

Abstract
Background
Cytosine methylation in plant genomes is important for the regulation of gene transcription and transposon activity. Genome-wide methylomes are studied upon mutation of the DNA methyltransferases, adaptation to environmental stresses or during development. However, from basic biology to breeding programs, there is a need to monitor multiple samples to determine transgenerational methylation inheritance or differential cytosine methylation. Methylome data obtained by sodium hydrogen sulfite (bisulfite)-conversion and next-generation sequencing (NGS) provide genome-wide information on cytosine methylation. However, a profiling method that detects cytosine methylation state dispersed over the genome would allow high-throughput analysis of multiple plant samples with distinct epigenetic signatures. We use specific restriction endonucleases to enrich for cytosine coverage in a bisulfite and NGS-based profiling method, which was compared to whole-genome bisulfite sequencing of the same plant material.
Methods
We established an effective methylome profiling method in plants, termed plant-reduced representation bisulfite sequencing (plant-RRBS), using optimized double restriction endonuclease digestion, fragment end repair, adapter ligation, followed by bisulfite conversion, PCR amplification and NGS. We report a performant laboratory protocol and a straightforward bioinformatics data analysis pipeline for plant-RRBS, applicable for any reference-sequenced plant species.
Results
As a proof of concept, methylome profiling was performed using an Oryza sativa ssp. indica pure breeding line and a derived epigenetically altered line (epiline). Plant-RRBS detects methylation levels at tens of millions of cytosine positions deduced from bisulfite conversion in multiple samples. To evaluate the method, the coverage of cytosine positions, the intra-line similarity and the differential cytosine methylation levels between the pure breeding line and the epiline were determined. Plant-RRBS reproducibly covers commonly up to one fourth of the cytosine positions in the rice genome when using MspI-DpnII within a group of five biological replicates of a line. The method predominantly detects cytosine methylation in putative promoter regions and not-annotated regions in rice.
Conclusions
Plant-RRBS offers high-throughput and broad, genome-dispersed methylation detection by effective read number generation obtained from reproducibly covered genome fractions using optimized endonuclease combinations, facilitating comparative analyses of multi-sample studies for cytosine methylation and transgenerational stability in experimental material and plant breeding populations.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-017-1070-y) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, Reduced representation bisulfite sequencing, RRBS, Oryza sativa, Epiline, Cytosine methylation, Rice, Plant
Background
In plants, DNA methylation at cytosines occurs in three sequence contexts, i.e. CG, CHG, CHH (H = A, T or C) [1–3], and is regulated by three pathways involving four DNA methyltransferases: the RNA-directed DNA methylation (RdDM) pathway with domains rearranged DNA methylase 2 (DRM2), the chromomethylase 2 (CMT2) and CMT3 pathway and the maintenance methyltransferase 1 (MET1) pathway [4]. The RdDM pathway controls de novo DNA methylation via small interfering RNAs (siRNAs) binding specific DNA sequences and guiding DRM2 to initiate methylation of cytosines in all three sequence contexts [5]. CMT3 maintains CHG methylation [6], while CMT2 mediates CHG and CHH methylation through binding to histone H3 lysine 9 (H3K9) methylation [7]. The methyltransferases CMT2, CMT3, and DRM2 redundantly control non-CG methylation, and are components of self-reinforcing loop mechanisms, which include histone H3K9 methylation and siRNAs [7]. MET1 maintains methylation in symmetric CG sites and is independent of siRNAs and histone modifications [8]. In addition to the activity of methyltransferases, the DNA methylation level is also shaped by demethylation processes which can be passive by cell division dilution or active through DNA glycosylases [9].
The methylation levels strongly vary between contexts and species (24 CG, 7 CHG, 2% CHH in Arabidopsis [1] and respectively 86, 74 and 5% in maize [10]). As exemplified in Arabidopsis, intensive methylation in all contexts acts to repress transcription at promoters and transcription start sites (TSSs) of silent genes and at inactive transposable elements (TEs) [11]. Methylated epialleles coincide with gene expression reduction [12–14], whereas cytosine demethylation is accompanied by activation of epiallele transcription [15] or retrotransposition [16]. CG methylation within the gene body of constitutively expressed genes is dispensable in expression regulation [17]. Therefore, DNA methylation has different effects on gene transcription depending on the genomic location and context. In a number of plant species, abiotic and biotic stresses induce changes in the DNA methylation level of specific DNA sequences, resulting in altered expression of stress- or defense-related genes and adaptation to environmental stress (reviewed by [18]). Spontaneous changes in DNA methylation (epimutations) contribute to heritable phenotypic variation [19, 20]. Flowering time and plant height phenotypes that are correlated with distinct cytosine methylation are stably inherited in Arabidopsis lines derived from a cross between the wild type and the nucleosome remodeler mutant ddm1 [21]. Brassica napus (canola) epilines have distinct epigenetic signatures of global cytosine methylation, histone H3 methylation and H4 acetylation, and show an enhanced drought stress tolerance, which remained stable for at least seven generations [22, 23].
Although the effects of epigenetic regulation are small compared with those of genetic variation [24], epigenetic breeding is an appealing approach to improve complex traits such as crop stress tolerance and yield stability by selecting putative changes in gene expression at multiple epialleles [25]. Epigenetic breeding requires high-throughput methods for the detection of cytosine methylation to facilitate the identification of individuals with interesting epialleles. In plants, cytosine methylation levels at nucleotide resolution [26] can be evaluated by sodium hydrogen sulfite (bisulfite) conversion-based techniques that are applied in whole-genome bisulfite sequencing (WGBS) using randomly sheared genomic DNA and next-generation DNA sequencing (NGS) [1, 2]. However, profiling methods would be more applicable for large breeding programs where high numbers of individuals are to be tested. Reduced representation bisulfite sequencing (RRBS) has been developed in which methylation-insensitive endonuclease restriction combined with size selection generates specific genome fractions for subsequent bisulfite conversion and NGS [27, 28]. Low cytosine coverage RRBS setups were established in plants, to study methylation in B. rapa subgenomes playing an important role in polyploid genome evolution [29] and at Quercus gene promoters in response to temperature regimes [30]. Methylation detection was combined with GBS (genotyping by sequencing) resulting in epiGBS, applicable also to non-model plant species lacking a reference genome, allowing to detect methylation polymorphisms from bisulfite-converted samples, but with the need to reconstruct the consensus sequence of the targeted genomic loci [31].
In order to design a high-throughput, cost-effective and reproducible methylome profiling method, we established an efficient workflow for RRBS in plants, referred to as plant-RRBS, using optimized double restriction endonuclease combinations and subsequent bisulfite conversion, followed by NGS and read data processing with conventional bioinformatics programs. The methylation level of tens of millions of cytosine positions was reproducibly detected in multiple biological replicates, which resulted in a broad coverage overlap and allowed the detection of differential cytosine methylation at a thousand CG sites, and less at CHG or CHH sites between lines, i.e. a pure breeding rice line (control) and a derived epiline.
Results and Discussion
Plant-RRBS methylome profiling–steps and workflow
A workflow was established for methylome profiling using a rice pure breeding seed lot of an inbred line (named control line) and an epiline, named LR2 with low cellular respiration and high energy use efficiency (EUE; Additional file 1: Table S1). The LR2 epiline was derived from the control seed lot by three selfings combined with testing for cellular respiration (Additional file 1: Table S2) and EUE; the identification and stabilization upon selfing was comparable with the procedure followed in B. napus [22, 23, 25]. The major steps in our plant-RRBS methylome profiling include double restriction endonuclease digestion, library construction for Illumina sequencing (Fig. 1a), large data set processing (Fig. 1b) and cytosine methylation detection (Fig. 1c), and are explained below.
Fig. 1.
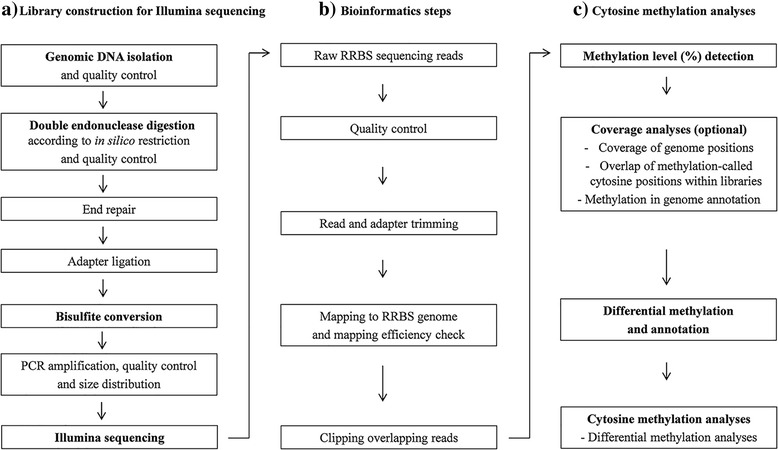
Workflow of plant-RRBS for plant science. (a) Main laboratory steps of library construction for Illumina sequencing, (b) main bioinformatics steps of plant-RRBS data analysis and (c) main steps for determination of coverage and cytosine methylation aspects
Effective endonuclease combinations and quality controls
First, genomic DNA was isolated from five individual plants per line and controlled for quality and quantity (Fig. 1a). Restriction endonuclease combinations were selected with cytosine-containing cutting sites that potentially enrich for fragments from C/G-rich regions containing all cytosine contexts (CG, CHG and CHH), aiming to obtain broad coverage of cytosine methylation detection in the plant genome. Appropriate restriction endonuclease combinations have been deduced from in silico-simulated complete digestion of the Oryza sativa ssp. indica nuclear reference genome that yielded high genome coverage by fragment sizes between 150 and 420 bp, representing the predicted library insert size range. Using MspI (C-CGG) in combination with DpnII (-GATC) or ApeKI (G-CWGC), representing an innovative double restriction endonuclease approach in plants, a high in silico genome coverage of approximately 37% (MspI-DpnII) or 25% (MspI-ApeKI), respectively, was observed in O. sativa ssp. indica, containing no distinct peaks of satellite DNA or other repeat classes, ideal for effective genome coverage by NGS. Double restriction endonuclease digestions were followed by digestion quality evaluations by gel electrophoresis. A high in silico genome coverage was also detected with MspI-DpnII and MspI-ApeKI in other plant species with nuclear reference genomes, representing different genome sizes and structural compositions. Indeed, in silico digestion of A. thaliana (TAIR 10), Beta vulgaris ssp. vulgaris, B. rapa, O. sativa ssp. japonica, and Zea mays (B73) resulted in respectively 36, 19, 33, 38, and 40% in silico genome coverage using MspI-DpnII and 15, 10, 15, 26, and 27% using MspI-ApeKI. Hence, the newly proposed combinations MspI-DpnII or MspI-ApeKI will be widely applicable for many plant genomes.
Library construction for Illumina sequencing and quality control
Upon double digestion, the fragments were end repaired, adapter ligated, bisulfite converted and PCR amplified, resulting in paired-end libraries. Purifications of the libraries were performed using solid-phase reversible immobilization (SPRI), followed by quality control using sub-cloning and sequencing, quantitative PCR (qPCR), and detection of library size distribution with the 2100 Bioanalyzer. The detected library size ranged from approximately 270 to 540 bp (Additional file 1: Fig. S1), containing the insert size range of 150 to 420 bp added with the forward and reverse primer length (sum here 119 bp). The quality-controlled RRBS libraries were sequenced using Illumina HiSeq 2500.
Bioinformatics pipeline–Raw read quality check, mapping and efficiency check
The bioinformatics pipeline with all parameters is described in more detail in the Methods section. In a first step, the raw read quality of the different libraries (Fig. 1b) was evaluated with FastQC to ensure read and nucleotide qualities. To ensure identical read lengths, read libraries were trimmed at the 3′ end using the FastX-Toolkit, to limit this confounding variable before starting the downstream bioinformatics pipeline, making direct comparisons between the mapping results possible. With sequencing adapters harnessing the downstream analysis, it was essential to perform adapter trimming, which was done with Trim Galore that by default trims nucleotides with a quality score lower than 20 and discards reads with a length smaller than 20. A low frequency of PCR duplicate reads between 0.08 and 0.76% was detected in the Illumina reads in both the RRBS and WGBS data sets (Additional file 1: Table S3.), thus no need to clean the PCR duplicates from the data sets. The resulting set of high-quality reads was subsequently mapped to the O. sativa ssp. indica reference genome using BSseeker [32] and bowtie2 [33]. Specific plant-RRBS genome indices were generated, as required by the software, to ensure mapping of the reads obtained from restriction endonuclease fragments. An index was generated for each of the defined cutting sites, C-CGG and -GATC, in the case of MspI-DpnII, and, C-CGG and G-CWGC, in the case of MspI-ApeKI. The mapping and its quality was evaluated using Qualimap [34]. Based on the mapping, the calculation of per-base genome coverage, i.e. how many nucleotides in the reference sequence are covered at least once by the set of reads, was performed using BEDTools genomecov [35].
Cytosine methylation detection
Cytosine methylation detection is defined as the determination of the methylation level at a cytosine position. The methylation detection was performed for cytosine positions in the reference sequence at which at least ten informative nucleotides (means C or T) were obtained, originating from mapped plant-RRBS reads, representing unconverted and converted cytosines in amplicons of fragments from genomic DNA. Processing of the mapping files was required before cytosine methylation detection, i.e. the obtained BAM files (Binary Alignment/Map, compressed binary version of the Sequence Alignment/Map format) were sorted by coordinate using Picard, and overlapping read pairs were clipped using bamUtil [36] to prevent biased detection. Methylation detection was performed using the well-established BSseeker software [32]. The genomic features were defined as genes, 2.0-kb upstream regions of TSSs (i.e. promoters) or not-annotated regions. The determination of cytosine coverage and methylation aspects were achieved by analyzing cytosine positions having detected methylation levels [26], followed by the determination of the genomic features that overlap with cytosine positions for which a methylation level was detected, and differential cytosine methylation level detection (Fig. 1c). Prior to the detection of differentially methylated cytosine positions, the BSseeker map.gz output files were converted to BED (Browser Extensible Data) format, as required by the often-used methylKit software, using custom scripting. Methylation detection supported by at least ten informative nucleotides (means C or T) at a cytosine position were retained to ensure a certain accuracy of methylation levels in the further analysis. Subsequently, normalization of the libraries was performed using standard parameters of the R package methylKit [37]. The detection of differentially methylated cytosine was performed through methylKit. To speed up NGS analysis, methylation detection, calculation of differential methylation, and determination of genomic features, a high-memory and multi-processor Linux grid server system was used.
Genome and cytosine coverage aspects of plant-RRBS methylome profiling and WGBS in rice
The MspI-DpnII and MspI-ApeKI double restriction endonuclease plant-RRBS setup consisted of biological replicates represented by five individual plants of the rice inbred control or the LR2 epiline. The covered genome fraction, the proportion of covered cytosine positions and the extent of library overlap were analyzed and a comparison between plant-RRBS and WGBS was made.
Genome coverage and cytosine coverage in individual plant-RRBS libraries
The average total read number per library was about 58 million paired reads (minimum 15 million paired reads) and read preprocessing and mapping quality of individual libraries are presented in Additional file 1: Table S4. The mapping quality of the different aligned libraries, which is denoted in the quality phred scale and gives the probability of having an incorrect read alignment, was on average 45.3 phred (Additional file 1: Table S4). The genome coverage by at least one read (also denoted per-base genome coverage) was on average approximately 31.0% (Table 1) based on a reference genome size of 427 Mbp [38]. The intersection of detected methylated sites between the individuals per line and per double digest ranges from ~40% for sites covered by all five samples, to ~80% for sites covered by at least three samples (Additional file 1: Fig. S2). This indicates that, in order to achieve a decent coverage of the methylated sites in the genome, the required number of samples should be three or more. The intersection of in silico fragments and mapped reads for each individual per line and per double digest varied between 54.84 and 77.72% (Additional file 1: Table S5) which underlines the robustness of our plant-RRBS approach (Fig. 1). Differences between observed and expected suggest reduced efficiency of some of the experimental steps in the procedure, such as double digestion, size selection, adaptor ligation, bisulfite conversion, etc. Their impact on RRBS was investigated in more detail in pigs [39]. A tendency for a higher genome coverage of MspI-DpnII compared with MspI-ApeKI, in agreement with the predicted coverage by the in silico digestion, is also visualized by the mapped reads of representative samples in the Integrative Genomics Viewer (IGV) at representative genome regions in the coverage data visualization (Additional file 1: Fig. S3). The proportion of covered cytosine positions was up to 48.7% of the genome (Table 1), using a threshold for sufficiently mapped cytosine positions in the reference genome by at least ten informative nucleotides (means C or T). Plant-RRBS generates an effective read number as information resource for broad methylation detection from the analyzed genome fraction (Table 1, Additional file 1: Table S4). Indeed, a maximum of covered cytosine positions of up to 17 million CG sites, 15 million CHG sites, and 56 million CHH sites was detected by the largest library (Table 1, plant 1 of LR2). The coverage varied for both restriction endonuclease combinations with a tendency for higher coverage by MspI-DpnII. MspI-DpnII covered 22.9 to 48.7% of the cytosine positions in the genome which was for the majority of libraries higher than the 24.5 to 34.1% covered by MspI-ApeKI (Table 1).
Table 1.
Genome and cytosine coverage in biological replicates of the control line and the LR2 epiline (fourth selfing) using plant-RRBS, and comparison to WGBS
| Line with biological replicatesa | Restriction endonuclease combination | Genome coverage (%)b | Cytosine coverage (%)c | Efficiencyd | Number of analyzed cytosine sites | ||
|---|---|---|---|---|---|---|---|
| CG | CHG | CHH | |||||
| (millions)e | |||||||
| Plant-RRBS | |||||||
| Control-1 | MspI-DpnII | 39.6 | 42.8 | 1.1 | 15.3 | 13.1 | 48.1 |
| Control-2 | MspI-DpnII | 42.3 | 44.9 | 1.1 | 15.6 | 13.6 | 51.1 |
| Control-3 | MspI-DpnII | 30.8 | 32.8 | 1.1 | 11.6 | 10.0 | 37.0 |
| Control-4 | MspI-DpnII | 35.0 | 37.1 | 1.1 | 13.0 | 11.3 | 42.0 |
| Control-5 | MspI-DpnII | 21.3 | 22.9 | 1.1 | 8.3 | 7.1 | 25.5 |
| LR2–1 | MspI-DpnII | 46.0 | 48.7 | 1.1 | 16.7 | 14.7 | 55.6 |
| LR2–2 | MspI-DpnII | 45.7 | 48.5 | 1.1 | 16.7 | 14.7 | 55.3 |
| LR2–3 | MspI-DpnII | 27.6 | 28.8 | 1.0 | 9.4 | 8.6 | 33.5 |
| LR2–4 | MspI-DpnII | 45.1 | 48.4 | 1.1 | 17.0 | 14.7 | 54.7 |
| LR2–5 | MspI-DpnII | 41.2 | 43.9 | 1.1 | 15.1 | 13.2 | 50.0 |
| Control-6 | MspI-ApeKI | 29.9 | 34.1 | 1.1 | 13.1 | 11.1 | 36.6 |
| Control-7 | MspI-ApeKI | 26.2 | 30.0 | 1.1 | 11.7 | 10.0 | 31.9 |
| Control-8 | MspI-ApeKI | 25.3 | 28.7 | 1.1 | 11.0 | 9.5 | 30.7 |
| Control-9 | MspI-ApeKI | 21.2 | 24.5 | 1.2 | 9.6 | 8.3 | 25.9 |
| Control-10 | MspI-ApeKI | 27.8 | 32.4 | 1.2 | 13.0 | 10.8 | 34.1 |
| LR2–6 | MspI-ApeKI | 23.0 | 26.0 | 1.1 | 9.8 | 8.8 | 27.9 |
| LR2–7 | MspI-ApeKI | 21.8 | 24.9 | 1.1 | 9.5 | 8.5 | 26.5 |
| LR2–8 | MspI-ApeKI | 22.3 | 25.4 | 1.1 | 9.6 | 8.7 | 27.1 |
| LR2–9 | MspI-ApeKI | 23.2 | 26.6 | 1.1 | 10.2 | 9.0 | 28.3 |
| LR2–10 | MspI-ApeKI | 24.0 | 27.1 | 1.1 | 10.0 | 9.0 | 29.3 |
| WGBS | |||||||
| Control −11 | - | 84.3 | 52.4 | 0.6 | 14.4 | 15.4 | 63.9 |
| LR2–11 | - | 83.7 | 52.6 | 0.6 | 14.5 | 15.6 | 63.9 |
Leaf material from five individual plants per line and per restriction endonuclease combination was used
The bisulfite conversion efficiency rate per biological replicate was higher than approximately 99%
a Name scheme: line–individual plant number (1–11) from selfing generation 4
b Genome coverage: coverage as number of genome nucleotide positions covered by at least one read *100% / 427,026,737 nucleotides in the reference genome [38]
c Cytosine coverage: proportion of analyzed (sufficiently covered) cytosine positions in the genome = sum of analyzed cytosines in CG, CHG and CHH context covered by at least ten informative nucleotides (means C or T) * 100% /178,637,468 cytosines in the reference genome for both strands [38]
d Ratio of cytosine coverage per genome coverage
e Millions of positions of a certain cytosine context (CG, CHG and CHH) in the reference genome for both strands that are sufficiently covered by at least ten informative nucleotides (means C or T) and therefore methylation level of cytosine sites was analyzed [38]
Plant-RRBS generates a broad overlap of detected cytosine positions within biological replicates
To investigate whether plant-RRBS generates sufficient overlap in covered regions for differential methylation analysis, we analyzed the cytosine methylation in the overlapping regions between the five biological replicates, i.e. libraries, per line for the two restriction endonuclease combinations (Table 2). The starting point was to determine the number of detected cytosine positions covered by at least one of the five biological replicates of a line, resulting in a total set of positions (union). Thus, the union of covered genome positions was high and up to 54.6% (Table 2, LR2, and MspI-DpnII). The MspI-DpnII combination covered more union positions in a group of biological replicates compared with MspI-ApeKI for both control line and LR2 epiline (Table 2). We conclude that plant-RRBS covers in total up to half of the cytosine positions in the rice genome using MspI-DpnII for a group of five biological replicates of a particular line.
Table 2.
Detected cytosine positions relative to the genome-wide cytosine positions per five biological replicates of the control line and the LR2 epiline (fourth selfing) discriminated by restriction endonuclease combination and cytosine context (CG, CHG and CHH)
| Group of biological replicates | Restriction endonuclease combination | Detected cytosine positions in genome | |||
|---|---|---|---|---|---|
| CG | CHG | CHH | C (%) | ||
| Union (collection of all covered positions in at least one replicate) | |||||
| Control | MspI-DpnII | 18,507,834 | 15,887,099 | 58,570,249 | 52.0 |
| LR2 | MspI-DpnII | 19,434,811 | 16,678,896 | 61,436,046 | 54.6 |
| Control | MspI-ApeKI | 15,715,023 | 12,901,771 | 42,018,210 | 39.5 |
| LR2 | MspI-ApeKI | 14,573,921 | 12,185,636 | 39,744,568 | 37.2 |
| Intersection (common positions in all replicates) | |||||
| Control | MspI-DpnII | 6,459,314 | 5,448,976 | 19,305,085 | 17.5 |
| LR2 | MspI-DpnII | 8,023,858 | 7,435,066 | 29,372,103 | 25.1 |
| Control | MspI-ApeKI | 7,053,953 | 6,410,114 | 19,063,516 | 18.2 |
| LR2 | MspI-ApeKI | 5,454,292 | 5,509,874 | 15,910,607 | 15.0 |
| Jaccard indexa (proportion of common on all detected positions) | |||||
| (%) | (%) | (%) | (%) | ||
| Control | MspI-DpnII | 34.9 | 34.3 | 33.0 | 33.6 |
| LR2 | MspI-DpnII | 41.3 | 44.6 | 47.8 | 46.0 |
| Control | MspI-ApeKI | 44.9 | 49.7 | 45.4 | 46.1 |
| LR2 | MspI-ApeKI | 37.4 | 45.2 | 40.0 | 40.4 |
a Jaccard index or Jaccard similarity coefficient = intersection / union
We proceeded with the detection of common cytosine positions in all five biological replicates of one line, resulting in common sites (intersection), which was relatively high with up to 25.1% commonly covered cytosine positions (up to 44.8 million) in the genome (Table 2). Filtering for common positions in an NGS analysis implies the loss of a fraction of reads but ensures comprehensive and accurate comparison between multiple samples. In conclusion, different aspects of the coverage were determined, showing the fractional enrichment of genome regions by the plant-RRBS method. Commonly occurring cytosine positions can be considered as a measure for the reproducibility of the plant-RRBS method in terms of the detectable proportion of cytosine sites. We conclude that plant-RRBS reproducibly covers commonly up to one fourth of the cytosine positions in the rice genome when using MspI-DpnII within a group of five biological replicates of a line.
Finally, we determined the overlap between libraries in terms of the Jaccard index, which is calculated by dividing the number of commonly covered positions within the libraries by all covered positions, in at least one of the libraries. A major fraction of one third to up to half (33.6–46.1%) of the total covered positions was found in the overlap of positions between five libraries per restriction endonuclease combination and per line (Table 2).
Comparison between plant-RRBS and WGBS in terms of genome coverage and coverage of cytosine positions
The analysis focused to compare the performance of both methods in detecting cytosine positions with deduced methylation levels, as this is the starting point of the analytic power of bisulfite sequencing-related techniques. Genome coverage is determined by the standard threshold of minimum one mapped read and was applied to the data of both evaluated methods. Plant-RRBS, using reproducible genomic DNA fragments generated by restriction endonucleases, allowed an average genome coverage of 31%, as compared with WGBS using randomly sheared genomic DNA, which covered 84.3 and 83.7% for the control line and the LR2 epiline, respectively (Table 1). The visualization of coverage data of plant-RRBS and WGBS in IGV confirms that those genome coverage percentages extent in representative regions (Additional file 1: Figure S2). The WGBS genome coverage in the control line and epiline is markedly better than the 76% observed in a previous study of an O. sativa ssp. indica plant [40]. The per-read cytosine coverage for both RRBS and WGBS indicates that the number of detected cytosine positions and the associated trend line is quite stable when taking positions into consideration that are covered by ten or more reads (Additional file 1: Fig. S4). No large dissimilarity is seen for either RRBS or WGBS, suggesting that the quality for both data sets is comparable. The ratio of cytosine coverage per genome coverage was for WGBS only 0.6 but for RRBS almost two times more efficient (≥1.0). This means that plant-RRBS obtains a better cytosine coverage for a lower genome coverage. Plant-RRBS increases the coverage of cytosine positions detected for their methylation levels with the advantage to analyze a reproducible genome fraction generated by double restriction endonuclease digestion. We conclude that plant-RRBS is beneficial to detect restriction endonuclease-specific genome fractions that are sufficiently covered by the NGS approach (Additional file 1: Figure S4). In consequence, the plant-RRBS produces reads more efficiently and requires much fewer reads for data analysis as compared with WGBS.
Genomic features at cytosine positions covered by plant-RRBS
We determined the genomic features of covered cytosine positions using the O. sativa ssp. indica annotation ASM465v1.27 as obtained from Ensembl Plants [41], which does not contain TE annotation features. We performed the annotation of covered cytosine positions on chromosomes of the rice reference genome, determined for gene-associated annotation features. The analysis was performed for both restriction endonuclease combinations and in both lines in all commonly detected CG, CHG and CHH sites of their biological replicates (Additional file 1: Fig. S5). Approximately 45 to 50% of detected cytosine positions were localized in not-annotated regions. A high percentage of approx. 35% of detected cytosine positions was localized in promoters, defined as 2000 nucleotides upstream of the TSS, as compared with protein-coding genes (approx. 15 to 20%). LR2 MspI-ApeKI common cytosine sites contained the highest percentage of protein-coding gene positions, including all detected annotation feature subclasses. Differences in percentages of annotated positions were detected for the two restriction endonuclease combinations, and the number of covered cytosine sites was different because the endonuclease restriction combinations cut specific genomic regions. In addition to the genome-wide determination, the visualization of coverage data in IGV indicates those aspects in representative regions (Additional file 1: Figure S3).
Despite current approaches, repeats collapse in reference genome sequences generated by NGS, due to limitations e.g. in read length and assembly procedure of sequence-similar and high-copy DNA elements, allowing very limited determination of repeat annotations [42]. Nonetheless, when additional sequence and annotation information of the genomic features become available, these data can be processed in the presented plant-RRBS data analysis pipeline for this research field. Currently, detailed cytosine methylation of repeats can be analyzed by cloned bisulfite-converted PCR products of repeat elements [43].
In summary, cytosine methylation levels in the individual biological replicates were detected for 22.9 up to 48.7% of the genome-wide cytosine positions, and tens of millions of cytosine positions in common between the five biological replicates of a particular line were subsequently annotated.
Cytosine methylation levels at genomic features
The available genomic features in O. sativa ssp. indica allowed to identify their cytosine methylation levels detected by plant-RRBS. The scattered distribution patterns of CG methylation levels in the different annotations were distinguishable between both MspI-DpnII and MspI-ApeKI combinations and we analyzed CG methylation levels linked with given genomic features such as promoter or genes in the rice control line and LR2 epiline (Fig. 2). Using the plant-RRBS setup, intronic and not-annotated cytosine positions were found to be rather frequently methylated at CG sites, in contrast to the low frequency of CG methylation detected in the promoters (Fig. 2). MspI-ApeKI detected CG methylated sites more frequently in different gene annotations (i.e. protein-coding genes, non-coding genes, transcript, exon and coding DNA sequence (CDS) annotation) as compared with MspI-DpnII. Non-coding genes were less frequently methylated when compared with the other annotation features such as transcript, exon and CDS annotation. Additionally, CG methylated cytosine sites in introns were slightly more frequently detected with MspI-ApeKI. The detection of methylation levels in different annotation features gives information about restriction endonuclease combinations enriching to a different extent methylated subfractions of the rice genome.
Fig. 2.
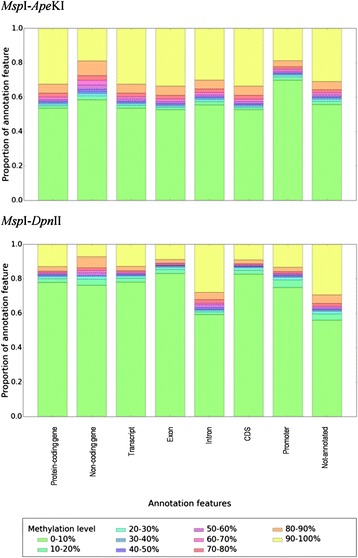
Detected cytosine methylation levels of CG sites in a given annotation within the control line and epiline LR2 (fourth selfing). The proportion of cytosine positions with a particular methylation level bin is displayed as a stacked bar diagram. Ten methylation level bins are considered with an interval of 10%. Proportions of methylation level bins are visualized for different annotation features (O. sativa ssp. indica ASM465v1.27): protein-coding gene, non-coding gene, transcript, exon, intron, coding DNA sequence (CDS) and promoter (2000-nucleotide region upstream of the TSS)
Differential cytosine methylation detection
Cytosine methylation level homogeneity within five biological replicates per line
The five biological replicates were used to investigate the homogeneity of cytosine methylation within the LR2 epiline. The comparison of cytosine methylation levels within five biological replicates per line and per restriction enzyme combination was performed by applying a 25% methylation level difference threshold as selection between the highest and the lowest detected methylation level at a common position in the biological replicates. The proportion of consistently measured cytosine methylation levels was high, because 96.8% or more were detected at the threshold of less than 25% methylation level difference between the biological replicates in the rice inbred line or the LR2 epiline (Additional file 1: Table S6). It can be concluded that the detected methylation levels of the vast majority of cytosine positions in one sample are consistent with that in the other replicates. In fact, less replicates may be used to investigate biological sample homogeneity.
Differential cytosine methylation in the LR2 epiline versus the control line
An important application of methylome profiling is the detection of differential cytosine methylation in certain genomic regions and genomic features between samples, which can assist epigenetic marker detection in breeding programs.
We determined the number of differentially methylated sites and their annotation in the LR2 epiline versus the inbred control line, each analyzed with five biological replicates, for the different cytosine sites (CG, CHG and CHH) by plant-RRBS using the two restriction endonuclease combinations. This allowed to detect more than one thousand differentially methylated positions representing distinct epiallelic states in the LR2 epiline (Table 3). MspI-ApeKI resulted in the detection of an order of magnitude more differentially methylated positions (i.e. 1295) in the methylome-profiled rice material as compared with MspI-DpnII (i.e. 142). The number of hypo-methylated cytosine positions detected by MspI-ApeKI exceeded that of hyper-methylated positions, whereas MspI-DpnII resulted in the detection of more hyper-methylated positions in the LR2 epiline (Table 3), confirming that the enrichment of specific genome positions depends on the used restriction endonuclease combination. The majority (70%) of differentially methylated sites were CG sites in the profiled LR2 epiline, mainly in not-annotated genomic regions and gene-associated regions (Table 3). The restriction endonuclease combination MspI-ApeKI detected a high number of differentially methylated sites in genes, and especially CG sites (Table 3). The 2000 nucleotides upstream of the TSS (i.e. promoters) oftentimes contained more differentially methylated cytosine positions than genes or all annotated regions (Table 3). Plant-RRBS using the rice inbred line and the LR2 epiline and specific restriction endonuclease combinations was therefore focusing on differentially methylated cytosine sites in not-annotated genomic regions, including gene promoters, and within genes. In conclusion, we demonstrate extensive detection of differentially methylated CG sites in the rice LR2 epiline compared with the control line. Integration of methylome with transcriptome will be part of future research.
Table 3.
Differential cytosine methylation and annotation of the control line versus the LR2 epiline (fourth selfing)
| Restriction endonuclease combination | Cytosine site | Number of differentially methylated sitesa | Number of diff. meth. sites within features | ||||
|---|---|---|---|---|---|---|---|
| Annotated region | Not-annotated region | Gene-associated region | |||||
| Hypo- | Hyper- | Gene | Promoterb | ||||
| MspI-ApeKI | CG | 541 | 463 | 182 | 822 | 160 | 362 |
| CHG | 169 | 94 | 28 | 235 | 28 | 81 | |
| CHH | 20 | 8 | 11 | 17 | 4 | 4 | |
| sum | 730 | 565 | 221 | 1074 | 192 | 447 | |
| MspI-DpnII | CG | 29 | 74 | 9 | 94 | 8 | 27 |
| CHG | 10 | 25 | 0 | 35 | 0 | 11 | |
| CHH | 2 | 2 | 1 | 3 | 1 | 0 | |
| sum | 41 | 101 | 10 | 132 | 9 | 38 | |
Cytosine positions, represented by the numbers above, can be counted to the different annotations multiple times
a hypo−/hyper-methylated means a 25% lower/higher methylation level in LR2 compared with the control line
b Promoter is defined as 2000 nucleotides upstream of the TSS and belongs to the not-annotated region in the used genomic features
Conclusions
A new, performant laboratory protocol and data analysis pipeline of plant-RRBS is reported for cytosine methylome profiling, allowing comparative analysis of multiple samples to detect differential methylation levels at nucleotide resolution in reference genome regions and genomic features. In plants, early RRBS setups using a single restriction endonuclease had a very limited number of covered cytosine positions hindering subsequent comparative analyses [29–31]. Our plant-RRBS protocol enriches for coverage of cytosine positions in reads by applying appropriate restriction endonuclease combinations, that were tested in silico at first. It offers broad, genome-dispersed methylation detection by more effective read number usage, as compared with WGBS. Plant-RRBS fulfills the need for an NGS-based cytosine methylome profiling method in large-scale studies in plant science and epigenetic marker-assisted plant breeding. It is applicable to any reference-sequenced plant species, as supported by the promising high in silico genome coverage observed for different plant genome sizes with different structural compositions and genomic features. Hence, it will be broadly applicable to profile the methylome of natural or experimental populations, like epilines and epigenetic recombinant inbred lines, in a user-friendly way.
Methods
Plant material and growth conditions
A rice pure breeding inbred line (Oryza sativa ssp. indica) of Bayer CropScience, named control line, and a derived epiline, named LR2 and selected for a higher EUE, were grown in a growth chamber at Bayer CropScience (Zwijnaarde, Belgium). Starting from a small population (n = 180) of a parental rice inbred line, individual plants were selected for lowest cellular respiration and improved EUE (see below). A number of selfings of selected plants followed by in vitro assays established the epiline LR2, which was selected over three generations for improved EUE.
The LR2 epiline and the control line were grown in soil in a growth chamber at 26 °C / 21 °C (day / night) for 24 days with a 16-h light/8-h dark regime (light intensity was 300 μmol m−2 s−1, the relative humidity was kept at 71%). The sample material for the methylome profiling was the fourth leaf of individual plants for each of the five biological replicates per line, which belongs to the fourth selfing generation.
Assay testing for cellular respiration and EUE analysis
Energy use efficiency, defined as the ratio between energy content and cellular respiration, and energy homeostasis, determined by the crosstalk of molecular networks, are significant components of crop yield stability under varying environmental conditions in the field [22, 25]. Both have an inheritable epigenetic layer of regulation (i.e. cytosine methylation and histone modification) that can be identified and stabilized, resulting in superior agronomical traits in crops [22, 25].
Selection implied a non-destructive assay on an explant per individual in order to identify better performing individuals and was basically done as described for B. napus [23] with specific adaptations for rice. Seedlings were in vitro-grown in the dark for 10 days on agar in half-strength Murashige and Skoog medium supplemented with 3% (w/v) sucrose. The first five centimeters above the coleoptiles (primary leaf rolls) were carefully cut in about 0.6-cm segments without damaging the meristem. Seven primary leaf roll explants of each seedling were cultured in the dark for 1 day on callus-inducing medium (Murashige and Skoog medium containing 2% (w/v) maltose, 3% (w/v) sorbitol, 2 mg/L 2,4D). The plantlets were cultured in the light to allow outgrowth of the meristem. For each seedling, the cellular respiration of the seven leaf roll explants was quantified by measuring the reduction of triphenyltetrazolium chloride as previously described [44]. The seven explants were transferred to 2 mL of 20 mM 2,3,5-triphenyltetrazolium (TTC) solution in 50 mM K-phosphate buffer (pH 7.4), incubated for 1.5 h in the dark at 26 °C, after which the TTC solution was removed and the explants were washed with water and freeze-thawed. Reduced TTC was extracted with 1 mL ethanol by shaking for about 1.5 h. Absorption of the extract was measured at 485 nm and 663 nm. The absorbance was calculated at OD485 due to the reduced TTC (TTC-H): OD485 TTC-H = a–(b/c) (a = OD485; b = OD663; c = constant determined by measuring the absorbance of chlorophyll extract (identical process as described above) at 485 nm and 663 nm → c = OD663/ OD485). Five to ten seedlings with the lowest cellular respiration were transferred to the greenhouse for seed production by self-fertilization. Both cellular respiration and NAD(P)H content of approximately 35 to 40 seedlings of the obtained progenies were measured as previously described [23]. Lines with the lowest cellular respiration and highest EUE were retained. Three rounds of selfing and testing for the cellular respiration and EUE parameter were sufficient to obtain the LR2 epiline with a distinct cellular respiration and EUE (Additional file 1: Table S1).
Rice inbred line and epiline used as material for plant-RRBS methylome profiling
Leaf roll explants from about 180 ten-day-old rice seedlings were evaluated for cellular respiration. A number of plantlets, of which the explants had the lowest cellular respiration, were transferred to the greenhouse for seed production by self-fertilization, and repeated three times (Additional file 1: Table S2), resulting in the LR2 epiline with a stable and significantly reduced cellular respiration level in the leaf rolls over at least two consecutive selfing generations (85 to 86%; Additional file 1: Table S2), an NAD(P)H content similar to the control line (100%; Additional file 1: Table S1), an EUE and photorespiration significantly increased to 118% and 106%, respectively, and a significantly reduced (to 80%) leaf respiration rate (Additional file 1: Table S1), that was used for methylome profiling.
Leaf respiration
For the determination of the leaf respiration, the plants were grown in soil for 4 weeks (temperature: day 26 °C–night 22 °C; light intensity: 400 μMol sec-1 m-2; light regime: 16-h light/8-h dark). The fifth leaf of six plants was harvested and put in 240 mL of buffer (25 mM K-phosphate, pH = 5.8; 2% sucrose; 0.1% Tween20) saturated with oxygen and contained in a closed, 200-mL, scaled bottle. Ten replications per line were performed. The amount of oxygen in the buffer was measured using an optode HQ-portable meter with LDO-electrode (Hach) after four hours of incubation at 24 °C (very gentle shaking). The consumed oxygen was determined by comparing with blank buffer containing no leaf material.
Plant-reduced representation bisulfite sequencing (plant-RRBS)
DNA isolation
Isolation of genomic DNA was performed from an optimal mass of about 60 mg leaf material to obtain a high quality and quantity of genomic DNA, using the Wizard Genomic DNA Purification Kit (Promega) according to manufacturer’s instructions, from five individual plants (biological replicates) of the LR2 epiline or the control line. Genomic DNA isolates were examined for high quality and sufficient quantity by NanoDrop spectrophotometry (concentration, A260/A280 of approximately 1.8 or more) and gel electrophoresis (high molecular weight, integrity, purity).
In silico and double restriction endonuclease digestion
In silico digestion was performed using biopieces v0.48 (www.biopieces.org) of the A. thaliana (TAIR 10), B. vulgaris ssp. vulgaris, B. rapa, O. sativa ssp. indica version 9311_BGF_2005, O. sativa ssp. japonica, and Z. mays (B73) nuclear reference genomes [38, 45–48]. The per-base genome coverage by digestion fragments with a length between 150 and 420 bp was expressed relative to the nuclear reference sequence size. About two micrograms of genomic DNA were either digested with firstly MspI (60 U, 37 °C) and secondly ApeKI (15 U, 75 °C), or with MspI (60 U, 37 °C) followed by DpnII (30 U, 37 °C) added in two half units portions and in Buffer 3.1 (all obtained from NEB, MA) in a final volume of 60 μL for 20 h each, according to manufacturer’s instructions. Successful digestion was confirmed by gel electrophoresis. For plant-RRBS samples, a smear of fragments of different sizes and no evidence of non-digested, high-molecular mass molecules were observed, indicating successful digestion. Genomic DNA control samples taken along without restriction endonucleases showed a discrete high-molecular mass, indicating the absence of contaminating nucleases and persistent DNA quality despite the incubation procedure.
Library preparation and sequencing
Plant-RRBS paired-end libraries for Illumina sequencing were constructed by Alpha Biolaboratory, Inc. Saratoga, CA according to Hsieh (2015) [49] and Pignatta et al. (2015) [50] with modifications. Approximately 300 ng of digested genomic DNA was purified, end repaired, and ligated to custom-synthesized methylated multiplex adapters (Eurofins MWG Operon, Huntsville, AL) according to the manufacturer’s (Illumina, San Diego, CA) instructions. To ensure recovery of shorter plant-RRBS library inserts, the SPRI method with 1.8 x (v/v) AMPure XP beads (Beckman Coulter, Brea, CA) was used for cleanup steps throughout the library construction procedure. Adaptor-ligated libraries were subjected to one round of bisulfite conversion with the EZ DNA Methylation-Lightning Kit (Zymo Research Corporation, Irvine, CA) as outlined in the manufacturer’s instructions. Five to ten nanograms of bisulfite-converted libraries were PCR-amplified with the following condition: 2.5 U of ExTaq DNA polymerase (Takara Bio), 5 μL of 10 x Extaq reaction buffer, 25 mM dNTPs, 1 μL of index primers (10 μM) in a 50-μL reaction. The thermocycling conditions were as follows: 95 °C for 3 min and then 12 cycles each of 95 °C for 30 s, 65 °C for 30 s, and 72 °C for 60 s. The enriched libraries were purified twice with 0.8× (v/v) AMPure XP beads to remove any adapter dimers. The library quality was assessed by randomly sub-cloning and sequencing 20 to 30 colonies to evaluate proper library construction, bisulfite conversion, and the presence of correct indexes. Final libraries were evaluated by qPCR for library size distribution and quantification in the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Called peaks ranged between approximately 270 and 540 bp (Additional file 1: Figure S1). The quality-controlled plant-RRBS libraries were then sequenced at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley on an Illumina HiSeq 2500 (PE50–75-100). Bisulfite conversion efficiency rates (approximately 99% or higher) were assessed by calculating cytosine methylation levels in the chloroplast genome.
Data processing
All sequencing libraries (ArrayExpress [51] accession numbers: E-MATB-4626 and E-MTAB-5002) were processed by scripts available via doi (10.5281/zenodo.168034) [52] and on GitHub [53], and evaluated for quality using FastQC v0.11.2 [54]. Due to different read lengths of the sequence libraries, reads were trimmed at their 3′ end to a uniform total read length of 50 nucleotides using the FASTX Toolkit v0.0.13 [55]. Adapters were subsequently removed using Trim Galore v0.3.3 with the options --paired --trim1 [56]. By default, Trim Galore also trims nucleotides with a quality lower than 20 (Phred ≥20; base call error rate ≤ 1.0%) prior to adapter trimming and discards reads with a length smaller than 20. Next, the reads were mapped to the reference genome of O. sativa ssp. indica cultivar 93–11 that was sequenced by the Beijing Genome Institute following a whole-genome shotgun strategy [38] [reference genome: 9311_BGF_2005 PLAZA [57]]. To be able to map reads to the reference genome, a plant-RRBS genome index was created for the defined cutting sites C-CGG,G-CWGC for the MspI-ApeKI enzyme combination and C-CGG,-GATC for the MspI-DpnII enzyme combination. Indexing was done using BSseeker v2.0.5 [32], bowtie v2.1.0 [33] and python 2.7.4 [58]. Read alignment was done using BSseeker v2.0.5 and bowtie v2.2.4 with the options --mismatches 2 -r -L 20 -U 500. Mapping quality was assessed using Qualimap v2.1 [34].
The mapping was visualized in Integrative Genomics Viewer (IGV) [59] and igv.js [60]. The per-base genome coverage of the different libraries was calculated using BEDTools genomecov v2.22.0 [35] and expressed relative to the reference genome size. The obtained BAM files were sorted by coordinate using Picard v1.129 [61]. Overlapping reads were clipped using bamUtil v20130118 [36].
PCR duplication information was retrieved from the mapped read files by using the SAM file flag properties and counting the mapped reads with a flag higher than or equal to 1024.
The number of cytosine positions covered per read was retrieved by mapping the reads using the normal procedure (see above), and then performing the calling procedure (see above) with the change that only a single read is necessary for a cytosine position to be called methylated. The intersection of the number of expected reads and the number of sequenced reads was performed using bedtools v2.26.0: a custom bed file was created from the in silico predicted fragments which was then intersected with the BAM file of mapped sequenced reads, returning only the fragments that are fully covered in both the in silico predictions and sequenced reads.
Methylation level detection was done using BSseeker v2.0.5. The calculation of cytosine site methylation level was performed with Ci/(Ci + Ti) at positions (i) in both DNA strands [26]. Also, the numbers of CG, CHG and/or CHH sites covered by the different libraries were extracted from the BSseeker CGmap.gz output files. Differential methylation was calculated using the R package methylKit v0.5.5 using default parameter settings [37] and R v2.15.1 [62]. Prior to this analysis, the CGmap.gz output files from BSseeker were converted by custom scripting to the BED format required by methylKit. Filtering was done with a threshold of ten informative nucleotides (means C or T) at a cytosine position (BSseeker options: −coverage 10) and normalization of the libraries was performed using standard settings in methylKit (median). Merging of all data was done in so-called ‘unite.txt’ files. Differential methylation of cytosine positions was defined by a threshold of minimum ten informative nucleotides (means C or T) per biological replicate, a pooled number of informative nucleotides for calculation of p-values using Fisher’s exact test and q-values <0.01 using the Sliding Linear Model, and percent methylation level differences > │25│% between the in the text indicated comparison pairs of each line calculated per CG, CHG and CHH site using custom scripting [37, 63]. Hypo- / hyper-methylated sites were determined as lower / higher methylation level in the LR2 epiline compared with the control line. A methylation difference analysis between the replicates of each line was performed based on the output of methylKit (i.e. unite files) using > │25│% methylation level differences threshold and custom Python scripting. Annotation of particular nucleotide positions was based on the Oryza_indica.ASM465v1.27.gff3 as obtained from Ensembl Plants [64]. Intronic regions were added using genometools v1.5.4 [65]. Promoter regions were defined as a window of 2000 nucleotides upstream of the TSS. Annotation of covered cytosine positions was done using custom Python scripting. Different features were considered: protein-coding gene, non-coding gene, transcript, exon, intron and CDS. Hence, a site can have multiple annotation features. For each considered contrast, visualization of the average methylation level (as extracted from the unite files) per annotation feature was done by a stacked bar plot using the Python libraries pandas and matplotlib. With non-coding genes possibly having identical child features as protein-coding genes (e.g. typically transcript and exon), child features were discarded when a position belongs to both a non-coding gene and a protein-coding gene. Hereby, the interpretation of protein-coding gene features is not skewed by identical non-coding gene features. One exception was however made for the feature ‘intron’. If a position fell within an intronic region, the intron feature was retained.
Whole-genome bisulfite sequencing (WGBS)
For genome-wide DNA methylome analysis by whole-genome bisulfite sequencing (WGBS), the isolation of genomic DNA was performed from 12 pooled fourth leaves of individual plants for the control line and for the LR2 epiline, which belong to the fourth selfing generation, using the CTAB (cetyltrimethyl/ammonium bromide) standard protocol [66]. Genomic DNA isolates were examined for high quality and sufficient quantity by NanoDrop spectrophotometry (concentration, A260/A280 of approximately 1.9 or more) and gel electrophoresis (high molecular weight, integrity, purity). For WGBS, paired-end bisulfite sequencing libraries were constructed by Alpha Biolaboratory, Inc. Saratoga, CA as described previously [67] with modifications. About 300 ng of genomic DNA was fragmented by sonication, end repaired and ligated to custom-synthesized methylated adapters (Eurofins MWG Operon, Huntsville, AL) according to the manufacturer’s (Illumina, San Diego, CA) instructions for genomic DNA library construction. Adaptor-ligated libraries were subjected to two successive treatments of sodium bisulfite conversion using the EpiTect Bisulfite kit (Qiagen, Hilden, Germany) as outlined in the manufacturer’s instructions. One quarter of the bisulfite-converted libraries was PCR amplified using the following conditions: 2.5 U of ExTaq DNA polymerase (Takara Bio), 5 μL of 10 x Extaq reaction buffer, 25 μM dNTPs, 1 μL primer 1.1, 1 μl primer 2.1 in a 50-μL reaction. The thermocyling conditions were as follows: 95 °C for 3 min, then 12–14 cycles of 95 °C for 30 s, 65 °C for 30 s and 72 °C for 60 s. The enriched libraries were purified twice with SPRI method using 0.8 x v/v AM-Pure beads (Beckman Coulter, Brea, CA) prior to quantification with a Bioanalyzer (Agilent Technologies, Santa Clara, CA). Sequencing on the Illumina platform (SE100 runs) was performed at the Vincent J. Coates Genomic Sequencing Laboratory at UC Berkeley. The WGBS data analysis was performed with the same workflow used for the plant-RRBS data, with the exception that no cut-site specific indices were build. Genome coverage and cytosine coverage were determined in the same manner as for the plant-RRBS data. Bisulfite conversion efficiency rates (approximately 99%) were assessed by calculating cytosine methylation levels in the chloroplast genome.
Additional file
Additional files may be found in the online version of this article: Figure S1. Examples of electropherograms of library quality. Figure S2. Normalized overlap percentage of number of detected methylated sites between different RRBS samples per restriction enzyme and group. Figure S3. Integrative Genomics Viewer (IGV) screenshot of a representative genome region of RRBS and WGBS coverage data and mapped reads. Figure S4. Cytosine coverage in representative RRBS and WGBS samples. Figure S5. Annotation of methylated and common cytosine positions located on chromosomes between the biological replicates of different restriction endonuclease combinations and the control line and the LR2 epiline LR2 of selfing generation 4. Table S1. Physiological properties of the rice LR2 epiline versus the control inbred line (%). Table S2. Cellular respiration in the LR2 epiline (% versus the control inbred line) during consecutive selfings of the epilines. Table S3. Percentage of PCR duplicates in the input data per sample. Table S4. Read preprocessing and mapping quality. Table S5. Intersection of in silico fragments and mapped reads (%). Table S6. Intra-line similarity between biological replicates per line of selfing generation 4 based on the methylation level difference of the cytosine sites CG, CHG and CHH detected in the replicates. (DOCX 1363 kb)
Acknowledgments
We thank Dr. Annick Bleys for the critical reading of the manuscript and the precious help to improve and finalize it.
Funding
This research is funded by the European Union Seventh Framework Programme though the Marie Curie Research Training Network ‘Chromatin in Plants–European Training and Mobility’ to M.V.L. and fellow M.S. (CHIP-ET, FP7-PEOPLE-2013-ITN607880), and the Intra-European Fellowship to M.W. (LIGHTER, FP7-PEOPLE-2010-IEF-273068), the Agency for Innovation by Science and Technology through the ‘IsisCode’ project to M.D.B. and M.V.L. (IWT 120100).
Availability of data and materials
The data sets supporting the conclusions of this article are included within the article and its additional files and are available in the ArrayExpress [68] repository: accession numbers: E-MATB-4626 and E-MTAB-5002. Scripts are available via doi (10.5281/zenodo.168034) [52] and on GitHub [53].
Abbreviations
- BAM
Binary Alignment/Map
- BED
Browser Extensible Data
- Bisulfite
Sodium hydrogen sulfite
- CDS
Coding DNA sequence
- CMT3
Chromomethylase 3
- Control
Pure breeding rice line
- DDM1
Decrease in DNA methylation 1
- DRD1
Defective in RNA-directed DNA methylation 1
- DRM1/2
Domains rearranged methylase 1/2
- EUE
Energy use efficiency
- GBS
Genotyping by sequencing
- Intersection
Common set
- Jaccard similarity coefficient
Jaccard index
- MET1
DNA methyltransferase 1
- NAD(P)H
Reduced Nicotinamide adenine dinucleotide phosphate
- NGS
Next-generation DNA sequencing
- Plant-RRBS
Plant reduced representation bisulfite sequencing
- qPCR
Quantitative PCR
- RRBS
Reduced representation bisulfite sequencing
- S4
Selfing generation 4
- SPRI
Solid-phase reversible immobilization
- TE
Transposable element
- TSS
Transcription start site
- TTC
2,3,5-triphenyltetrazolium
- TTC-H
Reduced TTC
- Union
Total set
- WGBS
Whole-genome bisulfite sequencing
Authors’ contributions
M.S., B.S., M.W. and M.V.L. wrote the manuscript. M.W. and M.S. performed the experiments. B.S., M.S., M.V.B., C.M. and F.C. carried out bioinformatics analyses. M.W., M.S., M.D.B. and M.V.L. designed the study. F.C., B.S., M.V.B., M.S. and M.D.B. provided helpful discussion. All authors revised the manuscript. M.V.L. coordinated the project. All authors approved the submission and revision of this manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-017-1070-y) contains supplementary material, which is available to authorized users.
Contributor Information
Martin Schmidt, Email: masch@psb.ugent.be.
Michiel Van Bel, Email: mibel@psb.ugent.be.
Magdalena Woloszynska, Email: mawol@psb.ugent.be.
Bram Slabbinck, Email: brsla@psb.ugent.be.
Cindy Martens, Email: cindy.martens@ugent.be.
Marc De Block, Email: marc.deblock@bayer.com.
Frederik Coppens, Email: frcop@psb.ugent.be.
Mieke Van Lijsebettens, Phone: +32 (0)9 331 39 70, Email: milij@psb.ugent.be.
References
- 1.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan S-W, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Finnegan EJ, Kovac KA. Plant DNA methyltransferases. Plant Mol Biol. 2000;43:189–201. doi: 10.1023/A:1006427226972. [DOI] [PubMed] [Google Scholar]
- 5.Cao X, Jacobsen SE. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002;12:1138–1144. doi: 10.1016/S0960-9822(02)00925-9. [DOI] [PubMed] [Google Scholar]
- 6.Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, Caro E, et al. Dual binding of chromomethylase domains to H3K9me2-containing nucleosomes directs DNA methylation in plants. Cell. 2012;151:167–180. doi: 10.1016/j.cell.2012.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schöb H, Grossniklaus U. The first high-resolution DNA “methylome”. Cell. 2006;126:1025–1028. doi: 10.1016/j.cell.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Zhu J-K. Active DNA demethylation in plants and animals. Cold Spring Harb Symp Quant Biol. 2012;77:161–173. doi: 10.1101/sqb.2012.77.014936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, et al. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013;23:628–637. doi: 10.1101/gr.146985.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saze H, Tsugane K, Kanno T, Nishimura T. DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol. 2012;53:766–784. doi: 10.1093/pcp/pcs008. [DOI] [PubMed] [Google Scholar]
- 12.Cubas P, Vincent C, Coen E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature. 1999;401:157–161. doi: 10.1038/43657. [DOI] [PubMed] [Google Scholar]
- 13.Jacobsen SE, Meyerowitz EM. Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science. 1997;277:1100–1103. doi: 10.1126/science.277.5329.1100. [DOI] [PubMed] [Google Scholar]
- 14.Manning K, Tör M, Poole M, Hong Y, Thompson AJ, King GJ, et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet. 2006;38:948–952. doi: 10.1038/ng1841. [DOI] [PubMed] [Google Scholar]
- 15.Xie HJ, Li H, Liu D, Dai WM, He JY, Lin S, et al. ICE1 demethylation drives the range expansion of a plant invader through cold tolerance divergence. Mol Ecol. 2015;24:835–850. doi: 10.1111/mec.13067. [DOI] [PubMed] [Google Scholar]
- 16.Liu ZL, Han FP, Tan M, Shan XH, Dong YZ, Wang XZ, et al. Activation of a rice endogenous retrotransposon Tos17 in tissue culture is accompanied by cytosine demethylation and causes heritable alteration in methylation pattern of flanking genomic regions. Theor Appl Genet. 2004;109:200–209. doi: 10.1007/s00122-004-1618-8. [DOI] [PubMed] [Google Scholar]
- 17.Bewick AJ, Ji L, Niederhuth CE, Willing E-M, Hofmeister BT, Shi X, et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc Natl Acad Sci U S A. 2016;113:9111–9116. doi: 10.1073/pnas.1604666113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elhamamsy AR. DNA methylation dynamics in plants and mammals: overview of regulation and dysregulation. Cell Biochem Funct. 2016;34:289–298. doi: 10.1002/cbf.3183. [DOI] [PubMed] [Google Scholar]
- 19.Becker C, Hagmann J, Müller J, Koenig D, Stegle O, Borgwardt K, et al. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–249. doi: 10.1038/nature10555. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz RJ, Schultz MD, Lewsey MG, O’Malley RC, Urich MA, Libiger O, et al. Transgenerational epigenetic instability is a source of novel methylation variants. Science. 2011;334:369–373. doi: 10.1126/science.1212959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N, et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009;5:e1000530. doi: 10.1371/journal.pgen.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hauben M, Haesendonckx B, Standaert E, Van Der Kelen K, Azmi A, Akpo H, et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc Natl Acad Sci U S A. 2009;106:20109–20114. doi: 10.1073/pnas.0908755106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verkest A, Byzova M, Martens C, Willems P, Verwulgen T, Slabbinck B, et al. Selection for improved energy use efficiency and drought tolerance in canola results in distinct transcriptome and epigenome changes. Plant Physiol. 2015;168:1338–1350. doi: 10.1104/pp.15.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng D, Dubin M, Zhang P, Osborne EJ, Stegle O, Clark RM, et al. Limited contribution of DNA methylation variation to expression regulation in Arabidopsis thaliana. PLoS Genet. 2016;12:e1006141. doi: 10.1371/journal.pgen.1006141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Block M, Van Lijsebettens M. Energy efficiency and energy homeostasis as genetic and epigenetic components of plant performance and crop productivity. Curr Opin Plant Biol. 2011;14:275–282. doi: 10.1016/j.pbi.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Schultz MD, Schmitz RJ, Ecker JR. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012;28:583–585. doi: 10.1016/j.tig.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Ge X, Wang J, Tan C, King GJ, Liu K. Genome-wide DNA methylation profiling by modified reduced representation bisulfite sequencing in Brassica rapa suggests that epigenetic modifications play a key role in polyploid genome evolution. Front Plant Sci. 2015;6:836. doi: 10.3389/fpls.2015.00836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gugger PF, Fitz-Gibbon S, Pellegrini M, Sork VL. Species-wide patterns of DNA methylation variation in Quercus lobata and its association with climate gradients. Mol Ecol. 2016;25:1665–1680. doi: 10.1111/mec.13563. [DOI] [PubMed] [Google Scholar]
- 31.van Gurp TP, Wagemaker NCAM, Wouters B, Vergeer P, JNJ O, KJF V. epiGBS: reference-free reduced representation bisulfite sequencing. Nat Methods. 2016;13:322–324. doi: 10.1038/nmeth.3763. [DOI] [PubMed] [Google Scholar]
- 32.Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, et al. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics. 2013;14:774. doi: 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okonechnikov K, Conesa A, García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32:292–294. doi: 10.1093/bioinformatics/btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.bamUtil. https://github.com/statgen/bamUtil. Accessed 25 Oct 2016.
- 37.Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu J, Hu S, Wang J, Wong GK-S, Li S, Liu B, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica) Science. 2002;296:79–92. doi: 10.1126/science.1068037. [DOI] [PubMed] [Google Scholar]
- 39.Choi M, Lee J, Le MT, Nguyen DT, Park S, Soundrarajan N, et al. Genome-wide analysis of DNA methylation in pigs using reduced representation bisulfite sequencing. DNA Res. 2015;22:343–355. doi: 10.1093/dnares/dsv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Zhu J, Hu F, Ge S, Ye M, Xiang H, et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics. 2012;13:300. doi: 10.1186/1471-2164-13-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ensembl Plants. http://plants.ensembl.org/index.html. Accessed 25 Oct 2016.
- 42.Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2012;13:36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt M, Hense S, Minoche AE, Dohm JC, Himmelbauer H, Schmidt T, et al. Cytosine methylation of an ancient satellite family in the wild beet Beta procumbens. Cytogenet Genome Res. 2014;143:157–167. doi: 10.1159/000363485. [DOI] [PubMed] [Google Scholar]
- 44.De Block M, De Brouwer D. A simple and robust in vitro assay to quantify the vigour of oilseed rape lines and hybrids. Plant Physiol Biochem. 2002;40:845–852. doi: 10.1016/S0981-9428(02)01439-0. [DOI] [Google Scholar]
- 45.The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 46.The Brassica rapa Genome Sequencing Project Consortium. Wang X, Wang H, Wang J, Sun R, Wu J, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–1039. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- 47.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 48.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 49.Hsieh T-F. Whole-genome DNA methylation profiling with nucleotide resolution. Methods Mol Biol. 2015;1284:27–40. doi: 10.1007/978-1-4939-2444-8_2. [DOI] [PubMed] [Google Scholar]
- 50.Pignatta D, Bell GW, Gehring M. Whole genome bisulfite sequencing and DNA methylation analysis from plant tissue. BioProtocol. 2015;5:e1407. [Google Scholar]
- 51.Parkinson H, Sarkans U, Kolesnikov N, Abeygunawardena N, Burdett T, Dylag M, et al. ArrayExpress update-an archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011;39:D1002–D10D4. doi: 10.1093/nar/gkq1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The DOI® System. https://doi.org/10.5281/zenodo.168034. Accessed 25 Oct 2016.
- 53.GitHub. https://github.com/VIB-PSB/PlantRRBS. Accessed 25 Oct 2016.
- 54.FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 25 Oct 2016.
- 55.FASTX Toolkit. http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 25 Oct 2016.
- 56.Trim Galore. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. Accessed 25 Oct 2016.
- 57.Reference genome: 9311_BGF_2005 PLAZA. ftp://ftp.psb.ugent.be/pub/plaza/plaza_public_monocots_03/Genomes/osaindica.con.gz. Accessed 25 Oct 2016.
- 58.Python. https://www.python.org/. Accessed 25 Oct 2016.
- 59.Integrative Genomics Viewer (IGV). http://software.broadinstitute.org/software/igv/. Accessed 25 Oct 2016.
- 60.igv.js. http://igv.org/doc/doc.html. Accessed 25 Oct 2016.
- 61.Picard. http://broadinstitute.github.io/picard/. Accessed 25 Oct 2016.
- 62.The R Project for Statistical Computing. https://www.r-project.org/. Accessed 25 Oct 2016.
- 63.Wang H-Q, Tuominen LK, Tsai C-J. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics. 2011;27:225–231. doi: 10.1093/bioinformatics/btq650. [DOI] [PubMed] [Google Scholar]
- 64.Oryza_indica.ASM465v1.27.gff3. ftp://ftp.ensemblgenomes.org/pub/plants/release-27/gff3/oryza_indica/Oryza_indica.ASM465v1.27.gff3.gz. Accessed 25 Oct 2016.
- 65.Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10:645–656. doi: 10.1109/TCBB.2013.68. [DOI] [PubMed] [Google Scholar]
- 66.Murray MG, Thompson WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980;8:4321–4325. doi: 10.1093/nar/8.19.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ibarra CA, Feng X, Schoft VK, Hsieh T-F, Uzawa R, Rodrigues JA, et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science. 2012;337:1360–1364. doi: 10.1126/science.1224839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.ArrayExpress. http://www.ebi.ac.uk/arrayexpress/browse.html. Accessed 25 Oct 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional files may be found in the online version of this article: Figure S1. Examples of electropherograms of library quality. Figure S2. Normalized overlap percentage of number of detected methylated sites between different RRBS samples per restriction enzyme and group. Figure S3. Integrative Genomics Viewer (IGV) screenshot of a representative genome region of RRBS and WGBS coverage data and mapped reads. Figure S4. Cytosine coverage in representative RRBS and WGBS samples. Figure S5. Annotation of methylated and common cytosine positions located on chromosomes between the biological replicates of different restriction endonuclease combinations and the control line and the LR2 epiline LR2 of selfing generation 4. Table S1. Physiological properties of the rice LR2 epiline versus the control inbred line (%). Table S2. Cellular respiration in the LR2 epiline (% versus the control inbred line) during consecutive selfings of the epilines. Table S3. Percentage of PCR duplicates in the input data per sample. Table S4. Read preprocessing and mapping quality. Table S5. Intersection of in silico fragments and mapped reads (%). Table S6. Intra-line similarity between biological replicates per line of selfing generation 4 based on the methylation level difference of the cytosine sites CG, CHG and CHH detected in the replicates. (DOCX 1363 kb)
Data Availability Statement
The data sets supporting the conclusions of this article are included within the article and its additional files and are available in the ArrayExpress [68] repository: accession numbers: E-MATB-4626 and E-MTAB-5002. Scripts are available via doi (10.5281/zenodo.168034) [52] and on GitHub [53].