

Abstract
Dietary fish oils have potential for prevention of colon cancer, and yet the mechanisms of action in normal and tumor colon tissues are not well defined. Here we evaluated the impact of the colonic fatty acid milieu on formation of prostaglandins and other eicosanoids. Distal tumors in rats were chemically induced to model inflammatory colonic carcinogenesis. After 21 weeks of feeding with either a fish oil diet containing an eicosapentaenoic acid:ω-6 fatty acid ratio of 0.4 or a Western fat diet, the relationships between colon fatty acids and prostaglandin E2 (PGE2) concentrations were evaluated. PGE2 is a key pro-inflammatory mediator in the colon tightly linked with the initiation and progression of colon cancer. The fish oil versus the Western fat diet resulted in reduced total fatty acid concentrations in serum but not in colon. In the colon, the effects of the fish oil on fatty acids differed in normal and tumor tissue. There were distinct lipodomic patterns consistent with a lipogenic phenotype in tumors. In tumor tissue, the eicosapentaenoic acid:arachidonic acid ratio, cyclooxygenase-2 expression and the mole percent of saturated fatty acids were significant predictors of inter-animal variability in colon PGE2 after accounting for diet. In normal tissues from either control rats or carcinogen-treated rats, only diet was a significant predictor of colon PGE2. These results show that the fatty acid milieu can modulate the efficacy of dietary fish oils for colon cancer prevention, and this could extend to other preventive agents that function by reducing inflammatory stress.
Keywords: Colon cancer, fatty acid metabolism, prostaglandin E2, diet, cancer prevention
1. Introduction
Many epidemiological and animal studies support the role of increased dietary ω-3 fatty acids, versus that of ω-6 fatty acids, for prevention of colon carcinogenesis [1-4]. The magnitude of this preventive effect can be large. For example, feeding ω-3 fatty acids reduces invasive colorectal cancer incidence and multiplicity in rodents by 35% to 70% [5-7]. This is similar in magnitude to the preventive effects of non-steroidal anti-inflammatory agents (NSAIDs). The cardiovascular and gastrointestinal toxicities of chronic NSAID use have rendered them unsuitable for long-term colon cancer prevention in healthy, normal risk populations [8].
Like NSAIDs, ω-3 fatty acids inhibit prostaglandin E2 (PGE2) synthesis. PGE2 in the colon is a pro-inflammatory mediator tightly linked with increased colon cancer risk [9]. Dietary ω-3 fatty acid supplementation increases the ratio of ω-3 to ω-6 fatty acids in membrane phospholipids. Increasing the ω-3 to ω-6 fatty acid ratio reduces the availability of ω-6 arachidonic acid (AA) for metabolism to PGE2, a process catalyzed by constitutive cyclooxygenase(COX)-1 and inducible COX-2 [10]. The ω-3 fatty acid, eicosapentaenoic acid (EPA), reduces PGE2 by reducing the availability of AA for the catalytic monomer of the COX-1 dimer and inhibiting COX-1 oxygenation of AA [11, 12]. EPA serves as a substrate for the COX-2 catalytic site; with the formation of PGE3 as the metabolic product [10]. EPA competes poorly with AA for the COX-2 catalytic site. For this reason, PGE2, the downstream catalytic product of AA predominates over PGE3.
Other common fatty acids cannot serve as substrates for the catalytic dimer of either COX-1 or COX-2 leading to the term “non-substrate fatty acids”. However, these common saturated and monounsaturated fatty acids (SFA, MUFA) such as stearic and palmitic acids bind to the COX-2 allosteric dimer and increase the activity of the catalytic dimer resulting in increased synthesis of PGE2 [12-14]. Binding to the allosteric dimer of COX-1 slightly inhibits the activity of the COX-1 catalytic dimer [15]. This effect of non-substrate fatty acids would take on particular importance when COX-2 is induced and/or when concentrations of non-substrate fatty acids are high, such as that in tumors (Figure 1).
Figure 1.
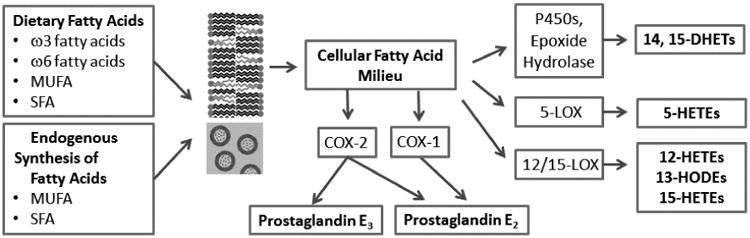
Schematic view of fatty acid pathways relevant to formation of eicosanoids in normal and tumor tissues. Fatty acids from endogenous synthesis and diet contribute to the fatty acid composition of cellular lipids stores in membranes and lipid droplets and modulate the relative proportion of arachidonic acid (ω-6) that is available to cyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P450 enzymes for production of oxygenated metabolites. Lipoxygenases selectively produce S-isomeric eicosanoids while lipid peroxidation produces R and S isomers in equal quantities. All of these enzymes use arachidonic acid as a substrate except that 13-HODE is produced from linoleic acid. Prostaglandin E2 is produced from arachidonic acid by both COX-1 and COX-2, and prostaglandin E3 is produced from eicosapentaenoic acid (ω-3) by COX-2, although eicosapentaenoic acid competes poorly with arachidonic acid as a substrate for COX-2. As discussed in the text, products that have been associated with beneficial effects in the colon include prostaglandin E3, 13-S-HODE and 15-S-HETE while the other eicosanoids shown generally have pro-tumorigenic effects. Prostaglandin E2 in particular is known to be promote colon tumorigenesis.
In rodents, we previously reported that dietary ω-3 fatty acids modify the fatty acid substrate pools to decrease PGE2 formation in the colon [16, 17]. There was a dose dependent response of dietary fish oil in reducing colonic PGE2 with dietary EPA:ω-6 fatty acid ratios of 0.4 or less [17]. We demonstrated that each 10% increase in serum EPA:AA ratio was associated with a 2% decrease in the geometric mean of colonic PGE2 concentration [17]. Consistent with this finding, fish oil fed rats treated with the carcinogen azoxymethane (AOM) had reduced colon tumor incidence and multiplicity compared to Western diet fed controls [7, 18]. In addition, fish oils are known to have a triglyceride lowering effect [19], but the effects of fish oils on colon lipids are currently less well defined.
In the present study, we treated rats with azoxymethane (AOM)/dextran sodium sulfate (DSS) which induces inflammatory and carcinogenic response in mainly the distal colon of rodents [20]. We evaluated how both a fish oil diet and the process of tumorigenesis affect fatty acid substrate pools and eicosanoid formation in normal and tumor tissue from distal colon. We anticipated that the relationships between fatty acids and prostaglandin formation might differ in normal and tumor colon tissue due to the induction of COX-2 in neoplastic tissue [9]. In addition, colon tumorigenesis was reported to be accompanied by increased endogenous synthesis of fatty acids that are mainly palmitate and oleate [21]. The goal of the study therefore was to quantify changes in colon lipids with dietary fish oils, as compared with a Western blend fat, on colon lipids and to determine the impact of these fatty acid alterations on prostaglandin production in normal and tumor colon tissues.
2. Materials and Methods
2.1. Animals and diets
Male Fisher F344 rats, 5 weeks of age, were purchased from Harlan Laboratories (Haslett, MI). The rats were given fresh food on a daily basis and water ad libitum throughout the experiment. The rats were maintained on a 12 h light/dark cycle. The animal experimental protocol was approved by the University Committee on Use and Care of Animals at the University of Michigan. Data from our published study evaluating different doses of fish oils [17] showed that the 57% reduction in PGE2 that can be achieved with a dietary EPA:ω-6 ratio of 0.4 can be detected with power > 90% using 10 animals/group.
After being acclimated to the standard AIN93-G diet for one week, rats were randomized into two dietary groups: control group (20 rats) and fish oil group (20 rats). Both the control and fish oil diets contained 34% energy from fat, 17% by weight. This fat content was achieved by decreasing cornstarch to accommodate the increased fat content versus the standard AIN93G diet. The Western fat diet contained a blend of fats and no EPA: coconut oil (45% by weight), olive oil (30% by weight), corn oil (15% by weight) and soybean oil (10% by weight). For the fish oil diet, the Western fat blend was mixed with menhaden oil to achieve an EPA:ω-6 ratio of 0.4 as previously described [17]. This resulted in a diet that was10.8% Western fat and 6.2% Menhaden oil, by weight of total diet. Both diets used in the experiment were prepared by Dyets Inc. (Bethlehem, PA). Diets were stored at -80°C and were used within three months after purchase. Small amounts of the diet were stored at -20°C for no more than one week and this was used to provide fresh food to the animals daily.
Half of the rats in each diet group were administered 15 mg/kg body weight of azoxymethane (AOM, acquired from the NCI Chemical Carcinogen Reference Standard Repository MRI Global, Kansas City, MO), by intra-peritoneal injection once after 7 days of starting the experimental diets. One week later, the AOM-treated rats were given 2% dextran sodium sulfate (DSS, catalog no. D6001, Sigma-Aldrich Co., St. Louis, MO) in the drinking water, which was replaced on daily basis for 7 days.
Rats were sacrificed using carbon dioxide inhalation and blood was obtained using cardiac puncture after opening the chest cavity. Sera were separated and stored at -80oC for fatty acid analysis. The whole colon was quickly removed, opened longitudinally and rinsed with ice-cold phosphate-buffered saline (PBS) containing indomethacin (5.6 μg/mL). Tumors in the distal colon were grossly visualized and dissected away from normal tissue. The tumors and grossly normal proximal, transverse and dissected distal sections of colon were snap frozen in liquid nitrogen and stored at -80oC before analysis. The normal and tumor distal colon tissues were subsequently pulverized in liquid nitrogen and stored at -80oC. A portion of this pulverized tissue was used for lipodomic analysis, a portion for RNA expression and another portion was used to prepare a homogenate for determination of both eicosanoids and fatty acids.
2.2. Fatty acid analysis by GC-MS
Approximately 60 mg of pulverized colon tissue was weighed out and added to 1 mL of cold phosphate-buffered saline containing 0.1mM indomethacin, 1mM EDTA. The homogenate was sonicated in ice water for 5 min (cycling between 20 seconds of sonication and 20 seconds of cooling). Aliquots were stored in a -80oC freezer until analysis of eicosanoids, triglycerides, fatty acids or protein. The protein concentration of homogenates was determined using the Bradford assay (Bio-Rad Laboratories Inc., Hercules, CA).
For quantitation of fatty acids, 150 μL of colon homogenate was added to 10 μL of internal standard (17:0, 1 mg/mL in hexane). The samples were then extracted with 1.0 mL of Folch reagent (chloroform: methanol 2:1), vortexed for 2 minutes, and centrifuged (1200 × g for 8 min). The organic layer was removed to another glass tube and dried in a SpeedVac. The samples were solubilized in 70 μL of hexane:chloroform (1:1), and vortexed. Fatty acid methyl esters (FAME) were prepared by adding 30 μL of METH-PREP II derivatization reagent (0.2 N methanolic (m-trifluoromethylphenyl) trimethylammonium hydroxide; Grace Science, Deerfield, IL). GC-MS was conducted as previously described using selected ion monitoring [22, 23].
Individual amounts of each fatty acid (in μg) were calculated from standard curves and converted to moles. The mole percentage of each fatty acid, of total moles of fatty acids, was calculated in serum and colon homogenates. For colon, μmoles of each fatty acid per μg protein were calculated. For serum, fatty acid concentrations were reported in μg/ml.
2.3. Eicosanoid analysis by chiral liquid chromatography tandem mass spectroscopy (LC-MS/MS)
For extraction of eicosanoids, 300 μL of the colonic homogenate was added to 12 × 75 mm glass tubes on ice, along with 700 μL de-ionized water, 20 μL of 0.75M citric acid-0.25 M ammonium acetate pH 2.7, 20 μL of 30 mM disodium EDTA, and 20 μL of a mixture of deuterated internal standards. The resulting solution was then extracted twice with 2 mL hexane:ethyl acetate (1:1 v/v, containing 0.1% BHT (w/v). The pooled extracts were evaporated under vacuum and reconstituted with 80 μL of HPLC-grade methanol. Aliquots (20 μL) were injected for analysis by LC-MS/MS. The LC-MS/MS method for eicosanoid analysis we published was adapted by adjusting our previously developed the HPLC program to include analysis of 14,15-S-dihydroxyeicosatrienoic acid (DHET) and 14,15-R-DHET [24]. Eicosanoid standards were obtained from Cayman Chemical (Ann Arbor, MI).
The HPLC separation was performed on a Waters 2695 separations module, using a Chiral-Pak AD-RH analytical column (2.1 × 150 mm, 3 μm particle size) (Chiral Technologies, West Chester, PA). The column was maintained at 40°C. Mobile phase A was 10 mM ammonium acetate pH 3.8 and mobile phase B was acetonitrile. The flow rate was 0.2 mL/min. The linear gradient program was: 30-50% B (0–5 min), 50-100% B (5–24 min), 100% B (25-30 min), and return to 30% B (30–37 min).The effluent was introduced into a Finnigan TSQ Quantum Ultra triple quadrupole mass spectrometer by electrospray ionization (ESI) and detection of negativeions in the single reaction monitoring mode was as previously described except that we added 14, 15-DHET which was monitored at the transition m/z 337.5 to 206.9 [24]. Eicosanoid concentrations in colonic tissue were normalized to protein concentrations.
2.4. Cyclooxygenase (COX) gene expression by real-time quantitative PCR
COX gene expression was performed using quantitative real-time PCR (RT-qPCR). RNA was extracted from 100 mg pulverized tissue using Trizol Plus Purification Kit, following the manufacturers' protocol (Thermo Fisher Scientific/Applied Biosystems, Waltham, MA). The recovered RNA was checked for purity and quantified spectrophotometrically. Real-Time PCR was performed with 10 ng cDNA using the TaqMan® Gene Expression Assay system (Applied Biosystems). A mixture of cDNA from the sample pool was used to construct a standard curve for each gene.
The primers and probes used for real time PCR were purchased from Applied Biosystems. The primers used were as follows: COX-1 (PTGS1, cat. no. 1428661), and COX-2 (PTGS2, cat. no. 1437258). Actin was used as an internal control for normalization (cat. no. 1429880). The real-time PCR was done with a CFX96 PC detection system (Bio-Rad Laboratories, Philadelphia, PA), and the thermal conditions were: 50°C 2 min, 95°C 10 min followed by 40 cycles of 95°C 15 sec and 60°C 1 min. All samples and standard curves were analyzed in duplicate and averaged. Duplicates that differed by more than 10% were repeated.
The mean efficiencies for the PCR standard curves for each primer were: PTGS1 (96%), PTGS2 (94%), and actin (98%). For quantification, the standard curve method was used, and the average amount of each target mRNA expression and actin mRNA expression was established from their respective standard curves. COX-1 and COX-2 gene expression was calculated relative to actin expression as a ratio.
2.5. Lipodomics of colon tissue
To investigate the effects of fish oil feeding and carcinogen treatment on colon lipid classes, two separate, untargeted, shotgun lipodomic experiments were carried out using aliquots of the same pulverized, flash-frozen colon tissue samples that were used for other assays. These assays utilized weighed aliquots of pulverized colon tissue of about 20 mg each. and results were normalized to tissue weight. In the first experiment, normal colon tissue from control rats fed fish oil or not and tumor colon tissue from rats treated with AOM/DSS fed fish oil or not were analyzed. Three samples from each of the four treatment/diet groups were selected based on colon PGE2 concentrations: one sample with the highest PGE2, one with the lowest PGE2 and one sample with mid-level PGE2 was selected within each treatment/diet group to maximize diversity in the samples.
Since the biology of tumors can modify colon lipids, a second lipodomic experiment was conducted to investigate the effects of fish oil and carcinogen treatment on grossly normal colon tissue. Samples of colon tissue from control rats and grossly normal colon tissue from rats treated with AOM/DSS were selected from rats fed Western fat or fish oil diets. Again three samples from each treatment/diet group were selected based on PGE2 concentrations to represent maximum diversity in PGE2 in the samples. The normal tissue from control rats in this experiment was not the same as that in the first experiment.
Extraction of lipids from pulverized tissue was carried out after spiking with internal standards from each lipid class and sonicating the samples in methanol briefly. Lipids were extracted using a modified Bligh-Dyer method with 2:2:2, by volume, of water: methanol: dichloromethane at room temperature. The organic layer was collected and dried completely under the stream of nitrogen before being re-suspended in 100 μL of HPLC solvent B (see below). The samples were analyzed by HPLC-MS-MS along with quality control samples. Separations were achieved with a 1.8 μm particle 50 × 2.1 mm ID Waters Acquity HSS T3 column (Waters, Milford, MA) which was heated to 55°C. Elution was performed using a gradient of 40% solvent A to 100% solvent B over 10 minutes. Solvent A was acetonitrile: water (40:60, by volume) with 10 mM ammonium acetate and solvent B was acetonitrile: water: isopropanol (10:5:85, by volume) with 10 mM ammonium acetate. Lipids were identified using the LIPIDBLAST library by matching the product ions MS/MS data using MultiQuant 1.1.0.26. Identified lipids were quantified by normalizing against their respective internal standard. Only lipids with a relative SD of <30% in quality control samples were analyzed.
2.6. Statistical analyses
Colon tissue eicosanoids were expressed as ng per mg protein. Colon fatty acids in μg/sample were summed to obtain μg of total fatty acids per sample and expressed relative to the protein content of the homogenate. Individual colon fatty acids were expressed as a mole percent of total. In serum, fatty acids were expressed as mole percent and a total weight amount of fatty acids per mL serum was also calculated.
All analyses of eicosanoids and fatty acids were performed using the IBM SPSS program, version 22 (IBM Corporation, Armonk, New York). Variables were transformed as needed to achieve normality, and the transformations used are given in the tables. The effect of fish oil feeding and carcinogen treatment on outcome variables was evaluated by two-way ANOVA. Pairwise analyses were conducted using Linear Distance Sampling. The same analyses were done to compare tumor tissue with normal tissue from control animals. In samples from animals for which both normal and tumor colon tissue was available, data were also analyzed using mixed models with diet (fish oil, Western fat), tissue type (normal, tumor) and their interaction as factors. To account for multiple comparisons, differences were considered significant when p<0.005.
Linear regression models with log PGE2 or log PGE3 as outcomes were constructed including predictor variables selected based on the biochemistry of cyclooxygenase function. We evaluated fatty acids that are substrates for COXs (EPA, AA, DHA, EPA:AA ratio) as well as non-substrates (MUFA, SFA, other ω-3 and ω-6 fatty acids). Although the fish oil diet had strong effects on prostaglandin formation, there was variability in the fatty acid concentrations and expression of COXs in each group. We optimized each regression model to obtain a maximal adjusted R2 for each tissue type: normal colon tissue, normal colon tissue from carcinogen-treated rats and tumor colon tissue from carcinogen-treated rats.
Lipidomic data for samples was normalized to tissue weight. Data was log transformed to stabilize the variance. A two-way ANOVA model was fit for each lipid with diet and tissue type as factors. After adjusting for false discovery rates, those lipids with p < 0.05 for the effect of diet (fish oil, Western fat) or tissue type (tumor, carcinogen-treated normal or normal) were subjected to principal component analysis using the pca3d package and hierarchical clustering analyses with the R program [25, 26]. Heatmaps were created using function “heatmap.2” in package “gplots” within the R program [27] and are shown in the Data in Brief accompanying article [28].
3. Results
3.1. Animal weight gain and tumors
Mean weight gain was similar in the four groups of ten rats while consuming the experimental diets from experimental week 3 to week 20. Weight gain in the animals fed the Western fat diet was 235 grams (SD 31) and with the fish oil diet it was 225 grams (SD 31). In the AOM-DSS treated animals, weight gain was 238 (SD 23) and 256 (SD 20) grams in animals fed the Western and fish oil diets, respectively. These differences were not statistically significant. All of the animals that received AOM and DSS and the Western diet developed tumors while nine of 10 animals on the fish oil diet developed tumors. None of the control animals (defined as no carcinogen treatment) developed colon tumors. Pathological evaluation of tumors was not possible since tissues were snap frozen for biochemical analyses. In parallel tumor experiments that will be reported separately, the fish oil diet reduced mean tumor volume and mean tumor multiplicity by about half, consistent with the reduction in PGE2 shown in Table 1 that was statistically significant in tumor tissue. These preventive effects of a fish oil diet are well documented and depend partially on the carcinogen dose [5-7].
Table 1.
Effects of a fish oil diet on colon eicosanoids and COX gene expression, given as mean (SD). Eicosanoids are in ng/mg protein and gene expression is relative to actin. The abbreviations used are: cyclooxygenase (COX), dihydroxyeicosatrienoic acid (DHET), hydroxyicosatetraenoic acid (HETE), hydroxyoctadecadienoic acid (HODE), prostaglandin (PG). The F344 rats were treated with AOM/DSS (“carcinogen”) or not and fed a 17% fat diet, by weight, for 21 weeks. Tissue labelled “normal” in carcinogen-treated rats is grossly normal. N=10 per group except n=9 for tumor tissue from fish oil fed rats since one rat in that group had no tumors.
| Eicosanoid or gene expression1 | Normal Tissue from Control Rats | Normal Tissue from Carcinogen-treated Rats | Tumor Tissue from Carcinogen-treated Rats | |||
|---|---|---|---|---|---|---|
| Western fat diet | Fish oil diet | Western fat diet | Fish oil diet | Western fat diet | Fish oil diet | |
| Eicosanoids (ng/mg protein) | ||||||
| PGE2 | 132 (64) | 78 (17) | 150 (74) | 92 (43) | 269 (153) | 85 (60)* |
| PGE3 | 0.6 (0.2) | 4.4 (1.1)* | 0.6 (0.1) | 7.0 (2.9)* | 0.9 (0.3) | 8.3 (5.8)* |
| 5-S-HETE | 3.4 (1.4) | 1.7 (0.6) | 8.0 (3.9) | 6.8 (4.0)^ | 16.6 (13.9)^ | 6.6 (3.5)^ |
| 5-R-HETE | 9.4 (6.6) | 2.6 (1.1)* | 11.0 (6.9) | 6.8 (2.5)^ | 14.7 (6.0) | 5.1 (3.3)* |
| 12-S-HETE | 21.4 (10.8) | 9.4 (3.0) | 60 (59) | 21 (10) | 419 (436)^ | 164 (229)^ |
| 12-R-HETE | 8.7 (5.9) | 1.6 (0.6)* | 13 (10) | 15 (7)^ | 32 (27) | 10 (9)*^ |
| 14,15-S-DHET | 1.6 (0.9) | 0.9 (0.2) | 2.4 (1.3) | 1.4 (0.3) | 2.5 (1.0) | 1.2 (0.4)* |
| 14,15-R-DHET | 1.6 (1.1) | 0.8 (0.2) | 2.2 (1.2) | 1.4 (0.4) | 2.8 (1.1)^ | 1.3 (0.5)* |
| 15-S-HETE | 601 (261) | 832 (264) | 280 (159)^ | 694 (200)* | 605 (305) | 714 (395) |
| 15-R-HETE | 68 (32) | 53 (9) | 73 (26) | 71 (21) | 111 (59) | 62 (25) |
| 13-S-HODE | 26 (12) | 15 (4) | 32 (13) | 28 (14) | 49 (28) | 18 (9)* |
| 13-R-HODE | 3.2 (2.5) | 6.2 (2.0) | 3.8 (3.1) | 9.5 (5.4) | 3.9 (3.9) | 6.6 (2.8) |
| Gene expression (relative to actin) | ||||||
| COX-1 | 1.81 (1.48) | 2.22 (1.93) | 1.19 (1.06) | 0.65 (0.64) | 1.07 (1.07) | 0.66 (0.52) |
| COX-2 | 0.29 (0.20) | 0.54 (0.38) | 0.56 (0.40) | 0.36 (0.23) | 1.09 (0.51) | 1.48 (1.17)^ |
| COX-2:COX-1 | 0.19 (0.08) | 0.33 (0.22) | 0.68 (0.41)^ | 0.69 (0.46) | 1.71 (1.02)^ | 3.17 (3.39)^ |
Significantly different for the fish oil diet versus Western fat diet (p<0.005), within a tissue type, from 2-way ANOVA. Eicosanoids and COX expression were log-transformed for these analyses except for 15-S-HETE which needed no transformation to achieve normality.
Significantly different versus normal tissue from control rats (p<0.005), within each diet type, from 2-way ANOVA.
3.2. Eicosanoids and cyclooxygenase expression in colon
Eicosanoids and cyclooxygenase expression were quantified in normal colon tissue from control animals and in both normal and tumor tissues from carcinogen-treated rats. PGE2 was decreased by fish oil feeding in all tissue types, consistent with preventive effects, but this was statistically significant only in the colon tumor tissue (Table 1). PGE3, however, was significantly higher in all three tissue types when animals were fed the fish oil diet. Fish oil feeding resulted in significantly lower 5-R-hydroxyicosatetraenoic acid (HETE) and 12-R-HETE in normal tissue from control rats and increased 15-S-HETE in normal tissue from carcinogen-treated rats relative to that with the Western fat diet. In tumor tissue, 15-S-HETE was not significantly increased by fish oil feeding, but there were a large number of significant differences with fish oil versus Western fat feeding in tumors as shown in Table 1. There was no significant effect of diet on expression of COX enzymes in the colon. Inter-animal variability was large for all of these measures.
The effect of carcinogen treatment and tissue type (normal versus tumor) was also evaluated by two-way ANOVA. In normal colon from the fish oil fed rats, 5-S-HETE, 5-R-HETE and 12-R-HETE were relatively higher in carcinogen-treated rats versus control rats. In the Western diet-fed rats, 15-S-HETE was lower and the COX-2:COX-1 expression ratio was higher in the carcinogen-treated rats versus the control rats that were not given carcinogen (Table 1). There were also several statistically significant differences between normal and tumor tissue eicosanoids evident from the two-way ANOVA as annotated in Table 1. The COX-2:COX-1 expression ratio was higher in tumors versus normal tissue from control rats fed either diet.
Additional analyses compared normal and tumor tissue from carcinogen-treated rats using mixed models since these tissues were obtained from the same animals, and these results are not annotated on Table 1. The models used tissue type (normal, tumor), diet (fish oil, Western fat), and their interaction as factors. Values significantly higher in tumor tissue versus normal tissue in pairwise contrasts were 5-S-HETE, 12-S-HETE, 12-R-HETE, and 13-S-HODE in rats fed the Western diet and 12-S-HETE in rats fed the fish oil diet. In addition, COX-2 expression and the ratio of COX-2 to COX-1 expression was significantly higher in tumor versus normal tissue from carcinogen-treated rats.
3.3. Fatty acids in serum and colon
In serum of control animals, the mole percent of ω-3 fatty acids increased while ω-6 fatty acids and monounsaturated fatty acids decreased with fish oil feeding (Table 2). The relative proportion of saturated fatty acids was not different by diet but the total concentration of fatty acids in serum was about half that in Western diet fed animals (p<0.005 from two-way ANOVA with post-hoc pairwise analyses). Similar effects of diet on serum fatty acids were found in control rats and carcinogen-treated rats (Table 2).
Table 2.
Fatty acids in serum of rats fed different diets. Shown are the mean and SD for saturated fatty acids (SFA), monounsaturated fatty acids (MUFA), polyunsaturated fatty acids (PUFA), arachidonic acid (AA), eicosapentaenoic acid (EPA) and select ratios. There were 10 rats per group.
| Fatty Acid (mole %, unless indicated otherwise) | Serum of Control Rats | Serum of Carcinogen-treated Rats | ||
|---|---|---|---|---|
| Western fat diet | Fish oil diet | Western fat diet | Fish oil diet | |
| SFA | 43 (2) | 44 (3) | 43 (1) | 44 (2) |
| MUFA | 26 (2) | 18 (2)* | 25 (2) | 17 (2)* |
| ω-3 PUFA | 3.2 (0.5) | 19 (2)* | 3.3 (0.4) | 19 (2)* |
| ω-6 PUFA | 27 (2) | 19 (1)* | 29 (2) | 20 (1)* |
| AA:linoleic acid ratio | 1.22 (0.15) | 0.72 (0.18)* | 1.20 (0.32) | 0.72 (0.07)* |
| 20:4, ω-6 (AA) | 14.12 (1.65) | 7.37 (1.39)* | 14.55 (1.90) | 7.96 (0.82)* |
| 20:5, ω-3 (EPA) | 0.23 (0.05) | 7.00 (0.70)* | 0.22 (0.06) | 6.85 (0.83)* |
| EPA:AA ratio | 0.02 (0.00) | 0.96 (0.14)* | 0.02 (0.00) | 0.86 (0.08)* |
| Total fatty acids (μg/ml serum) | 43 (7) | 21 (5)* | 38 (5) | 17 (2)* |
Significantly different for the fish oil diet versus Western fat diet (p<0.005), within a treatment group from 2-way ANOVA with pairwise post-tests. All variables were log transformed before analysis. Full fatty acid profiles are shown in the accompanying Data in Brief article [28].
In colon tissues, fish oil feeding resulted in significant higher total and individual ω-3 fatty acids in all three tissue types, and the lower relative proportion of total ω-6 PUFA in rats fed fish oil was statistically significant only in tumor tissue (Table 3). Total fatty acid amount, per mg protein, tended to be lower after fish oil versus Western fat feeding, but this was not significant in colon tissue.
Table 3.
Fatty acids in distal colon of rats fed different diets. Shown are the mean and SD; variables are the same as in Table 2. There were 10 rats per group and 9 per group for tumor tissue from carcinogen-treated rats fed the fish oil diet.
| Fatty Acid (mole %, unless indicated otherwise) | Normal Tissue from Control Rats | Normal Tissue from Carcinogen-treated Rats | Tumor Tissue from Carcinogen-treated Rats | |||
|---|---|---|---|---|---|---|
| Western fat diet | Fish oil diet | Western fat diet | Fish oil diet | Western fat diet | Fish oil diet | |
| SFA | 36 (4) | 34 (4) | 36 (3) | 38 (5) | 40 (1)^ | 40 (1)^ |
| MUFA | 40 (8) | 39 (5) | 42 (6) | 36 (7) | 28 (4)^ | 26 (7)^ |
| ω-3 PUFA | 3.3 (1.5) | 7.5 (2.4)* | 3.0 (2.2) | 8.1 (1.9)* | 5 (2) | 11 (4)* |
| ω-6 PUFA | 21 (3) | 19 (2) | 19 (2) | 18 (1) | 27 (4)^ | 22 (4)* |
| AA:linoleic acid ratio | 0.49 (0.38) | 0.31 (0.17) | 0.28 (0.18) | 0.34 (0.18) | 1.17 (0.42)^ | 0.61 (0.30) |
| 20:4, ω-6 (AA) | 5.98 (3.61) | 4.16 (2.08) | 3.82 (2.26) | 4.14 (1.79) | 12.6 (3.7)^ | 7.18 (3.23) |
| 20:5, ω-3 (EPA) | 1.44 (0.80) | 2.61 (1.03) | 1.34 (1.31) | 3.31 (1.65)* | 2.12 (1.48) | 5.19 (2.74)* |
| EPA:AA ratio | 0.25 (0.08) | 0.69 (0.18)* | 0.36 (0.21) | 0.81 (0.24)* | 0.20 (0.23) | 0.78 (0.33)* |
| Total fatty acids(μg/mg proteinfor colon) | 97 (55) | 73 (38) | 98 (51) | 87 (93) | 56 (26) | 43 (38) |
Two separate ANOVA models were constructed, one for tumor versus normal tissue from control rats and another for normal tissue from carcinogen-treated rats versus that from control rats. Significant differences in fatty acids for the fish oil diet versus Western fat diet (p<0.005), within a treatment group, from two-way ANOVA, are starred. All variables were log-transformed before analysis except that natural log was used for AA, EPA and the AA:linoleic acid ratio. ^ Marked values differ significantly in tumor or in normal tissue from carcinogen-treated rats versus normal tissue of control rats, within each diet group (p<0.005), from two-way ANOVA. Full fatty acid profiles are shown in the accompanying Data in Brief article [28].
Fatty acids in normal colon of carcinogen-treated rats were similar to that in normal colon from control rats. Tumor tissue did, however, have higher SFA and lower MUFA than normal tissue from control rats with either kind of diet (Table 3). In addition, arachidonic acid (AA) and the AA:linoleic acid ratio were higher in tumor than normal tissue for rats fed the Western diet. These differences between normal and tumor tissue were not significant in rats fed the fish oil diet. The complete fatty acid profiles are shown in the accompanying Data in Brief article [28].
3.4. Lipodomic analyses of colon tissues
A representative subset of normal and tumor colon tissue samples was analyzed using un-targeted shotgun lipodomic methods to identify differences in specific lipid classes between samples defined by diet and tissue type. Principal component analyses of the results are shown in Figure 2 showing clear segregation of lipids by diet and tissue type. In comparing lipodomic patterns between tumor tissue from carcinogen-treated rats and normal tissue from control rats (Figure 2A), the four groups defined by diet (fish oil or Western) and tissue type (tumor or normal) segregated completely. The main effect of tissue type was evident (PC1, 32%), although the diet effect was still strong in PC2 (28%), with a smaller contribution of PC3 that was similar to the diet effect (17%). In tumor tissue, there was more variability between samples in tumor tissue (depicted by triangles) versus that in normal tissues (depicted as curcles) as shown in Figure 2A.
Figure 2.
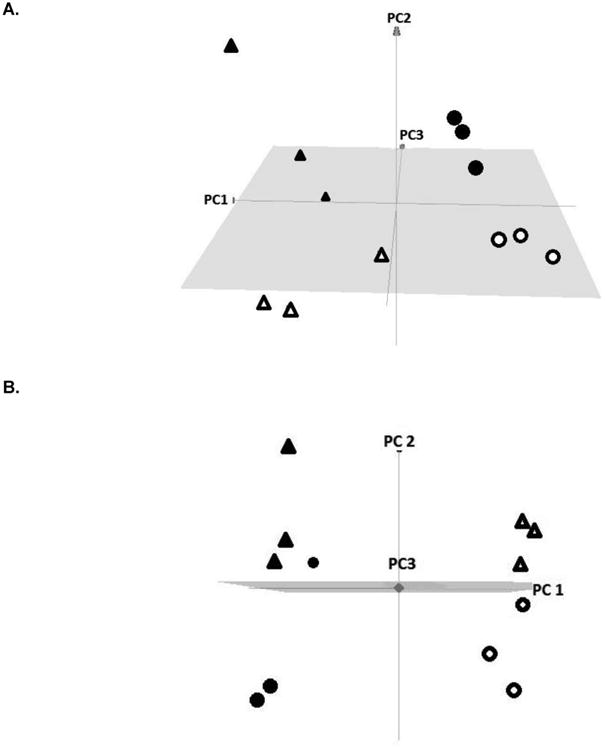
Principal Component analyses of lipodomic results.
A. Experiment comparing normal colon tissue from control rats and tumor tissue from rats treated with carcinogen. In each group, rats were fed either the Western fat or fish oil diet. Principal component 1 (PC1, tissue type) accounted for 32% of the variance, PC2 (diet) for 28% of the variance and PC3 for 17% of the variance. Legend: Solid sphere, normal tissue from control rats fed the fish oil diet; Open circle, normal tissue from control rats fed the Western fat diet; Solid triangle, tumor tissue from rats fed the fish oil diet; Open triangle, tumor tissue from rats fed the Western fat diet.
B. Experiment comparing grossly normal colon tissue from rats treated with carcinogen (AOM and DSS) or not, fed either the Western fat or fish oil diet. Principal component 1 (PC1, diet) accounted for 43% of the variance, PC2 (carcinogen treatment) for 20% of the variance and PC3 for 9% of the variance. Legend: Open circle, normal colon from control rats fed the Western fat diet; Open triangle, normal colon from carcinogen-treated rats fed the Western fat diet; Solid circle, normal colon from control rats fed the fish oil diet; Solid triangle, normal colon from carcinogen-treated rats fed the fish oil diet.
In a second lipodomic experiment, normal tissues from both control and carcinogen-treated rats were analyzed. Principal component analysis indicated a major effect of diet on lipids in PC1 accounting for 43% of the variance. Carcinogen treatment (PC2) accounted for another 20% of the variance and PC3 for 9% of the variance (Figure 2B). In rats fed the Western fat diet, the carcinogen effect on lipids was distinct. In normal tissue from animals fed the fish oil diet, however, the carcinogen effect was not as pronounced and one sample from a control rat was indistinguishable from the samples obtained from carcinogen-treated rats.
The hierarchical clustering analyses of the lipodomic data allowed for comparisons of specific lipids that differed by tissue type or by diet, and those lipids that consistently differed by diet type or tissue type are listed in Table 4. Tumor tissue versus normal (Experiment A), regardless of diet, had higher amounts of lipids containing saturated and monounsaturated fatty acids in the free fatty acids, lysophosphatidyl choline (PC), lyso phosphatidylethanolamine (PE) lipids and several plasmenyl PC and PE lipids. Carcinogen treatment of rats also resulted in elevated saturated and monounsaturated fatty acids in normal tissue. This was mainly in the free fatty acids and not in other lipid classes (Table 4, Experiment B). Notably, free arachidonic acid was elevated in both carcinogen-treated normal tissue and in tumor tissue relative to normal tissue of control rats. Fish oil feeding markedly resulted in higher long chain, highly unsaturated fatty acids in mainly TG, DG, PC, PE and plasmenyl PE lipids. Fish oil feeding also resulted in lipids that were lower versus that in the Western fat diet: this included several lipids that likely contain arachidonic acid (eg. PC 40:4), as shown in Table 4. More detailed lipodomic data is available in the accompanying Data in Brief article [28].
Table 4.
Major colon lipid groups identified in heirarchical cluster analysis to differ by diet or carcinogen treatment of rats. The lipid groups for Experiment A refer to lipodomic analysis of tumor tissues from carcinogen-treated rats and normal tissues from control rats fed either Western fat or fish oils diet. Lipid groups for Experiment B refer to lipodomic analysis of normal tissues from control rats and normal tissues from carcinogen-treated rats. These lipids groups are annotated in the heat maps generated from hierarchical cluster analysis as shown in the accompanying Data in Brief article [28]). The lipids in each group are sorted alphabetically. Abbreviations: cardiolipin, CL; diglycerides, DG; free fatty acids, FFA; monoglycerides, MG; phosphatidylcholine, PC; phosphatidylethanolamine, PE; PG; phosphatidylinositol, PI; phosphatidylserine, PS; Sphingomyelin, SM; triglycerides, TG.
| Experiment A: Group 1 Increased in tumors vs. normal tissue of control rats | Experiment A: Group 2 Decreased by fish oil vs. Western fat diet | Experiment A: Group 3 & 4 Increased by fish oil vs. Western fat diet | Experiment B: Group 1 Increased by fish oil diet vs. Western fat diet | Experiment B: Group 2 Decreased by fish oil vs. Western fat diet | Experiment B: Group 3 Increased by carcinogen treatment vs. no carcinogen |
|---|---|---|---|---|---|
| CL 74:3 | CE 22:4 | CE 22:1 | CL 72:8 | CL 68:2 | FFA 16:0 |
| CL 74:6 | lysoPE 22:4 | CL 68:1 | CL 74:9 | CL 74:6 | FFA 18:0 |
| CL 78:3 | plasmenyl-PE 40:4 | CL 72:7 | CL 82:9 | CL 76:9 | FFA 18:1 |
| FFA 16:0 | plasmenyl-PE 40:4 | CL 72:8; | DG 32:6 | DG 40:4 | FFA 18:2 |
| FFA 18:0 | plasmenyl-PE 38:4 | CL 76:10 | DG 34:5 | FFA 22:3 | FFA 20:0 |
| FFA 18:1 | PC 40:4 | CL 78:9 | DG 34:6 | FFA 24:3 | FFA 20:1 |
| FFA 18:2 | DG 40:4 | DG 34:6 | DG 36:5 | lysoPC 22:4 | FFA 20:2 |
| FFA 20:0 | PE 40:4 | DG 36:6 | DG 38:6 | lysoPE 20:4 | FFA 20:4 |
| FFA 20:1 | PC 36:4 | DG 38:6 | DG 40:6 | lysoPE 22:4 | FFA 22:1 |
| FFA 20:2 | PC 38:4 | DG 38:7 | DG 40:7 | PC 36:4 | FFA 22:2 |
| FFA 20:4 | DG 40:7 | lysoPC 14:0 | PC 38:4 | FFA 24:1 | |
| FFA 22:0 | lysoPC 20:5 | lysoPC 15:0 | PC 40:4 | FFA 24:2 | |
| FFA 22:1 | lysoPE 20:5 | lysoPC 16:1 | PC 42:5 | lysoPC 18:0 | |
| FFA 22:2 | PA 38:5 | lysoPC 17:1 | PE 38:2 | lysoPC 18:1 | |
| FFA 24:0 | PC 31:1 | lysoPC 18:2 | PE 40:4 | lysoPC 20:2 | |
| FFA 24:1 | PC 34:5 | lysoPC 19:0 | PG 38:5 | MG 18:1 | |
| FFA 24:2 | PC 35:5 | lysoPC 23:0 | PG 40:5 | MG 18:2 | |
| FFA 24:3 | PC 36:5 | lysoPE 19:0 | PG 40:6 | plasmenyl-PC 18:0 | |
| lysoPE 17:1 | PC 36:6 | lysoPE 22:6 | PG 42:8 | plasmenyl-PC 20:0 | |
| lysoPE 18:0 | PC 36:7 | lysoPE 23:0 | PG 44:10 | ||
| lysoPE 20:0 | PC 37:6 | PC 31:0 | PG 44:9 | ||
| lysoPE 21:0 | PC 38:6 | PC 32:1 | PI 36:4 | ||
| lysoPE 22:1 | PC 38:7 | PC 33:0 | plasmenyl-PE 36:4 | ||
| lysoPE 24:1 | PC 38:8 | PC 33:1 | plasmenyl-PE 38:3 | ||
| lysoPE 26:0 | PC 38:9 | PC 34:2 | plasmenyl-PE 38:4 | ||
| PC 29:0 | PC 40:10 | PC 34:5 | plasmenyl-PE 40:3 | ||
| PC 30:0 | PC 44:12 | PC 36:2 | plasmenyl-PE 40:4 | ||
| PC 30:1 | PE 35:1 | PC 36:5 | PS 36:4 | ||
| PC 32:0 | PE 36:5 | PC 36:6 | PS 38:4 | ||
| PC 32:1 | PE 36:6 | PC 37:5 | PS 40:4 | ||
| PC 33:0 | PE 37:5 | PC 37:6 | PS 42:5 | ||
| PC 33:1 | PE 37:6 | PC 38:5 | TG 42:0 | ||
| PC 34:1 | PE 38:6 | PC 38:6 | TG 54:0 | ||
| PC 34:2 | PE 40:5 | PC 38:7 | |||
| PC 35:1 | PE 40:6 | PC 40:7 | |||
| PC 36:0 | PG 34:2 | PC 40:8 | |||
| PC 36:1 | PG 42:11 | PC 42:1 | |||
| PC 36:3 | PG 44:12 | PC 42:10 | |||
| PC 38:1 | PI 38:5 | PC 42:11 | |||
| PC 38:2 | PI 38:6 | PC 42:2 | |||
| PC 40:0 | plasmenyl-PE 30:0 | PE 36:5 | |||
| PC 40:1 | plasmenyl-PE 36:2 | PE 36:6 | |||
| PC 40:3 | plasmenyl-PE 36:5 | PE 38:5 | |||
| PC 40:7 | plasmenyl-PE 38:6 | PE 38:6 | |||
| PC 40:8 | plasmenyl-PE 40:5 | PE 38:7 | |||
| PC 42:1 | plasmenyl-PE 40:6 | PE 40:5 | |||
| PC 42:2 | plasmenyl-PE 42:6 | PE 40:6 | |||
| PC 42:3 | PS 38:4 | PG 33:0 | |||
| PC 44:3 | SM 36:2 | PG 40:7 | |||
| PC 44:5 | SM 40:2 | PG 44:11 | |||
| PE 30:0 | TG 49:1 | PG 44:12 | |||
| PE 32:0 | TG 49:2 | PI 32:1 | |||
| PE 32:1 | TG 51:4 | PI 34:1 | |||
| PE 33:0 | TG 52:6 | PI 34:2 | |||
| PE 36:0 | TG 52:7 | PI 36:1 | |||
| PE 38:1 | TG 53:5 | PI 36:2 | |||
| PE 40:1 | TG 53:6 | PI 38:3 | |||
| PE 40:2 | TG 54:7 | PI 38:5 | |||
| PE 41:1 | TG 54:8 | PI 40:6 | |||
| PE 42:1 | TG 55:6 | plasmenyl-PE 34:1 | |||
| PE 42:2 | TG 55:7 | plasmenyl-PE 34:2 | |||
| PE 42:3 | TG 56:7 | plasmenyl-PE 36:1 | |||
| PE 43:1 | TG 56:8 | plasmenyl-PE 36:2 | |||
| PE 44:2 | TG 56:9 | plasmenyl-PE 36:5 | |||
| PE 44:3 | TG 57:6 | plasmenyl-PE 38:5 | |||
| PE 44:4 | TG 57:7 | plasmenyl-PE 38:6 | |||
| PG 32:0 | TG 58:1 | plasmenyl-PE 40:5 | |||
| PG 34:1 | TG 58:10 | plasmenyl-PE 40:6 | |||
| PG 40:7 | TG 58:11 | plasmenyl-PE 42:6 | |||
| PG 40:8 | TG 58:7 | PS 34:1 | |||
| PG 42:10 | TG 58:8 | PS 35:1 | |||
| PG 44:11 | TG 58:9 | PS 36:2 | |||
| PI 34:2 | TG 60:1 | PS 38:3 | |||
| PI 36:1 | TG 60:10 | PS 38:5 | |||
| PI 36:2 | TG 60:13 | PS 38:6 | |||
| PI 36:4 | TG 60:7 | PS 40:5 | |||
| PI 38:3 | TG 60:8 | PS 40:6 | |||
| plasmenyl-PE 32:0 | TG 60:9 | SM 37:1 | |||
| plasmenyl-PE 32:1 | SM 39:1 | ||||
| plasmenyl-PE 34:1 | SM 44:2 | ||||
| plasmenyl-PE 36:1 | TG 49:1 | ||||
| plasmenyl-PE 38:1 | TG 49:2 | ||||
| plasmenyl-PE 42:5 | TG 51:4 | ||||
| TG 52:6 | |||||
| TG 52:6 | |||||
| TG 52:7 | |||||
| TG 53:4 | |||||
| TG 53:5 | |||||
| TG 53:6 | |||||
| TG 53:7 | |||||
| TG 54:7 | |||||
| TG 54:8 | |||||
| TG 55:7 | |||||
| TG 56:10 | |||||
| TG 56:6 | |||||
| TG 56:7 | |||||
| TG 56:8 | |||||
| TG 56:9 | |||||
| TG 58:10 | |||||
| TG 58:11 | |||||
| TG 58:12 | |||||
| TG 58:7 | |||||
| TG 58:8 | |||||
| TG 58:9 | |||||
| TG 60:11 | |||||
| TG 60:12 | |||||
| TG 60:9 | |||||
| TG 62:13 |
3.5. Fatty acid predictors of colon prostaglandins
Although we did observe differences in colon prostaglandins and fatty acids by diet and carcinogen treatment, there was variability in these measures within each animal group. We therefore used linear regression analysis to evaluate the impact of inter-individual variability in colon fatty acids and COX expression among animals on the log of both PGE2 and PGE3 concentrations. In normal tissue of control or carcinogen-treated rats, diet alone was a significant predictor of colon PGE2, although in the case of carcinogen-treated rats a larger proportion of the variability in PGE2 (25% versus 16%) was accounted for by including fatty acid and COX expression variables (Table 5). In tumor tissue, EPA:AA fatty acid ratio, COX-2 expression and the mole percent of SFA in the colon were all significant predictors, with the final model accounting for 68% of the variance in colon PGE2. The beta coefficient was negative for the EPA:AA ratio and for exposure to a fish oil diet, indicating lower PGE2 with higher EPA:AA, and the beta coefficient was positive for COX-2 expression and mole percent of SFA. Including COX-1 expression along with COX-2 expression did not improve the model (adjusted R2 in that case was 67%). Other fatty acid variables, such as mole percent of MUFA, other ω-3 fatty acid variables, or total fatty acid weight per mg tissue protein did not improve the models shown in Table 5. The mole percent of EPA in colon was highly correlated with diet and did not yield a reliable regression model. Similar models using the same variables were constructed for the log of colon PGE3 in tumor tissue, but in this case diet explained a much larger proportion of the variance in PGE3 (54%) as compared with the models for PGE2 (30% of the variance in PGE2). The regression model for PGE3 was improved another 7.2% by including EPA:AA ratio, 11.1% by including COX-2 expression, and 8.2% by including mole percent of SFA. This generated a final model that accounted for 80.6% of the variance in log of tumor PGE3 concentration.
Table 5.
Predictors of the log of colon prostaglandin E2 and E3 in rats fed different diets (fish oil or Western fat) and treated with carcinogen or not, from linear regression analyses (n=20 per tissue type except for tumor tissue where n=19).
| Tissue | Predictor of colon prostaglandin | Adjusted R2 | P-value of F-change | P-value of overall model |
|---|---|---|---|---|
| Prostaglandin E2 | ||||
| Tumor colon, carcinogen-treated rats | Diet | 0.303 | 0.009 | 0.009 |
| + EPA:AA ratio | 0.498 | 0.014 | 0.002 | |
| + COX2 expression | 0.606 | 0.035 | 0.001 | |
| + Mole % SFA | 0.684 | 0.048 | <0.001 | |
| Normal colon, carcinogen-treated rats | Diet | 0.157 | 0.047 | 0.047 |
| EPA:AA ratio | 0.166 | 0.288 | 0.083 | |
| + COX1 and COX2 | 0.227 | 0.222 | 0.097 | |
| Expression + Mole % SFA | 0.245 | 0.263 | 0.109 | |
| Normal colon, control rats | Diet1 | 0.176 | 0.037 | 0.037 |
| Prostaglandin E3 | ||||
| Tumor colon, carcinogen-treated rats | Diet | 0.541 | <0.001 | <0.001 |
| EPA:AA mole ratio | 0.613 | 0.059 | <0.001 | |
| + COX2 expression | 0.724 | 0.016 | <0.001 | |
| + Mole % SFA | 0.806 | 0.017 | <0.001 | |
| Normal colon, carcinogen-treated rats | Diet1 | 0.909 | <0.001 | <0.001 |
| Normal colon control rats | Diet1 | 0.929 | <0.001 | <0.001 |
Adding fatty acid variables and/or COX expression did not improve these models for prediction of prostaglandin concentrations in normal colon tissues.
4. Discussion
Dietary fish oils are thought to be part of a healthful diet. Many rodent studies point to the cancer preventive effects of fish oils [5-7]. Human studies have, however, been less consistent [29]. Data on the biochemistry of fish oil fatty acid metabolism to PGE2 show that both expression of COXs and the composition of the fatty acid milieu, not just AA concentration, will govern rates of PGE2 formation [11, 13-15, 30]. This is highly relevant in tumors where it is well-established that COX-2 is induced and fatty acid synthesis is elevated to support active tumor growth [9, 31-34]. In the present study, we therefore examined fish oil effects on fatty acids and eicosanoids in a rodent colon cancer model.
As could be expected, diets containing fish oil resulted in increased formation of PGE3 in normal and tumor tissues (Table 1). PGE3 was higher in colon of carcinogen-treated rats fed fish oil than in normal colon from control rats, consistent with higher COX-2 expression levels. PGE3 is produced by COX-2 mediated oxygenation of EPA, and EPA is not a substrate for COX-1 [10, 11, 13]. PGE3 has been shown to have protective effects in the colon, reducing colon stem cell expansion [35]. Unlike with EPA, both COX-1 and COX-2 utilize AA as a substrate to result in formation of PGE2 [10]. Induction of COX-2 in tumors therefore could account for the somewhat higher PGE2 concentrations in tumor versus normal tissue (Table 1).
Other eicosanoids also exhibited beneficial changes with fish oil feeding. AA oxygenation by cytochrome P450 forms epoxyeicosatrienoic acids, and epoxide hydrolase subsequently results in formation of the more stable 14,15-DHETs (Figure 1). These compounds have been associated with pro-inflammatory processes [36, 37]. Neither 14,15-S and 14,15-R DHET were significantly altered by diet in normal tissues, but both products were reduced in colon tumors of fish oil-fed animals versus that of Western fat-fed animals (Table 1). 12-S-HETE also was lower in tumor tissue of fish oil fed rats. This would presumably be protective given its role in stimulating tumor cell proliferation, although the role of 12-S-HETE in colon cancer specifically has not been well-studied [38, 39]. Finally, 15-S-HETE and 13-S-HODE that are products of 15-lipoxygenase action from oxygenation of AA and linoleic acid, respectively, also were quantified (Figure 1). In the colon, 15-lipoxygenase appears to have anti-tumor effects [40]. In rodents, 12/15-lipoxygenase action is carried out by a single enzyme. It is therefore interesting that 15-S-HETE tended to increase with fish oil feeding while 12-S-HETE tended to decrease (Table 1). In addition, 13-S-HODE was not significantly changed by diet in any of the three tissue types (Table 1). This contrasts with our previous results in rats fed varying amounts of fish oil, although the previously observed the reduction in 15-S-HETE in colon with a fish oil diet was modest [17].
The S-isomers of HETEs and 13-HODE are formed enzymatically while the R-isomers are thought to be formed by both enzymatic and non-enzymatic oxygenation of substrates. Quantifying R-isomers using chiral chromatography therefore offers a method to assess potential pro-oxidant effects of fish oils. These products were either unchanged or reduced by fish oils: this is important since fish oils can be easily peroxidized [41]. The R isomers do appear to have biological effects that would enhance carcinogenesis. For example, 9-R-HODE and 13-R-HODE increased monocyte migration and calcium influx [42]. In Caco-2 cells, 13-S-HODE was shown to be a ligand for proliferator-activated receptor-γ and to induce apoptosis of undifferentiated cells while 13-R-HODE activated inflammatory signaling and PGE2 synthesis [43].
Fatty acid changes also exhibited important differences by diet and tissue type that were evident in both the fatty acid and lipodomic profiles (Tables 3 and 4). In serum, changes in fatty acids reflected diet. The relative proportion of ω-3 PUFA was higher and of ω-6 PUFA was lower in animals fed the fish oil versus Western fat diets, and fish oil feeding resulted in lower total fatty acid serum concentrations as well (Table 2). In colon, the fish oil diet resulted in relatively higher total ω-3 PUFA in all tissues, but the lower proportions of total ω-6 PUFA and AA were significant only in tumor tissue (Table 3). As a result, the ω-6 PUFA were higher in colon tumors versus normal tissue of animals fed the Western fat diet, but not in animals fed the fish oil diet. This indicates that a fish oil diet drove increases in the EPA:AA ratios differently in normal and tumor tissue: altering both in tumors but only EPA in normal tissue.
Lipodomic analysis of colonic lipid fractions provided more specific information than the fatty acids in total lipid extracts analyzed by GC-MS. The lipodomic analysis were done on a subset of tissues, and this data shows that the most commonly elevated lipids in tumor versus normal tissues contain saturated and monounsaturated fatty acids, including many free fatty acid moieties (Table 4). This is consistent with the reported lipogenic environment of tumors [31-34]. Indeed, increased fatty acid synthase expression appears to be an early marker of colorectal neoplasia [21], and may even be prognostic in humans [44]. The data is also consistent with active fatty acid cycling in tumors, releasing lyso lipids and free fatty acids from phospholipids (Table 4). Certain lyso PC have pro-inflammatory properties and stimulate monocyte attraction [42, 45-47]. Tumors actively cycle fatty acids via lysoPC to free FFA and PG in the extracellular space, followed by uptake of the released FFA into membranes [48]. Active cycling between TAG and free fatty acids, and a high rate of glycolysis provide glycerol for TAG synthesis and accumulation, allowing for cell survival even when tumors are serum starved [49].
Although fish oil did have significant effects on colonic eicosanoid concentrations, there was considerable variability among animals in each treatment group. We investigated whether colon tissue fatty acids that were assayed in all samples by GC-MS could explain at least part of the inter-animal variability in colon PGE2 concentrations, after controlling for diet type using linear regression models. The biochemical regulation of COX activity by fatty acids has been reported in detail which guided the statistical analyses. We focused on the EPA:AA ratio since EPA binding to COX-1 inhibits AA oxygenation [11]. Other long chain ω-3 fatty acids (22:6 and 22:5) are poor COX-1 substrates and very modest COX-1 inhibitors [11, 15]. In normal tissue from carcinogen-treated rats, fatty acids and COX expression were weak, non-significant predictors of colonic PGE2 after controlling for diet. In tumors, however, the EPA:AA ratio, COX-2 expression and the mole percent of SFA all were significant predictors of PGE2 concentrations (Table 5). This is consistent with biochemical data showing that SFA and MUFA slightly reduce COX-1 activity but greatly increase oxygenation of AA by COX-2 (Figure 3): COX-2 is induced in tumors, and biochemical data show that non-substrate fatty acids modulate the activity of COX-2 to a much greater extent than that of COX-1 [9, 12-15]. In the linear regression models, MUFA was not a significant predictor of colon PGE2, perhaps because in tumors the relative proportion of total SFA was higher than that of total MUFA (Table 3).
Figure 3.
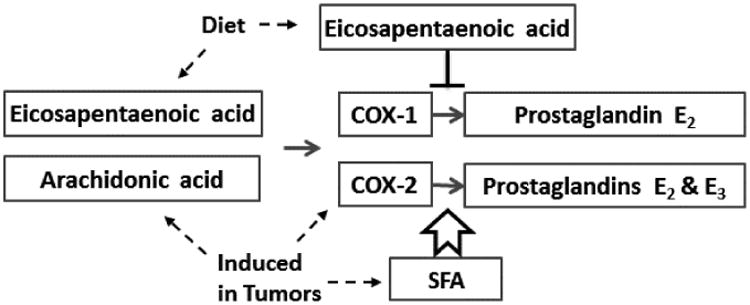
Summary of fatty acid modulation of prostaglandin E production. Arachidonic acid is a substrate for both cyclooxygenase (COX) isoforms resulting in production of prostaglandin E2. Eicosapentaenoic acid is an inhibitor of COX-1 and a weak substrate for COX-2, resulting in production of small amounts of prostaglandin E3. Saturated and monounsaturated fatty acids (SFA, MUFA) stimulate COX-2 activity [12-15]. In tumors, COX-2 is induced and the fatty acid profile is altered to increase SFA, as shown in Tables 3 and 4. Fish oil feeding reduced the tumor-associated elevation in both arachidonic acid and prostaglandin E2 concentrations. In normal colon tissue, the type of diet explains the majority of the inter-animal variability in prostaglandin E2 concentrations (Table 5). In tumor tissue the EPA:AA ratio, COX-2 expression, and the relative abundance of saturated fatty acids contributes significantly to the inter-animal variability in prostaglandin E2 concentrations.
The linear regression results showing a significant influence of fatty acid variables on tumor tissue PGE2 but not in normal tissue PGE2 are interesting (Table 5). Tumor colon tissue had a fatty acid milieu consistent with a lipogenic phenotype of higher saturated and monounsaturated fatty acids in many lipid subclasses and higher free fatty acids relative to normal colon tissue (Table 4). Interestingly, the fish oil diet resulted in the enrichment of triglycerides with ω-3 fatty acids to a relatively greater extent in tumors versus that in normal tissue, consistent with elevated fatty acid flux into intracellular lipid droplets (Table 4). Lipid droplets in tumors are a storage depot for fatty acids in the triglyceride form, likely supporting proliferative needs [50, 51]. Despite good uptake of ω-3 fatty acids into tumor tissues, an increased proportion of SFA in total lipid extracts still had a significant stimulatory effect on formation of PGE2 in tumor tissue. This accounted for an additional 8% of the inter-animal variance in PGE2, after accounting for diet, EPA:AA ratio and COX-2 expression in the linear regression models (Table 5). Although this is a modest fraction of the inter-animal variability, it was statistically significant and similar in magnitude to the effects of COX-2 expression that accounted for 11% of the variability after taking into account diet type and EPA:AA ratio. This could be even more important in human populations that have greater variability in diet and in enzyme expression due to polymorphisms and environmental exposures.
A similar scenario could occur in obesity. In condition of caloric excess, fatty acid uptake and synthesis in cells would mimic the endogenous synthesis of fatty acids in tumors, and this has been shown to interfere with the cancer therapeutic effects of fatty acid synthase inhibitors [52]. In our recent clinical trial of omega-3 fatty acid supplementation, obese versus normal weight individuals exhibited smaller decreases in colonic PGE2 concentrations post supplementation [53]. It is tempting to speculate that the lipogenic environment in the case of a positive energy balance would result in a similar scenario as in tumors where increased concentrations of saturated fatty acids enhance COX-2 activity and attenuate the efficacy of dietary preventive agents for reducing PGE2. Given the central role of PGE2 in colon cancer risk [9], inter-individual variation in fatty acid metabolism could have major implications for optimizing the dosing of fish oils and other anti-inflammatory agents for colon cancer prevention in human populations.
Acknowledgments
We thank ElKhansa Sidahmed for assistance with the PCR analyses.
Funding: This work was supported by grants P50 CA130810 (Gastrointestinal Specialized Program of Research Excellence), P30 CA046592 (University of Michigan Comprehensive Cancer Center), DK097153 (Michigan Regional Comprehensive Metabolomics Resource Core) and Cancer Biology Training grant T32 CA009676 (support for B.S. and R.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu S, Feng B, Li K, Zhu X, Liang S, Liu X, et al. Fish consumption and colorectal cancer riskin humans: a systematic review and meta-analysis. Am J Med. 2012;125:551–9 e5. doi: 10.1016/j.amjmed.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Reddy BS. Dietary fat and colon cancer: animal model studies. Lipids. 1992;27:807–13. doi: 10.1007/BF02535855. [DOI] [PubMed] [Google Scholar]
- 3.Baena R, Salinas P. Diet and colorectal cancer. Maturitas. 2015;80:258–64. doi: 10.1016/j.maturitas.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 4.Oh K, Willett WC, Fuchs CS, Giovannucci E. Dietary marine n-3 fatty acids in relation to risk of distal colorectal adenoma in women. Cancer Epidemiol Biomarkers Prev. 2005;14:835–41. doi: 10.1158/1055-9965.EPI-04-0545. [DOI] [PubMed] [Google Scholar]
- 5.Rose DP, Connolly JM. Omega-3 fatty acids as cancer chemopreventive agents. Pharmacology & Therapeutics. 1999;83:217–44. doi: 10.1016/s0163-7258(99)00026-1. [DOI] [PubMed] [Google Scholar]
- 6.Reddy BS, Sugie S. Effect of different levels of omega-3 and omega-6 fatty acids on azoxymethane-induced colon carcinogenesis in F344 rats. Cancer Res. 1988;48:6642–7. [PubMed] [Google Scholar]
- 7.Rao CV, Hirose Y, Indranie C, Reddy BS. Modulation of experimental colon tumorigenesis by types and amounts of dietary fatty acids. Cancer Research. 2001;61:1927–33. [PubMed] [Google Scholar]
- 8.Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. Bmj. 2011;342:c7086. doi: 10.1136/bmj.c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69(1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 10.Smith WL. Nutritionally essential fatty acids and biologically indispensable cyclooxygenases. Trends Biochem Sci. 2008;33:27–37. doi: 10.1016/j.tibs.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, et al. Enzymes and receptorsof prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derivedsubstrates and products. J Biol Chem. 2007;282:22254–66. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- 12.Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, et al. Cyclooxygenase allosterism: Fatty Acid Mediated Cross-talk Between Monomers of Cyclooxygenase homodimers. J Biol Chem. 2009;284:10042–55. doi: 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong L, Zou H, Yuan C, Hong YH, Kuklev DV, Smith WL. Different Fatty Acids Compete with Arachidonic Acid for Binding to the Allosteric or Catalytic Subunits of Cyclooxygenases to Regulate Prostanoid Synthesis. J Biol Chem. 2016;291:4069–78. doi: 10.1074/jbc.M115.698001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong L, Yuan C, Orlando BJ, Malkowski MG, Smith WL. Fatty Acid Binding to the Allosteric Subunit of Cyclooxygenase-2 Relieves a Tonic Inhibition of the Catalytic Subunit. J Biol Chem. 2016;291:25641–55. doi: 10.1074/jbc.M116.757310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou H, Yuan C, Dong L, Sidhu RS, Hong YH, Kuklev DV, et al. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J Lipid Res. 2012;53:1336–47. doi: 10.1194/jlr.M026856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neilson AP, Djuric Z, Ren J, Hong YH, Sen A, Lager C, et al. Effect of cyclooxygenase genotype and dietary fish oil on colonic eicosanoids in mice. J Nutr Biochem. 2012;23:966–76. doi: 10.1016/j.jnutbio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y, Djuric Z, Sen A, Ren J, Kuklev D, Waters I, et al. Biomarkers for personalizing omega-3 fatty acid dosing. Cancer Prev Res (Phila) 2014;7:1011–22. doi: 10.1158/1940-6207.CAPR-14-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujise T, Iwakiri R, Kakimoto T, Shiraishi R, Sakata Y, Wu B, et al. Long-term feeding of various fat diets modulates azoxymethane-induced colon carcinogenesis through Wnt/beta-catenin signaling in rats. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1150–6. doi: 10.1152/ajpgi.00269.2006. [DOI] [PubMed] [Google Scholar]
- 19.Leslie MA, Cohen DJ, Liddle DM, Robinson LE, Ma DW. A review of the effect of omega-3 polyunsaturated fatty acids on blood triacylglycerol levels in normolipidemic and borderline hyperlipidemic individuals. Lipids Health Dis. 2015;14:53. doi: 10.1186/s12944-015-0049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, et al. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruz MD, Wali RK, Bianchi LK, Radosevich AJ, Crawford SE, Jepeal L, et al. Colonic mucosal fatty acid synthase as an early biomarker for colorectal neoplasia: modulation by obesity and gender. Cancer Epidemiol Biomarkers Prev. 2014;23:2413–21. doi: 10.1158/1055-9965.EPI-14-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Djuric Z, Ren J, Blythe J, VanLoon G, Sen A. A Mediterranean dietary intervention in healthy American women changes plasma carotenoids and fatty acids in distinct clusters. Nutr Res. 2009;29:156–63. doi: 10.1016/j.nutres.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren J, Mozurkewich E, Sen A, Vahratian A, Ferreri T, Morse A, et al. Total Serum Fatty Acid Analysis by GC-MS: Assay Validation and Serum Sample Stability. Current Pharmaceutical Analysis. 2013;9 doi: 10.2174/1573412911309040002. Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neilson AP, Ren J, Hong YH, Sen A, Smith WL, Brenner DE, et al. Effect of fish oil on levels of R- and S-enantiomers of 5-, 12-, and 15-hydroxyeicosatetraenoic acids in mouse colonic mucosa. Nutr Cancer. 2012;64:163–72. doi: 10.1080/01635581.2012.630168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiner J. Three Dimensional PCA Plots. pca3d. 2015 Available online at https://cran.r-project.org/web/packages/pca3d/index.html.
- 26.R Development Core Team. Vienna, Austria: R Foundation for Statistical Computing; Team R: A Language and Environment for Statistical Computing; p. 2014. Available online at http://www.R-project.org. [Google Scholar]
- 27.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R Programming Tools for Plotting Data. 2015 Available online at http://CRAN.R-project.org/package=gplots.
- 28.Djuric Z, Aslam MN, Simon BR, Sen A, Jiang Y, Ren J, et al. Fatty Acid and Lipodomic Data in Normal and Tumor Colon Tissue of Rats Fed Diets with and without Fish Oils. Data in Brief. :2017. doi: 10.1016/j.dib.2017.06.032. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen GC, Qin LQ, Lu DB, Han TM, Zheng Y, Xu GZ, et al. N-3 polyunsaturated fatty acids intake and risk of colorectal cancer: meta-analysis of prospective studies. Cancer Causes Control. 2015;26:133–41. doi: 10.1007/s10552-014-0492-1. [DOI] [PubMed] [Google Scholar]
- 30.Yuan C, Sidhu RS, Kuklev DV, Kado Y, Wada M, Song I, et al. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J Biol Chem. 2009;284:10046–55. doi: 10.1074/jbc.M808634200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cifkova E, Holcapek M, Lisa M, Vrana D, Gatek J, Melichar B. Determination of lipidomic differences between human breast cancer and surrounding normal tissues using HILIC-HPLC/ESI-MS and multivariate data analysis. Anal Bioanal Chem. 2015;407:991–1002. doi: 10.1007/s00216-014-8272-z. [DOI] [PubMed] [Google Scholar]
- 32.Szachowicz-Petelska B, Dobrzynska I, Figaszewski Z, Sulkowski S. Changes in physico-chemical properties of human large intestine tumour cells membrane. Mol Cell Biochem. 2002;238:41–7. doi: 10.1023/a:1019946718876. [DOI] [PubMed] [Google Scholar]
- 33.Zhou X, Mao J, Ai J, Deng Y, Roth MR, Pound C, et al. Identification of plasma lipid biomarkers for prostate cancer by lipidomics and bioinformatics. PLoS One. 2012;7:e48889. doi: 10.1371/journal.pone.0048889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones SF, Infante JR. Molecular Pathways: Fatty Acid Synthase. Clin Cancer Res. 2015;21:5434–8. doi: 10.1158/1078-0432.CCR-15-0126. [DOI] [PubMed] [Google Scholar]
- 35.Fan YY, Davidson LA, Callaway ES, Goldsby JS, Chapkin RS. Differential effects of 2- and 3-series E-prostaglandins on in vitro expansion of Lgr5+ colonic stem cells. Carcinogenesis. 2014;35:606–12. doi: 10.1093/carcin/bgt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang T, Peng R, Guo Y, Shen L, Zhao S, Xu D. The role of 14,15-dihydroxyeicosatrienoic acid levels in inflammation and its relationship to lipoproteins. Lipids Health Dis. 2013;12:151. doi: 10.1186/1476-511X-12-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oni-Orisan A, Deng Y, Schuck RN, Theken KN, Edin ML, Lih FB, et al. Dual modulation of cyclooxygenase and CYP epoxygenase metabolism and acute vascular inflammation in mice. Prostaglandins Other Lipid Mediat. 2013:104–105. 67–73. doi: 10.1016/j.prostaglandins.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YQ, Duniec ZM, Liu B, Hagmann W, Gao X, Shimoji K, et al. Endogenous 12(S)-HETE production by tumor cells and its role in metastasis. Cancer Res. 1994;54:1574–9. [PubMed] [Google Scholar]
- 39.Powell WS, Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta. 2015;1851:340–55. doi: 10.1016/j.bbalip.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuo X, Shureiqi I. Eicosanoid profiling in colon cancer: emergence of a pattern. Prostaglandins Other Lipid Mediat. 2013:104–105. 139–43. doi: 10.1016/j.prostaglandins.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Else PL, Kraffe E. Docosahexaenoic and arachidonic acid peroxidation: It's a within molecule cascade. Biochim Biophys Acta. 2015;1848:417–21. doi: 10.1016/j.bbamem.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 42.Rolin J, Vego H, Maghazachi AA. Oxidized lipids and lysophosphatidylcholine induce the chemotaxis, up-regulate the expression of CCR9 and CXCR4 and abrogate the release of IL-6 in human monocytes. Toxins (Basel) 2014;6:2840–56. doi: 10.3390/toxins6092840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabral M, Martin-Venegas R, Moreno JJ. Differential cell growth/apoptosis behavior of 13-hydroxyoctadecadienoic acid enantiomers in a colorectal cancer cell line. Am J Physiol Gastrointest Liver Physiol. 2014;307:G664–71. doi: 10.1152/ajpgi.00064.2014. [DOI] [PubMed] [Google Scholar]
- 44.Long QQ, Yi YX, Qiu J, Xu CJ, Huang PL. Fatty acid synthase (FASN) levels in serum of colorectal cancer patients: correlation with clinical outcomes. Tumour Biol. 2014;35:3855–9. doi: 10.1007/s13277-013-1510-8. [DOI] [PubMed] [Google Scholar]
- 45.Iwase M, Sonoki K, Sasaki N, Ohdo S, Higuchi S, Hattori H, et al. Lysophosphatidylcholine contents in plasma LDL in patients with type 2 diabetes mellitus: relation with lipoprotein-associated phospholipase A2 and effects of simvastatin treatment. Atherosclerosis. 2008;196:931–6. doi: 10.1016/j.atherosclerosis.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 46.Sevastou I, Kaffe E, Mouratis MA, Aidinis V. Lysoglycerophospholipids in chronic inflammatory disorders: the PLA(2)/LPC and ATX/LPA axes. Biochim Biophys Acta. 2013;1831:42–60. doi: 10.1016/j.bbalip.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 47.Carneiro AB, Iaciura BM, Nohara LL, Lopes CD, Veas EM, Mariano VS, et al. Lysophosphatidylcholine triggers TLR2- and TLR4-mediated signaling pathways but counteracts LPS-induced NO synthesis in peritoneal macrophages by inhibiting NF-kappaB translocation and MAPK/ERK phosphorylation. PLoS One. 2013;8:e76233. doi: 10.1371/journal.pone.0076233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raynor A, Jantscheff P, Ross T, Schlesinger M, Wilde M, Haasis S, et al. Saturated and mono-unsaturated lysophosphatidylcholine metabolism in tumour cells: a potential therapeutic target for preventing metastases. Lipids Health Dis. 2015;14:69. doi: 10.1186/s12944-015-0070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Przybytkowski E, Joly E, Nolan CJ, Hardy S, Francoeur AM, Langelier Y, et al. Upregulation of cellular triacylglycerol - free fatty acid cycling by oleate is associated with long-term serum-free survival of human breast cancer cells. Biochem Cell Biol. 2007;85:301–10. doi: 10.1139/o07-001. [DOI] [PubMed] [Google Scholar]
- 50.Zaytseva YY, Harris JW, Mitov MI, Kim JT, Butterfield DA, Lee EY, et al. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget. 2015;6:18891–904. doi: 10.18632/oncotarget.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, et al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008;68:1732–40. doi: 10.1158/0008-5472.CAN-07-1999. [DOI] [PubMed] [Google Scholar]
- 52.Kinlaw WB, Baures PW, Lupien LE, Davis WL, Kuemmerle NB. Fatty Acids and Breast Cancer: Make them on Site or have them Delivered. J Cell Physiol. 2016;231:2128–41. doi: 10.1002/jcp.25332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Djuric Z, Turgeon DK, Sen A, Ren J, Herman K, Ramaswamy D, et al. The Anti-inflammatory Effect of Personalized Omega-3 Fatty Acid Dosing for Reducing Prostaglandin E2 in the Colonic Mucosa is Attenuated in Obesity. Cancer Prev Res. 2017 doi: 10.1158/1940-6207.CAPR-17-0091. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]