. Nucleic Acids Res. 2004;32(12):3724–33. doi: 10.1093/nar/gkh686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy SW, Penny D. On the incidence of intron loss and gain in paralogous gene families. Mol Biol Evol. 2007;24(8):1579–1581. doi: 10.1093/molbev/msm082. [DOI] [PubMed] [Google Scholar]
- 50.Kumar A, Bhandari A, Sinha R, Goyal P, Grapputo A. Spliceosomal intron insertions in genome compacted ray-finned fishes as evident from phylogeny of MC receptors, also supported by a few other GPCRs. PloS ONE. 2011;6(8):22046. doi: 10.1371/journal.pone.0022046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar A. Bayesian phylogeny analysis of vertebrate serpins illustrates evolutionary conservation of the intron and indels based six groups classification system from lampreys for ∼500 MY. PeerJ. 2015;3:1026. doi: 10.7717/peerj.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coulombe-Huntington J, Majewski J. Characterization of intron loss events in mammals. Genome Res. 2007;17(1):23–32. doi: 10.1101/gr.5703406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sverdlov AV, Rogozin IB, Babenko VN, Koonin EV. Reconstruction of ancestral protosplice sites. Curr Biol. 2004;14(16):1505–8. doi: 10.1016/j.cub.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 54.Yenerall P, Zhou L. Identifying the mechanisms of intron gain: progress and trends. Biol Direct. 2012;7:29. doi: 10.1186/1745-6150-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal Structure of beta-Arrestin at 1.9 Å. Structure. 2001;9(9):869–80. doi: 10.1016/S0969-2126(01)00644-X. [DOI] [PubMed] [Google Scholar]
- 56.Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J Biol Chem. 2012;287(35):29495–505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Promponas VJ, Iliopoulos I, Ouzounis CA. Annotation inconsistencies beyond sequence similarity-based function prediction - phylogeny and genome structure. Standards Genomic Sci. 2015;10:108. doi: 10.1186/s40793-015-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koepfli KP, Paten B, O’Brien SJ. The Genome 10K Project: a way forward. Annu Rev Animal Biosci. 2015;3:57–111. doi: 10.1146/annurev-animal-090414-014900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larhammar D, Nordström K, Larsson TA. Evolution of vertebrate rod and cone phototransduction genes. Philos Trans R Soc B Biol Sci. 2009;364(1531):2867–80. doi: 10.1098/rstb.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fried C, Prohaska SJ, Stadler PF. Independent Hox-cluster duplications in lampreys. J Exp Zool B Mol Dev Evol. 2003;299(1):18–25. doi: 10.1002/jez.b.37. [DOI] [PubMed] [Google Scholar]
- 61.Mehta TK, Ravi V, Yamasaki S, Lee AP, Lian MM, Tay BH, Tohari S, Yanai S, Tay A, Brenner S, Venkatesh B. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum) Proc Natl Acad Sci U S A. 2013;110(40):16044–9. doi: 10.1073/pnas.1315760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuraku S, Meyer A, Kuratani S. Timing of genome duplications relative to the origin of the vertebrates: did cyclostomes diverge before or after? Mol Biol Evol. 2009;26(1):47–59. doi: 10.1093/molbev/msn222. [DOI] [PubMed] [Google Scholar]
- 63.Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, Yandell MD, Manousaki T, Meyer A, Bloom OE, Morgan JR, Buxbaum JD, Sachidanandam R, Sims C, Garruss AS, Cook M, Krumlauf R, Wiedemann LM, Sower SA, Decatur WA, Hall JA, Amemiya CT, Saha NR, Buckley KM, Rast JP, Das S, Hirano M, McCurley N, Guo P, Rohner N, Tabin CJ, Piccinelli P, Elgar G, Ruffier M, Aken BL, Searle SMJ, Muffato M, Pignatelli M, Herrero J, Jones M, Brown CT, Chung-Davidson YW, Nanlohy KG, Libants SV, Yeh CY, McCauley DW, Langeland JA, Pancer Z, Fritzsch B, de Jong PieterJ, Zhu B, Fulton LL, Theising B, Flicek P, Bronner ME, Warren WC, Clifton SW, Wilson RK, Li W. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet. 2013;45(4):415–2142112. doi: 10.1038/ng.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomizuka J, Tachibanaki S, Kawamura S. Phosphorylation-independent suppression of light-activated visual pigment by arrestin in carp rods and cones. J Biol Chem. 2015. doi:10.1074/jbc.M114.634543. [DOI] [PMC free article] [PubMed]
- 65.Mahabaleshwar H, Tarbashevich K, Nowak M, Brand M, Raz E. beta-arrestin control of late endosomal sorting facilitates decoy receptor function and chemokine gradient formation. Development (Cambridge, England) 2012;139(16):2897–902. doi: 10.1242/dev.080408. [DOI] [PubMed] [Google Scholar]
- 66.Materna SC, Berney K, Cameron RA. The S. purpuratus genome: a comparative perspective. Dev Biol. 2006;300(1):485–95. doi: 10.1016/j.ydbio.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 67.Raible F, Tessmar-Raible K, Arboleda E, Kaller T, Bork P, Arendt D, Arnone MI. Opsins and clusters of sensory G-protein-coupled receptors in the sea urchin genome. Dev Biol. 2006;300(1):461–75. doi: 10.1016/j.ydbio.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 68.Byrum CA, Walton KD, Robertson AJ, Carbonneau S, Thomason RT, Coffman JA, McClay DR. Protein tyrosine and serine-threonine phosphatases in the sea urchin, Strongylocentrotus purpuratus: identification and potential functions. Dev Biol. 2006;300(1):194–218. doi: 10.1016/j.ydbio.2006.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fitzpatrick DA, O’Halloran DM, Burnell AM. Multiple lineage specific expansions within the guanylyl cyclase gene family. BMC Evol Biol. 2006;6:26. doi: 10.1186/1471-2148-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jarvis ED, Mirarab S, Aberer A, Houde P, Li C, Ho S, Faircloth BC, Nabholz B, Howard JT, Suh A, Weber CC, Fonseca RR, Alfaro-Nunez A, Narula N, Liu L, Burt DW, Ellegren H, Edwards SV, Stamatakis A, Mindell DP, Cracraft J, Braun EL, Warnow T, Jun W, Gilbert MTP, Zhang G. Phylogenomic analyses data of the avian phylogenomics project. GigaScience Database. 2014. doi:10.5524/101041.
- 71.Zhang G, Li B, Li C, Gilbert MTP, Jarvis ED, Wang J. Comparative genomic data of the Avian Phylogenomics Project. GigaScience. 2014;3(1):26. doi: 10.1186/2047-217X-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardner PP, Fasold M, Burge SW, Ninova M, Hertel J, Kehr S, Steeves TE, Griffiths-Jones S, Stadler PF, Mariño-Ramírez L. Conservation and Losses of Non-Coding RNAs in Avian Genomes. PloS ONE. 2015;10(3):0121797. doi: 10.1371/journal.pone.0121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109(3):421–36. doi: 10.1016/S0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- 75.Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269(6):4613–9. [PubMed] [Google Scholar]
- 76.Song X, Gurevich EV, Gurevich VV. Cone arrestin binding to JNK3 and Mdm2: conformational preference and localization of interaction sites. J Neurochem. 2007;103(3):1053–62. doi: 10.1111/j.1471-4159.2007.04842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuniga FI, Craft CM. Deciphering the structure and function of Als2cr4 in the mouse retina. Investig Ophthalmol Vis Sci. 2010;51(9):4407–415. doi: 10.1167/iovs.10-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Craft CM, Huang J, Possin DE, Hendrickson A. Primate Short-Wave cones share molecular markers with rods In: Ash J, Grimm C, Hollyfield JG, Anderson RE, LaVail MM, Rickman CB, editors. Retinal Degenerative Diseases: Mechanisms and Experimental Therapy. New York: Springer. p. 49–56.
- 79.Deming JD, Pak JS, Brown BM, Kim MK, Aung MH, Eom YS, Shin J-a, Lee EJ, Pardue MT, Craft CM. Visual Cone Arrestin 4 Contributes to Visual Function and Cone Health. Investig Ophthalmol Vis Sci. 2015;56(9):5407. doi: 10.1167/iovs.15-16647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi G, Yau KW, Chen J, Kefalov VJ. Signaling properties of a short-wave cone visual pigment and its role in phototransduction. J Neurosci Off J Soc Neurosci. 2007;27(38):10084–93. doi: 10.1523/JNEUROSCI.2211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol. 2016;33(6):1635–8. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Indrischek H, Wieseke N, Stadler PF, Prohaska SJ. The paralog-to-contig assignment problem: High quality gene models from fragmented assemblies. Algorithms Mol Biol. 2016;11(1):199. doi: 10.1186/s13015-016-0063-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7(10):1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 85.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, Haussler D, Miller W. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14(4):708–15. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Washietl S, Findeiss S, Müller SA, Kalkhof S, Bergen MV, Hofacker IL, Stadler PF, Goldman N. RNAcode: robust discrimination of coding and noncoding regions in comparative sequence data. RNA (New York, NY) 2011;17(4):578–94. doi: 10.1261/rna.2536111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang G, Li B, Gilbert MTP, Jarvis ED, Wang J. Avian Phylogenomics Project. 2014. http://avian.genomics.cn/en/jsp/database.shtml. [DOI] [PMC free article] [PubMed]
- 88.NIHNational Center for Biotechnology Information. Translated BLAST: tblastn. (19 November 2015). http://blast.ncbi.nlm.nih.gov/blast/Blast.cgi?PROGRAM=tblastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome. Accessed 9 Nov 2016.
- 89.Ranwez V, Harispe S, Delsuc F, Douzery EJP, Murphy WJ. MACSE: multiple alignment of coding SEquences accounting for frameshifts and stop codons. PloS ONE. 2011;6(9):22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol. 2007;24(8):1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 91.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 92.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–5. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Waterhouse AM, Procter JB, Martin DavidMA, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10(4):1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol. 2012;29(9):2157–67. doi: 10.1093/molbev/mss084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baele G, Li WLS, Drummond AJ, Suchard MA, Lemey P. Accurate model selection of relaxed molecular clocks in bayesian phylogenetics. Mol Biol Evol. 2013;30(2):239–43. doi: 10.1093/molbev/mss243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Drummond AJ, Bouckaert RR. Bayesian Evolutionary Analysis with BEAST. Cambridge: Cambridge University Press; 2015. [Google Scholar]
- 98.Rambaut A, Suchard MA, Drummond AJ. Tracer. 2014. http://beast.bio.ed.ac.uk/Tracer. Accessed 30 June 2017.
- 99.Raumbaut A. FigTree: Tree Figure Drawing Tool. 2006. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 30 June 2017.
- 100.Pirovano W, Feenstra KA, Heringa J. Sequence comparison by sequence harmony identifies subtype-specific functional sites. Nucleic Acids Res. 2006;34(22):6540–8. doi: 10.1093/nar/gkl901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feenstra KA, Pirovano W, Krab K, Heringa J. Sequence harmony: detecting functional specificity from alignments. Nucleic Acids Res. 2007;35(Web Server issue):495–8. doi: 10.1093/nar/gkm406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ye K, Feenstra KA, Heringa J, Ijzerman AP, Marchiori E. Multi-RELIEF: a method to recognize specificity determining residues from multiple sequence alignments using a Machine-Learning approach for feature weighting. Bioinformatics. 2008;24(1):18–25. doi: 10.1093/bioinformatics/btm537. [DOI] [PubMed] [Google Scholar]
- 103.Brandt BW, Feenstra KA, Heringa J. Multi-Harmony: detecting functional specificity from sequence alignment. Nucleic Acids Res. 2010;38(Web Server issue):35–40. doi: 10.1093/nar/gkq415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pazos F, Rausell A, Valencia A. Phylogeny-independent detection of functional residues. Bioinformatics. 2006;22(12):1440–8. doi: 10.1093/bioinformatics/btl104. [DOI] [PubMed] [Google Scholar]
- 105.Rausell A, Juan D, Pazos F, Valencia A. Protein interactions and ligand binding: from protein subfamilies to functional specificity. Proc Natl Acad Sci U S A. 2010;107(5):1995–2000. doi: 10.1073/pnas.0908044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brandt BW, Feenstra KA, Heringa J. multi-Harmony: multi-group Sequence Harmony & multi-Relief. 2016. http://www.ibi.vu.nl/programs/shmrwww/. Accessed 30 June 2016.
- 107.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24(8):1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 109.Bielawski JP, Baker JL, Mingrone J. Inference of episodic changes in natural selection acting on protein coding sequences via CODEML. Curr Protoc Bioinformatics. 2016;54:6–15161532. doi: 10.1002/cpbi.2. [DOI] [PubMed] [Google Scholar]
- 110.Karlin S, Brocchieri L. Evolutionary conservation of RecA genes in relation to protein structure and function. J Bacteriol. 1996;178(7):1881–94. doi: 10.1128/jb.178.7.1881-1894.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Manning JR, Jefferson ER, Barton GJ. The contrasting properties of conservation and correlated phylogeny in protein functional residue prediction. BMC Bioinforma. 2008;9:51. doi: 10.1186/1471-2105-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt S, Johnson N, Juettemann T, Kähäri AK, Keenan S, Kulesha E, Martin FJ, Maurel T, McLaren WM, Murphy DN, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ruffier M, Sheppard D, Taylor K, Thormann A, Trevanion SJ, Vullo A, Wilder SP, Wilson M, Zadissa A, Aken BL, Birney E, Cunningham F, Harrow J, Herrero J, Hubbard TimJP, Kinsella R, Muffato M, Parker A, Spudich G, Yates A, Zerbino DR, Searle SMJ. Ensembl 2014. Nucleic Acids Res. 2014;42(Database issue):749–55. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schroedinger LLC. The PyMOL Molecular Graphics System, Version 1.8, Schroedinger, LLC. 2015.
- 114.Stover BC, Muller KF. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinforma. 2010;11:7. doi: 10.1186/1471-2105-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61(6):1061–7. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 116.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Betancur-R R, Broughton RE, Wiley EO, Carpenter K, López JA, Li C, Holcroft NI, Arcila D, Sanciangco M, Cureton Ii JamesC, Zhang F, Buser T, Campbell MA, Ballesteros JA, Roa-Varon A, Willis S, Borden WC, Rowley T, Reneau PC, Hough DJ, Lu G, Grande T, Arratia G, Ortí G. The tree of life and a new classification of bony fishes. PLoS Currents. 2013; 5. doi:10.1371/currents.tol.53ba26640df0ccaee75bb165c8c26288. [DOI] [PMC free article] [PubMed]
- 118.Zhuang T, Vishnivetskiy SA, Gurevich VV, Sanders CR. Elucidation of inositol hexaphosphate and heparin interaction sites and conformational changes in arrestin-1 by solution nuclear magnetic resonance. Biochemistry. 2010;49(49):10473–10485. doi: 10.1021/bi101596g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Felsenstein J. PHYLIP (Phylogeny Inferernce Package): version 3.2 (20.10.2015). http://evolution.genetics.washington.edu/phylip/. Accessed 30 June 2017.
- 120.Indrischek H, Prohaska SJ, Gurevich VV, Gurevich EV, Stadler PF. Dataset: Uncovering missing pieces. Duplication and deletion history of arrestins in deuterostomes. 2017. https://doi.org/10.5281/zenodo.820867. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file includes Figures S1–S7, Table S1 (Genomes used) and Table S2 (Transcriptome/EST data used) as well as the following Appendices: Appendix 1 — Arrestin inventories in lampreys. Appendix 2 — Annotation of arrestins in cartilaginous fish. Appendix 3 — Evolution of visual arrestins in different orders of teleosts. Appendix 4 — Investigation of the ARR3 locus in Afrotheria, Xenarthra and common shrew. Appendix 5 — Investigation of loss of ARRB2 in Sauropsids. Appendix 6 — Domains of deuterostome arrestins. Appendix 7 — Isoforms and changes of the conserved exon-intron structure. Appendix 8 — Parsimonious reconstruction of intron gain/loss events. Appendix 9 — Annotation of arrestins in mammals. (PDF 2380 kb)
Approximate ML tree of the arrestin fold family as extracted from UniProtKB (depicted in Additional file 1: Figure S1). Hits were assigned to the arrestin fold family if they contained at least one arrestin_N or arrestin_C domain (see Methods). The tree was generated with the FastTree software and bootstrapping was performed 1000 times with SeqBoot [119]. The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (TREE 78.3 kb)
ML tree of arrestins (depicted in Fig. 4). Starting information for tree reconstruction was an alignment of arrestins investigated in this study, for which sequence information was close to complete. Sequences derived from genomic annotations were substituted by sequences with experimental evidence available if these were more complete. The tree was constructed using PhyML from the amino acid sequences of arrestin paralogs from an alignment of nucleotide sequences aligned with MACSE (model JTT+G+I with α 1.04, p-invariable 0.05, 200x bootstrapping). The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (NW 1340 kb)
Maximum clade credibility tree of arrestins. Starting from the same alignment as used for Additional file 3, we constructed a phylogenetic tree with BEAST2 [94] under the Birth Death model with a relaxed molecular clock (log normal) using the gamma site model (JTT+G+I with α 1.04, p-invariable 0.05). The tree can be visualized with a tree viewer, e.g. Figtree [99]. (TREE 253 kb)
ML tree of arrestins excluding columns known to confer receptor binding. The tree was constructed from the same alignment as Additional file 3 deleting the columns known to confer receptor specificity according to [2, 17, 19, 23, 31–33] (model JTT+G+I with α 1.05, p-invariable 0.05, 200x bootstrapping). The tree can be visualized with a tree viewer, e.g. Dendroscope [115]. (NW 7.42 kb)
Chromosomal locations: Chromosomal locations of arrestin genes. Table of chromosomal locations of arrestin genes in species with genomes assembled on chromosome-level. The columns “SAG(a)” contain the location of SAG for non-duplicated species (SAGb does not exist) or of SAGa for teleosts. This applies to all columns. Note that ARR3 is located on the sex chromosome X in all mammals.
SDP ARR0: SDPs distinguishing ARR0.1 from sea urchin ARR0.2 and other ARR0. The classification is based on unsupervised multi-correspondence analysis of all ARR0 with S3det. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARRB1 are listed. SDP SAG: SDPs distinguishing SAGa and SAGb in teleosts. The classification is based on unsupervised MCA of all filtered teleost SAG with S3det. During filtering, SAGb from pufferfish and stickleback were excluded due to sequence coverage < 70%. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine SAG are listed.
SDP ARRB2: SDPs distinguishing ARRB2a and ARRB2b in teleosts. The classification was given in supervised MCA of all filtered teleost ARRB2. Due to redundancy > 98%, ARRB2a of platyfish (Xiphophorus maculatus), pufferfish and medaka were excluded from the analysis. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARRB2 are listed.
SDP ARR3: SDPs distinguishing ARR3a and ARR3b in teleosts. The classification is based on unsupervised MCA of all teleost ARR3 with S3det. SDPs are listed that were predicted by at least two out of the four following methods: Xdet, S3det, SH and multi-RELIEF. Additionally, the functional annotations of homologous positions in bovine ARR3 are listed.
Codeml: Analysis of positive selection after arrestin duplication. Specific branches within the arrestin gene tree were tested for positive selection using the branch-site model of codeml, part of the PAML program. The null-hypothesis assumes purifying or neutral selection on the foreground and background branches, while the alternative model allows for positive selection on the foreground branch. Fractions of sites are given, that are predicted to belong to the respective classes (p) together with their dN/dS ratios (w). If the null hypothesis was rejected, sites that were under positive selection under BEB were mapped onto the respective bovine ortholog. (XLSX 44.5 kb)
Alignment of deuterostome arrestins with functional annotation known from experimental studies of arrestins in mammals. Note that the naming differs from the naming used throughout the manuscript with ARR1, ARR2, ARR3 and ARR4 used instead of SAG, ARRB1, ARRB2 and ARR3, respectively. Please load alignment with annotation file (provided at http://dx.doi.org/10.5281/zenodo.820866) in Jalview alignment viewer. (FA 44.5 kb)
Data Availability Statement
The data sets supporting the results of this article are available in the zenodo repository, DOI 10.5281/zenodo.820866. [120].