

Abstract
Vitiligo, characterized by progressive melanocyte death, can be initiated by exposure to vitiligo-inducing phenols (VIPs). VIPs generate oxidative stress in melanocytes and activate the master antioxidant regulator NRF2. While NRF2-regulated antioxidants are reported to protect melanocytes from oxidative stress, the role of NRF2 in the melanocyte response to monobenzone, a clinically relevant VIP, has not been characterized. We hypothesized that activation of NRF2 may protect melanocytes from monobenzone-induced toxicity. We observed that knockdown of NRF2 or NRF2-regulated antioxidants NQO1 and PRDX6 reduced melanocyte viability, but not viability of keratinocytes and fibroblasts, suggesting that melanocytes were preferentially dependent upon NRF2 activity for growth compared to other cutaneous cells. Furthermore, melanocytes activated the NRF2 response following monobenzone exposure and constitutive NRF2 activation reduced monobenzone toxicity, supporting NRF2’s role in the melanocyte stress response. In contrast, melanocytes from individuals with vitiligo (vitiligo melanocytes) did not activate the NRF2 response as efficiently. Dimethyl fumarate-mediated NRF2 activation protected normal and vitiligo melanocytes against monobenzone-induced toxicity. Given the contribution of oxidant-antioxidant imbalance in vitiligo, modulation of this pathway may be of therapeutic interest.
Keywords: Monobenzyl ether of hydroquinone, Dimethyl Fumarate, NQO1, KEAP1, depigmentation
Introduction
Vitiligo is an acquired disorder characterized by depigmented skin lesions that spread due to autoimmune targeting of melanocytes. Vitiligo pathogenesis is complex; however, one widely held hypothesis is that stress responses are compromised in melanocytes from individuals with vitiligo (vitiligo melanocytes). Vitiligo can be initiated following exposure to certain phenolic compounds (vitiligo-inducing phenols or VIPs), which include 4-tertiary butyl phenol (4-TBP) and monobenzone (monobenzyl ether of hydroquinone, MBEH) (1, 2). Both VIPs promote depigmentation in mice engineered to have interfollicular epidermal melanocytes (3). MBEH, the more potent VIP (3), is currently the only drug approved by the US FDA to treat vitiligo and is used for complete depigmentation of extensive vitiligo (4). We investigated the effects of MBEH exposure on normal melanocytes, as a clinically relevant in vitro model for studying melanocyte toxicity, which may underlie vitiligo initiation and lesion spreading (1, 5, 6). Our goal was to identify survival pathways activated in normal melanocytes and determine if they were dysregulated in vitiligo melanocytes, and thus contribute to disease onset.
We previously reported that expression of the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2), which regulates cellular response to oxidative stress, is increased in melanocytes dosed with VIPs (6), similar to responses elicited by other redox-balance disruptors (7–10). In the absence of oxidative stress, the Kelch-like ECH-associated protein 1 (KEAP1) repressor complex binds cytoplasmic NRF2, causing ubiquitination and proteasomal degradation of NRF2 (11). Oxidative stress triggers KEAP1 dissociation and nuclear translocation, where NRF2 binds antioxidant response elements (AREs), promoting expression of cytoprotective antioxidants, detoxification proteins, and phase II drug metabolizing enzymes, including heme oxygenase 1 (HMOX1) and NAD(P)H dehydrogenase, quinone 1 (NQO1) (12, 13). Moreover, NRF2 activation can prevent or treat oxidative stress- and inflammation-mediated diseases (14, 15). For example, dimethyl fumarate (DMF) induces NRF2 stabilization, has anti-inflammatory properties, is neuroprotective (16, 17), and is approved for treatment of multiple sclerosis (18). DMF, the most commonly prescribed systemic therapy for psoriasis in Germany (19), is in clinical trials for treatment of alopecia areata (20). Although the NRF2-mediated effects of DMF on astrocytes (17) and keratinocytes (21, 22) have been elucidated, DMF effects on melanocytes remain uncharacterized.
Several studies described a cytoprotective role for NRF2 in melanocytes. Upregulation of HMOX1 protected melanocytes against hydrogen peroxide (H2O2) (7, 9, 10), while melanoma cells overexpressing NQO1 were resistant against the depigmenting agent rhododendrol (23). Thus, NRF2 contributes to the melanocyte antioxidant response and may play an important role protecting against MBEH-induced toxicity.
MBEH has pleiotropic effects that contribute to depigmentation in vitiligo (3, 24, 25), and delineating the protective responses against MBEH-induced cytotoxicity are of clinical importance. In this study, we explored the role NRF2 plays in melanocyte survival following MBEH exposure, a model for vitiligo initiation. Our results demonstrate that NRF2 activation protects melanocytes against MBEH and is a potential therapeutic target for vitiligo.
Materials and Methods
Cell culture and dosing
Human cells were used in all experiments. Epidermal melanocytes from neonatal foreskin (NHEM, 3 lines established from normally pigmented, unrelated individuals) and an adult epidermal melanocyte (AHEM) line were purchased and cultured in DermaLife-M culture medium (Lifeline Cell Technology, Frederick, MD). Neonatal epidermal keratinocytes (NHEK) were purchased and cultured in DermaLife-K medium (Lifeline Cell Technology). Dermal fibroblasts were isolated from neonatal foreskins (NHDF) and cultured in DMEM plus 10% serum (MediaTech, Manassas, VA). Melanocytes were isolated from two unrelated individuals with stable vitiligo. Two sites, non-lesional and perilesional (<3mm from depigmented lesion) were biopsied and melanocytes cultured in DermaLife-M. (NYU Institutional Review Board approved, Study i15-00445). Immortalized, normal epidermal (PIG1) and vitiligo melanocytes (PIG3V) (generous gift from Dr. Le Poole, Loyola University Chicago, IL) were also used (26, 27). Cells were treated with 4-TBP or MBEH as previously described (6). 4-TBP, MBEH, DMF, N-acetyl cysteine (NAC), and buthionine sulfoximine (BSO) (purchased from Sigma-Aldrich, St. Louis, MO) were dissolved in DMSO. Cells were treated with indicated concentrations of tert-butyl hydroperoxide (t-BHP) for 1 hour followed by incubation in culture medium as a positive control for oxidative stress.
Cell Viability
Melanocytes (20,000 cells/well), fibroblasts and keratinocytes (5,000 cells/well) were plated in 96-well plates and dosed after 24 hours. Viability was determined using the Cell-Titer 96 Cell Proliferation Assay (MTS) (Promega, Madison, WI) per manufacturer’s instructions and confirmed by crystal violet assays as previously reported (28). Relative viability of vitiligo patient-derived melanocytes and normal control adult melanocytes was measured after 96 hours to account for slower proliferation (75% proliferation of control neonatal melanocytes).
Determination of sub-toxic 4-TBP and MBEH concentrations
Sub-toxic concentrations (< 20% decrease in viability) of 4-TBP or MBEH were determined based on viability of NHEM following exposure to increasing concentrations of VIPs for 24 and 72 hours (6). For normal melanocytes, 250 μM 4-TBP or 300 μM MBEH were used unless otherwise specified. siRNA-transfected melanocytes were more sensitive to MBEH, therefore 200 μM MBEH (equivalent sub-toxic dose) was used when dosing for longer than 48 hours. Vitiligo melanocytes were also more sensitive to MBEH compared to normal adult melanocytes; therefore 100 μM MBEH was used when dosing for longer than 48 hours. 300 μM MBEH was well tolerated by all lines dosed for less than 24h.
Statistical analysis
Data are presented as mean ± SD except where indicated. Statistical significance was assessed using unpaired 2-tailed t-test using GraphPad Prism. P < 0.05 was considered statistically significant.
Results
NRF2 promotes melanocyte viability
Universally expressed antioxidants such as superoxide dismutase (SOD) and catalase neutralize reactive oxygen species (ROS) generated during oxidative stress in virtually all cell types. SOD converts superoxide anion radicals to H2O2, which catalase reduces to water and oxygen. Additional antioxidants, such as NRF2-targets NQO1 and PRDX6, participate in ROS neutralization through scavenging or reducing reactions (Fig. 1a). NQO1, in addition to having quinone reductase activity, can scavenge superoxide anions directly (29), while PRDX6 can reduce H2O2 to water (30). The predominant antioxidant response varies between cell types and is context dependent; thus, we first sought to compare intrinsic NRF2-regulated antioxidant expression in unstressed cutaneous cells.
Figure 1. NRF2 promotes melanocyte viability.
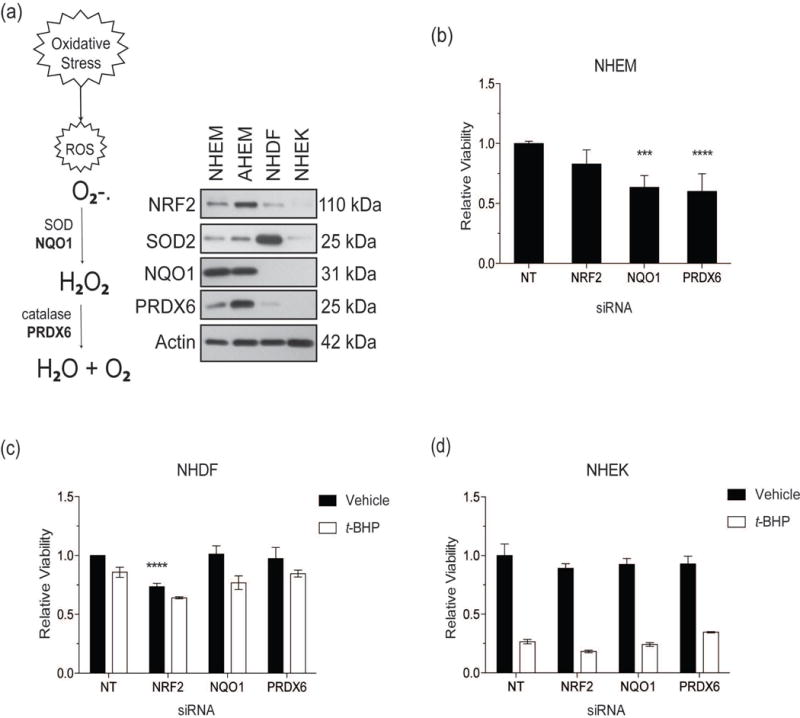
(a) The schema describes the process of ROS neutralization and highlights NRF2-targets, NQO1 and PRDX6 (in bold), that perform the same functions as generalized antioxidants. Total protein was extracted from four untreated cell lines derived from human skin. Cutaneous lines compared consisted of neonatal human epidermal melanocytes (NHEM), adult human epidermal melanocytes (AHEM), neonatal human dermal fibroblasts (NHDF) and neonatal human epidermal keratinocytes (NHEK). Protein levels of NRF2, SOD2, NQO1, PRDX6, and Actin (loading control) were measured by Western blot analysis. (b) NHEM were transfected with siRNAs against NRF2, NQO1, and PRDX6 and viability measured relative to non-target (NT) siRNA control. (c) NHDF and (d) NHEK were treated with siRNAs as in (b) in addition to t-BHP (250 μM) for 24 hours and viability measured relative to untreated NT siRNA control. Relative viability data are presented as mean ± SD, n=4 *** p < 0.001; **** p < 0.0001.
Antioxidant expression profiles were established utilizing melanocyte lines (NHEM and AHEM), fibroblasts (NHDF) and keratinocytes (NHEK). We compared antioxidant expression levels in each line in the absence of exogenous oxidative stress (“unstressed”). Strikingly, although KEAP1-mediated degradation of NRF2 typically maintains NRF2 at undetectable levels (11), both unstressed melanocyte lines expressed NRF2 (Fig. 1a). Comparison of nuclear and cytoplasmic expression revealed that NRF2 was predominantly nuclear, while cytoplasmic NRF2 was undetectable (data not shown). Furthermore, melanocytes express abundant basal levels of NQO1 and PRDX6 compared to fibroblasts or keratinocytes (Fig. 1a). We observed basal expression of NRF2 in two additional NHEM lines and an immortalized NHEM line (data not shown). In contrast, unstressed fibroblasts expressed very low NRF2 and PRDX6 levels, did not express NQO1, but had abundant SOD2 expression (Fig. 1a). NRF2, NQO1 and PRDX6 expression was undetectable in keratinocytes, while SOD2 expression was low (Fig. 1a). Expression of NRF2-target HMOX1 protein was not detected in any unstressed cells (Fig. S1).
To determine if NRF2 and its targets impact viability, we downregulated NQO1 and PRDX6 in each neonatal line. Downregulation of NQO1 and PRDX6 in unstressed NHEM resulted significant reduction of viable cells (P < 0.001 and p < 0.0001 respectively) (Fig. 1b). Downregulation of NQO1 and PRDX6 did not affect viability of NHDF or NHEK except with exogenous oxidative stress induced by t-BHP, a cell permeable oxidant (Fig. 1c–d). Equivalent doses of t-BHP triggered the death of all melanocytes (data not shown). Thus, the NRF2-cascade is critical for melanocyte survival. We next investigated a role for the response in VIP-induced melanocyte stress.
VIPs activate an antioxidant gene signature in melanocytes
We hypothesized that since both 4-TBP and MBEH induce vitiligo (1), commonalities between melanocyte transcriptional responses activated would provide insights into genes and biological processes involved in vitiligo initiation. We sought to identify genes upregulated in response to stress by analysis of data from gene expression microarrays (6). Hierarchical clustering was performed to enhance statistical power. We then compared gene expression in 4-TBP (pooled: 3h/6h) and MBEH (pooled: 6h/24h)-treated melanocytes using sub-toxic concentrations sufficient to induce stress and elicit a molecular response without greatly affecting viability (described in (6)). We identified 354 commonly upregulated genes (Fig. S2) and evaluated this list using the Database for Annotation, Visualization and Integrated Discovery, which revealed significant (p < 0.05) enrichment of oxidative stress responses (Biocarta and Panther pathway analyses) and enrichment of Gene Ontology (GO) terms related to oxidative stress (Table S1). Upregulated genes from the “Response to Oxidative Stress” GO term included NRF2-targets HMOX1 and PRDX6, and genes encoding small mafs (MAFK and MAFF), which heterodimerize with NRF2 localized at AREs (12, 13) (Table S2). MBEH induced higher expression of oxidative stress genes than 4-TBP (Table S2), consistent with documented differences in potency (3, 31). Pathway analysis results suggested a role for NRF2-mediated antioxidant pathways in melanocyte response to VIP-induced stress, which we further characterized.
VIP exposure activates melanocyte NRF2 antioxidant responses, while constitutive NRF2 activation reduces MBEH-induced toxicity
Melanocytes were treated with 4-TBP (250 μM) or MBEH (300 μM) as previously described (6) to determine the expression time course of NRF2 and its targets. Nuclear localization of NRF2 increased within 1 hour of 4-TBP (Fig. S3a) exposure and 3 hours of MBEH exposure (Fig. 2a). The increase was greater after exposure to MBEH than 4-TBP (Fig. 2a and Fig. S3a). Cytoplasmic NRF2 was not detected following VIP treatment (Fig. 2a and Fig. S3a). Notably, vehicle-treated melanocytes expressed nuclear NRF2 protein at baseline (Fig. S3a), similar to unstressed melanocytes (Fig. 1a). VIPs also promoted increased mRNA expression of NRF2-target HMOX1 (Fig. S3b). HMOX1 levels returned to baseline by 24 hours of 4-TBP exposure, while levels were substantially higher with MBEH exposure and were sustained, even at 24 hours (Fig. S3b).
Figure 2. VIP exposure activates melanocyte NRF2 antioxidant responses, while constitutive NRF2 activation reduces MBEH-induced toxicity.
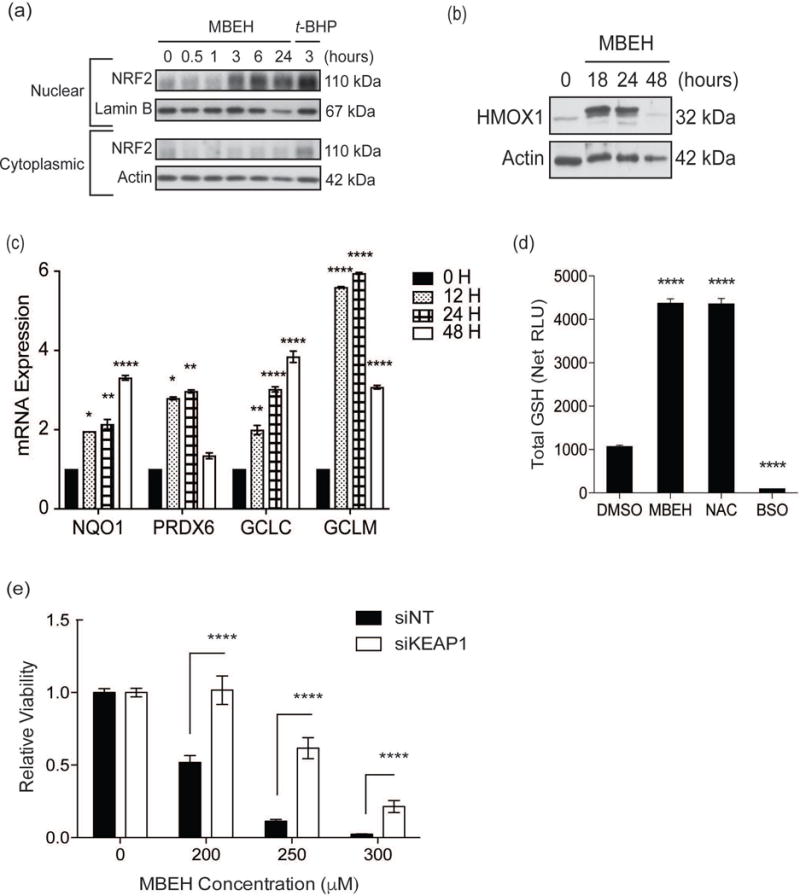
(a) Melanocytes were treated with MBEH (300 μM) for increasing periods up to 24 hours. Treatment with t-BHP (100 μM) for 3 hours served as positive control. Nuclear and cytoplasmic fractions were extracted and protein expression measured with Western blot analysis. Melanocytes were stimulated with MBEH (300 μM) for increasing periods up to 48 hours. (b) Total protein lysate was extracted for Western blot analysis of HMOX1 protein expression. (c) mRNA expression of NRF2-targets, NQO1, PRDX6, GCLC, and GCLM, was measured relative to housekeeping gene RPLP0 using quantitative RT-PCR. (d) Melanocytes were treated with MBEH (300 μM) for 20 hours and total GSH levels measured using a luminescence based assay. NAC (20 mM) and BSO (2 mM) treatment for 20 hours were used as positive and negative controls for GSH increase and depletion respectively. (e) Melanocytes were transfected with KEAP1 siRNA or NT siRNA for 48 hours followed by exposure to increasing concentrations of MBEH and relative viability measured after 24 hours. mRNA expression data and relative luminescence data are presented as mean ± SEM, n=3. The relative viability data is presented as mean ± SD. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. RLU, relative luminescence units; GSH, glutathione; NAC, N-acetyl cysteine; BSO, buthionine sulfoximine.
4-TBP-induced NRF2 activity (nuclear NRF2 and HMOX1 mRNA expression levels) peaked and returned to baseline within 24 hours (Fig. S3a–b), suggesting resolution of stress and restored homeostasis. In contrast, MBEH triggered slower, but more robust NRF2 activation, which was sustained at 24 hours (Fig. 2a and Fig. S3b). The MBEH time course was thus extended to 48 hours (Fig. 2b and Fig. S3c–d). HMOX1 mRNA expression peaked at 12 hours of MBEH exposure and decreased by 48 hours (Fig. S3c) consistent with HMOX1 protein expression, which increased within 18 hours of MBEH exposure and returned to basal levels by 48 hours (Fig. 2b). MBEH-induced NQO1 mRNA expression also increased (Fig. 2c); but not NQO1 protein levels (Fig. S3e). Baseline NQO1 protein levels are already elevated, thus an increase may not be readily determined. MBEH further induced upregulation of NRF2-targets PRDX6, and the catalytic and regulatory/modifier subunits of γ-glutamylcysteine ligase (GCLC and GCLM, respectively) (Fig. 2c and Fig. S3d). GCLC and GCLM catalyze the rate-limiting step during glutathione (GSH) biosynthesis (32). Glutathione peroxidase 1 (GPX1), a potent antioxidant that oxidizes reduced-GSH and simultaneously neutralizes H2O2, was also upregulated with MBEH exposure (data not shown). This suggests that the cytoprotective response to MBEH-induced stress also involves GSH biosynthesis. In support of this hypothesis, MBEH increased total GSH to levels comparable those of cells treated with the GSH precursor N-acetylcysteine (NAC) (Fig. 2d). The increase was accompanied by a shift towards oxidized GSH (data not shown). Buthionine sulfoximine (BSO), which inhibits GSH synthesis, effectively depleting GSH (Fig. 2d). Thus, the melanocyte response to VIP-induced stress may involve multiple NRF2 targets that participate in parallel antioxidant activities.
Since sub-toxic MBEH doses stimulated NRF2-mediated expression of antioxidants with restoration of homeostasis by 48 hours (Fig. S3d), we next investigated whether constitutive activation of NRF2 reduced melanocyte sensitivity to MBEH. Deletion of the negative regulator KEAP1 gene results in constitutive NRF2 activation (33). We therefore performed siRNA-mediated KEAP1 knockdown (Fig. S3f), which increased NRF2 activity and promoted a significant increase in HMOX1 (p < 0.05) and NQO1 (p < 0.01) expression (Fig. S3g). Notably, we observed a significant (p < 0.001) increase in melanocyte viability with KEAP1 downregulation (Fig. S3h) and significantly reduced MBEH-induced toxicity (p < 0.0001) (Fig. 2e). Thus, the NRF2 response protects melanocytes against MBEH-induced toxicity.
An impaired NRF2 response contributes to increased MBEH-induced toxicity in vitiligo melanocytes
We observed that normal melanocytes activate the NRF2 and GSH-synthesis pathways in response to MBEH exposure, while constitutive NRF2 activation reduced MBEH toxicity. We next investigated whether these pathways were compromised in vitiligo melanocytes. Both perilesional vitiligo melanocytes (PLVM) and non-lesional vitiligo melanocytes (NLVM) were pigmented and maintained their melanogenic capacity. The immortalized vitiligo PIG3V line however displayed decreased melanogenic capacity and reduced pigmentation. Intriguingly, untreated PIG3V had increased expression of NRF2-targets HMOX1 and NQO1 compared to normal immortalized melanocytes (PIG1) (Fig. S4). However, when exposed to increasing concentrations MBEH for 96 hours, NLVM, PLVM, and PIG3V were more sensitive to MBEH when compared to normal control melanocytes (Fig. 3a–b). NLVM and PLVM were equally sensitive to MBEH (Fig. 3a).
Figure 3. An impaired NRF2 response contributes to increased MBEH-induced toxicity in vitiligo melanocytes.
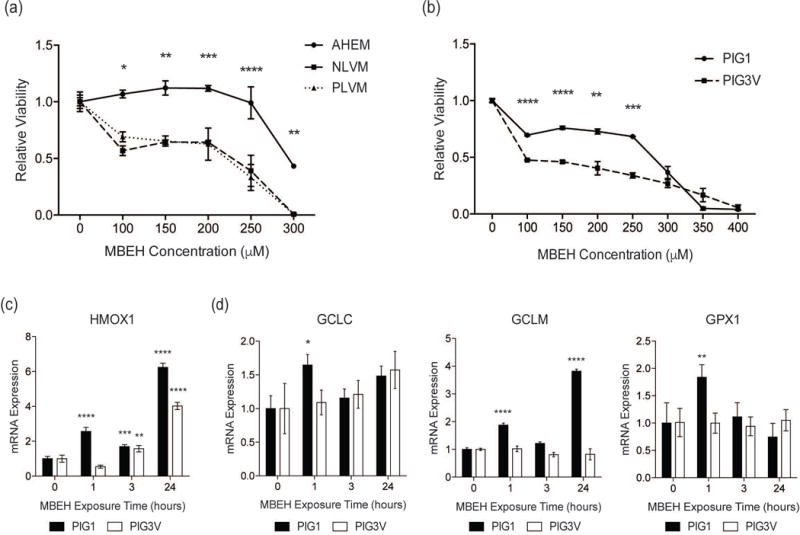
(a) Relative viability of normal adult human epidermal melanocytes (AHEM) and melanocytes derived from perilesional (PLVM) and non-lesional (NLVM) skin from a vitiligo patient were compared after exposure to increasing concentrations of MBEH for 96 hours. (b) Relative viability of immortalized control melanocytes (PIG1) and immortalized vitiligo melanocytes (PIG3V) compared after exposure to increasing concentrations of MBEH for 96 hours. PIG1 and PIG3V were stimulated with MBEH (300 μM) for increasing periods up to 24 hours and mRNA expression of (c) HMOX1 and (d) GSH pathway members GCLC, GCLM, and GPX1 was measured. The relative viability data is presented as mean ± SD. mRNA expression data are presented as mean ± SEM, n=3. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
PIG1 and PIG3V were treated with MBEH (300 μM) to measure the time-course of NRF2 and GSH-pathway activation. PIG1 cells upregulated HMOX1 within 1 hour of MBEH exposure, whereas PIG3V upregulated HMOX1 after 3 hours (Fig. 3c). HMOX1 expression was higher in PIG1 than PIG3V following exposure (Fig. 3c). PIG1 upregulated GSH synthesis pathway genes GPX1, GCLC, and GCLM within 1 hour of MBEH exposure, however expression of these genes was not significantly upregulated in PIG3V (Fig. 3d). Therefore, vitiligo melanocytes are more sensitive to MBEH, and NRF2 and GSH pathway activation, which is critical for protection against MBEH toxicity, is impaired.
NRF2 activation by DMF protects normal and vitiligo melanocytes against MBEH-induced toxicity
As constitutive NRF2 activation protects normal melanocytes against MBEH and vitiligo melanocytes have impaired NRF2 activation, identification of a pharmacologic agent that activates NRF2 for clinical use in vitiligo is of importance. We investigated whether NRF2 activation using the drug DMF protects melanocytes against toxic MBEH doses. DMF was not toxic in normal melanocytes up to 100 μM; instead, there was a significant (p < 0.05) increase in cell number following treatment (Fig. S5a). As expected, treatment of melanocytes with increasing DMF concentrations promoted increased nuclear NRF2 localization (Fig. 4a) and HMOX1 mRNA expression (Fig. 4b).
Figure 4. DMF, an NRF2 activator, protects normal and vitiligo melanocytes against MBEH-induced toxicity.
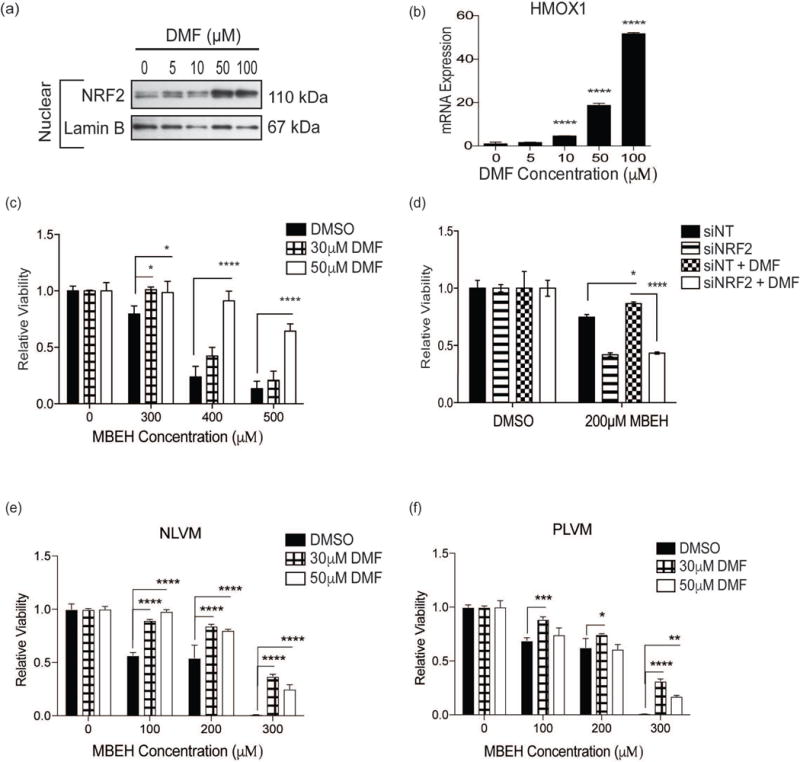
(a) Melanocytes were treated with increasing concentrations of DMF (0, 5, 10, 50, and 100 μM) for 4 hours and nuclear protein was extracted from cell lysate. Nuclear NRF2 expression was measured with Western blot analysis. (b) HMOX1 mRNA expression was measured by quantitative RT-PCR analysis of total RNA extracted from melanocytes exposed to increasing concentrations of DMF for 4 hours. The mRNA expression data is presented as mean ± SEM, n=3 **** p < 0.0001. (c) Melanocytes were pre-treated with DMF (30 μM or 50 μM) for 4 hours followed by exposure to increasing concentrations of MBEH (300, 400, and 500 μM) and relative viability compared to vehicle control after 24 hours of MBEH exposure. (d) Melanocytes were transfected with NT control siRNA or NRF2 siRNA and exposed to MBEH (200 μM) with or without DMF (50 μM) pre-treatment (as in (c)) and relative viability measured after 24 hours. (e) NLVM and (f) PLVM were pre-treated with DMF (30 μM or 50 μM) for 4 hours followed by exposure to increasing concentrations of MBEH (100, 200, and 300 μM). Relative viability measured after 96 hours. The relative viability data is presented as mean ± SD, * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. NLVM, non-lesional vitiligo melanocytes; PLVM, perilesional vitiligo melanocytes.
Remarkably, pretreatment with a low dose of DMF (30 μM) was sufficient to increase NRF2 activation and protect normal melanocytes against exposure to 300 μM MBEH. Moreover, there was a dose-dependent protective effect against high MBEH doses (400–500 μM) (Fig. 4c). siRNA-mediated NRF2 depletion abolished DMF-mediated protection (200 μM MBEH; p < 0.0001) (Fig. 4d).
Vitiligo melanocytes have a disrupted oxidant-antioxidant balance and impaired NRF2 response (8, 34) (Fig 3c–d). We therefore investigated whether NRF2 activation by DMF reduced MBEH-induced toxicity in vitiligo melanocytes. Pretreatment with 30 μM DMF conferred significant protection, with greatest efficacy at 300 μM MBEH (NLVM = 44.6% and PLVM = 45.1% protection; p < 0.0001) (Fig. 4e–f). Pretreatment with 50 μM DMF also significantly protected vitiligo melanocytes (NLVM = 29.8% and PLVM = 24.4% protection, 300 μM MBEH; p < 0.0001), but was less effective than the 30 μM DMF dose (Fig. 4e–f).
Discussion
In this study, we sought to identify cytoprotective pathways activated by melanocytes in response to agents known to trigger vitiligo. We identified a key role for the NRF2 pathway, in protecting melanocytes against MBEH exposure. Current vitiligo therapies aim to promote repigmentation by targeting autoimmune responses or stimulating melanocyte migration into lesions (35–37). While the use of antioxidants to improve repigmentation is documented (38), therapies that effectively ameliorate melanocyte oxidative stress—a known mediator of disease progression (39, 40)—are not currently approved.
Melanocytes activate the NRF2 response when exposed to oxidative stress (7, 41) and vitiligo melanocytes have been reported to exhibit compromised activation of the NRF2 response (8, 42). Comparison of the antioxidant profile of cutaneous cells revealed that NRF2 and NRF2-regulated antioxidants promoted melanocyte viability, but played a lesser role in determining keratinocyte and fibroblast viability. Multiple studies reported more robust NRF2 activation in melanocytes compared to keratinocytes after exposure to chemical stressors (41) or ultraviolet radiation (43). Comparison of melanocytes to other cutaneous cells in the absence of exogenous stressors revealed that while melanocytes had lower basal expression of SOD2, they expressed abundant NQO1, which can alternatively scavenge superoxide radicals. Most cell types use SOD as their first line of defense against superoxide anions because it has better reducing enzyme activity. However, cells that have low SOD levels, such as cardiovascular tissues (44) and perhaps melanocytes, utilize NQO1. NQO1 reduces quinone compounds such as DOPA-quinone and indole 5,6 quinone, which are intermediates of melanin synthesis (45); therefore, melanocytes may maintain high basal NQO1 levels to quickly scavenge quinone byproducts that leak into the cytosol should the melanosome be compromised (46). Moreover, melanocytes preferentially expressed PRDX6, which reduces hydrogen and lipid peroxides. Melanocytes may also require PRDX6 as an immediate defense against melanogenesis by-products. Interestingly, NQO1 and PRDX6 downregulation did not affect fibroblasts and keratinocytes viability except in following exposure to exogenous oxidative stress, suggesting that in the absence of oxidative stress, NRF2 targets have a cytoprotective function in melanocytes, but not in fibroblasts or keratinocytes.
In addition to NRF2 expression in unstressed melanocytes, melanocytes further activated NRF2 upon exposure to sub-lethal doses of VIPs. We employed an in vitro model for vitiligo initiation, in which normal melanocytes were exposed to sub-toxic 4-TBP and MBEH doses, and evaluated the cellular responses. Exposure to 4-TBP initiated a rapid NRF2/HMOX1 response which quickly resolved to basal levels. However, MBEH exposure triggered a more expansive NRF2 response, upregulating additional NRF2-targets including HMOX1, NQO1, PRDX6, GCLC, and GCLM. As MBEH can form semi-quinone radicals, we anticipated upregulation of NQO1, a quinone reductase (29, 47). Another mechanism for quinone reduction is through GSH conjugation (48). As GCLC and GCLM catalyze the rate-limiting step in GSH synthesis, we hypothesized their increased expression would promote increased GSH levels to combat MBEH-induced toxicity. As predicted, GSH levels increased in response to sub-toxic MBEH doses. MBEH, which is more potent than 4-TBP (3), induces depigmentation through numerous mechanisms including cytotoxic T cell-mediated autoimmune destruction of melanocytes, ROS production, quinone haptenization, and necrotic cell death (25). Our findings suggest that increased expression of NRF2 and its targets is more dramatic following MBEH exposure as compared to 4-TBP exposure. An amplified NRF2 response may be necessary to respond to MBEH-mediated inflammatory and oxidative stress. Furthermore, constitutive NRF2 activation, by silencing of its repressor KEAP1, increased melanocyte proliferation and markedly reduced MBEH toxicity.
Upon establishing the importance of the NRF2 response in protecting normal melanocytes against MBEH, we investigated the impact on vitiligo melanocytes. They were more sensitive to MBEH and displayed impaired activation of the NRF2-regulated GSH response. Pharmacological NRF2 activation with DMF, an FDA-approved drug, was remarkably protective against toxic levels of MBEH in normal and vitiligo melanocytes. This protection is NRF2-dependent, since DMF did not protect normal melanocytes when NRF2 was silenced. While the effect of DMF was dose dependent in the normal melanocytes, in the vitiligo melanocytes, 50 μM DMF was less effective than the 30 μM DMF dose. NRF2 activation is impaired in vitiligo melanocytes (7–10), it is therefore possible that the pathway is maximally activated with 30 μM DMF, and that 50 μM DMF is not more protective and instead excess DMF reduces efficacy. Of clinical importance, DMF was protective at doses that were not themselves toxic to melanocytes. This is in contrast to the inherent toxicities of other NRF2 activators, such as phytochemical carnosic acid (23) and curcumin (our unpublished observation). Although clinical trials will be required to establish the safest dose and efficacy of DMF in vitiligo patients, our study showed a protective effect against a potent vitiligo inducer using lower concentrations of DMF than those currently approved for psoriasis or multiple sclerosis. In addition, constitutive and chronic NRF2 activation can be detrimental (33); thus promoting a transient NRF2 increase using DMF, would translate into clinical practice more effectively. DMF has additional immunomodulatory properties (16) that may further protect melanocytes in vivo and prove beneficial for modulating autoimmune-mediated progression of vitiligo.
Monotherapies targeting oxidative stress have been ineffective in vitiligo to date. This is potentially due to the focus on general oxidative stress amelioration, rather than targeting specific pathways activated by melanocytes in response to stressors with pleiotropic effects, such as MBEH. For instance, despite reports suggesting that catalase protects melanocytes undergoing stress (5), topical treatment with pseudocatalase has not prevented disease progression. Although melanocytes from individuals with vitiligo demonstrate compromised antioxidant activity (34, 49, 50), dysfunctional NRF2 activity (8, 42) may be more disruptive than the imbalance of other antioxidants. Vitiligo melanocytes have low nuclear NRF2 expression compared to normal melanocytes (42). Thus, further activating the NRF2 response in vitiligo melanocytes using DMF may promote increased nuclear NRF2. DMF may be a useful adjuvant therapy that reduces active depigmentation and protects melanocytes that are repopulating lesional areas following treatment with modalities such as UVB. In summary, we demonstrated that the NRF2 response contributes to melanocyte survival and protects against MBEH in both normal and vitiligo melanocytes. We thus introduce a potential therapeutic strategy - activating the NRF2 response using DMF - for the treatment of vitiligo.
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Award AR41880, SJO), part of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Funding was received from the American Skin Association (Mulvaney Family Foundation Research Grant, PM). OAA was supported by the NYU Medical Scientist Training Program, UNCF-Merck Science Initiative Graduate Fellowship and Molecular Pharmacology NIH-T32 (GM066704, Bach). OAA, SJO, NE, and PM designed the studies. OAA conducted the experiments. NE performed the clinical studies. OAA, SJO, and PM analyzed the data. OAA, SJO, and PM wrote the manuscript. We thank Martha Vega and Genevieve Torres for technical assistance, and Dr. Caroline Le Poole for providing the PIG1 and PIG3V lines.
Footnotes
Conflict of Interest
None reported.
References
- 1.Boissy RE, Manga P. On the etiology of contact/occupational vitiligo. Pigment Cell Res. 2004;17:208–214. doi: 10.1111/j.1600-0749.2004.00130.x. [DOI] [PubMed] [Google Scholar]
- 2.Oliver EA, Schwartz L, Warren LH. Occupational leukoderma: Preliminary report. Journal of the American Medical Association. 1939;113:927–928. [Google Scholar]
- 3.Hariharan V, Toole T, Klarquist J, et al. Topical application of bleaching phenols; in-vivo studies and mechanism of action relevant to melanoma treatment. Melanoma Res. 2011;21:115–126. doi: 10.1097/CMR.0b013e328343f542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.AlGhamdi KM, Kumar A. Depigmentation therapies for normal skin in vitiligo universalis. J Eur Acad Dermatol Venereol. 2011;25:749–757. doi: 10.1111/j.1468-3083.2010.03876.x. [DOI] [PubMed] [Google Scholar]
- 5.Manga P, Sheyn D, Yang F, et al. A role for tyrosinase-related protein 1 in 4-tert-butylphenol-induced toxicity in melanocytes: Implications for vitiligo. Am J Pathol. 2006;169:1652–1662. doi: 10.2353/ajpath.2006.050769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol. 2012;132:2601–2609. doi: 10.1038/jid.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jian Z, Li K, Liu L, et al. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J Invest Dermatol. 2011;131:1420–1427. doi: 10.1038/jid.2011.56. [DOI] [PubMed] [Google Scholar]
- 8.Bellei B, Pitisci A, Ottaviani M, et al. Vitiligo: a possible model of degenerative diseases. PloS one. 2013;8:e59782. doi: 10.1371/journal.pone.0059782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elassiuty YE, Klarquist J, Speiser J, et al. Heme oxygenase-1 expression protects melanocytes from stress-induced cell death: implications for vitiligo. Experimental dermatology. 2011;20:496–501. doi: 10.1111/j.1600-0625.2010.01232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jian Z, Tang L, Yi X, et al. Aspirin induces Nrf2-mediated transcriptional activation of haem oxygenase-1 in protection of human melanocytes from H O -induced oxidative stress. J Cell Mol Med. 2016 doi: 10.1111/jcmm.12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 14.Pall ML, Levine S. Nrf2, a master regulator of detoxification and also antioxidant, anti-inflammatory and other cytoprotective mechanisms, is raised by health promoting factors. Sheng Li Xue Bao. 2015;67:1–18. [PubMed] [Google Scholar]
- 15.Meissner M, Valesky EM, Kippenberger S, et al. Dimethyl fumarate – only an anti-psoriatic medication? JDDG: Journal der Deutschen Dermatologischen Gesellschaft. 2012;10:793–801. doi: 10.1111/j.1610-0387.2012.07996.x. [DOI] [PubMed] [Google Scholar]
- 16.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain: a journal of neurology. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 17.Scannevin RH, Chollate S, Jung MY, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. The Journal of pharmacology and experimental therapeutics. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 18.Bomprezzi R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: an overview. Therapeutic advances in neurological disorders. 2015;8:20–30. doi: 10.1177/1756285614564152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mrowietz U, Asadullah K. Dimethylfumarate for psoriasis: more than a dietary curiosity. Trends in Molecular Medicine. 11:43–48. doi: 10.1016/j.molmed.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Das RK, Brar SK, Verma M. Recent advances in the biomedical applications of fumaric acid and its ester derivatives: The multifaceted alternative therapeutics. Pharmacol Rep. 2016;68:404–414. doi: 10.1016/j.pharep.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Stoof TJ, Flier J, Sampat S, et al. The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. British Journal of Dermatology. 2001;144:1114–1120. doi: 10.1046/j.1365-2133.2001.04220.x. [DOI] [PubMed] [Google Scholar]
- 22.Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. doi: 10.1146/annurev-immunol-032713-120225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okubo A, Yasuhira S, Shibazaki M, et al. NAD(P)H dehydrogenase, quinone 1 (NQO1) protects melanin-producing cells from cytotoxicity of rhododendrol. Pigment Cell Melanoma Res. 2016 doi: 10.1111/pcmr.12461. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y, Wang S, Xu A. A mouse model of vitiligo induced by monobenzone. Experimental dermatology. 2013;22:499–501. doi: 10.1111/exd.12184. [DOI] [PubMed] [Google Scholar]
- 25.van den Boorn JG, Melief CJ, Luiten RM. Monobenzone-induced depigmentation: from enzymatic blockade to autoimmunity. Pigment Cell Melanoma Res. 2011;24:673–679. doi: 10.1111/j.1755-148X.2011.00878.x. [DOI] [PubMed] [Google Scholar]
- 26.Le Poole IC, van den Berg FM, van den Wijngaard RM, et al. Generation of a human melanocyte cell line by introduction of HPV16 E6 and E7 genes. In Vitro Cell Dev Biol Anim. 1997;33:42–49. doi: 10.1007/s11626-997-0021-6. [DOI] [PubMed] [Google Scholar]
- 27.Le Poole IC, Boissy RE, Sarangarajan R, et al. PIG3V, an immortalized human vitiligo melanocyte cell line, expresses dilated endoplasmic reticulum. In Vitro Cell Dev Biol Anim. 2000;36:309–319. doi: 10.1290/1071-2690(2000)036<0309:PAIHVM>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Cheng T, Orlow SJ, Manga P. Loss of Oca2 disrupts the unfolded protein response and increases resistance to endoplasmic reticulum stress in melanocytes. Pigment Cell Melanoma Res. 2013;26:826–834. doi: 10.1111/pcmr.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegel D, Gustafson DL, Dehn DL, et al. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol. 2004;65:1238–1247. doi: 10.1124/mol.65.5.1238. [DOI] [PubMed] [Google Scholar]
- 30.Fisher AB. Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid Redox Signal. 2011;15:831–844. doi: 10.1089/ars.2010.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hariharan V, Klarquist J, Reust MJ, et al. Monobenzyl ether of hydroquinone and 4-tertiary butyl phenol activate markedly different physiological responses in melanocytes: relevance to skin depigmentation. J Invest Dermatol. 2010;130:211–220. doi: 10.1038/jid.2009.214. [DOI] [PubMed] [Google Scholar]
- 32.Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 33.Wakabayashi N, Itoh K, Wakabayashi J, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nature genetics. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 34.Ines D, Sonia B, Riadh BM, et al. A comparative study of oxidant-antioxidant status in stable and active vitiligo patients. Archives of dermatological research. 2006;298:147–152. doi: 10.1007/s00403-006-0680-2. [DOI] [PubMed] [Google Scholar]
- 35.Picardo M, Dell’Anna ML, Ezzedine K, et al. Vitiligo Nature Reviews Disease Primers. 2015;15011 doi: 10.1038/nrdp.2015.11. [DOI] [PubMed] [Google Scholar]
- 36.Rashighi M, Harris JE. Interfering with the IFN-gamma/CXCL10 pathway to develop new targeted treatments for vitiligo. Ann Transl Med. 2015;3:343. doi: 10.3978/j.issn.2305-5839.2015.11.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris JE, Rashighi M, Nguyen N, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata (AA) J Am Acad Dermatol. 2016;74:370–371. doi: 10.1016/j.jaad.2015.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dell’Anna ML, Mastrofrancesco A, Sala R, et al. Antioxidants and narrow band-UVB in the treatment of vitiligo: a double-blind placebo controlled trial. Clin Exp Dermatol. 2007;32:631–636. doi: 10.1111/j.1365-2230.2007.02514.x. [DOI] [PubMed] [Google Scholar]
- 39.Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond) 2011;120:99–120. doi: 10.1042/CS20090603. [DOI] [PubMed] [Google Scholar]
- 40.Schallreuter KU, Moore J, Wood JM, et al. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J Investig Dermatol Symp Proc. 1999;4:91–96. doi: 10.1038/sj.jidsp.5640189. [DOI] [PubMed] [Google Scholar]
- 41.Natarajan VT, Singh A, Kumar AA, et al. Transcriptional upregulation of Nrf2-dependent phase II detoxification genes in the involved epidermis of vitiligo vulgaris. J Invest Dermatol. 2010;130:2781–2789. doi: 10.1038/jid.2010.201. [DOI] [PubMed] [Google Scholar]
- 42.Jian Z, Li K, Song P, et al. Impaired activation of the Nrf2-ARE signaling pathway undermines H2O2-induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo. J Invest Dermatol. 2014;134:2221–2230. doi: 10.1038/jid.2014.152. [DOI] [PubMed] [Google Scholar]
- 43.Marrot L, Jones C, Perez P, et al. The significance of Nrf2 pathway in (photo)-oxidative stress response in melanocytes and keratinocytes of the human epidermis. Pigment Cell Melanoma Res. 2008;21:79–88. doi: 10.1111/j.1755-148X.2007.00424.x. [DOI] [PubMed] [Google Scholar]
- 44.Zhu H, Li Y. NAD(P)H: quinone oxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovascular toxicology. 2012;12:39–45. doi: 10.1007/s12012-011-9136-9. [DOI] [PubMed] [Google Scholar]
- 45.Hochstein P, Cohen G. The cytotoxicity of melanin precursors. Ann N Y Acad Sci. 1963;100:876–886. [PubMed] [Google Scholar]
- 46.Borovanský J, Riley PA. Melanins and Melanosomes. Wiley-VCH Verlag GmbH & Co. KGaA; 2011. Physiological and Pathological Functions of Melanosomes; pp. 343–381. [Google Scholar]
- 47.Siegel D, Yan C, Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochemical pharmacology. 2012;83:1033–1040. doi: 10.1016/j.bcp.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi N, Schreiber J, Fischer V, et al. Formation of glutathione-conjugated semiquinones by the reaction of quinones with glutathione: An ESR study. Archives of Biochemistry and Biophysics. 1987;252:41–48. doi: 10.1016/0003-9861(87)90006-3. [DOI] [PubMed] [Google Scholar]
- 49.Yildirim M, Baysal V, Inaloz HS, et al. The role of oxidants and antioxidants in generalized vitiligo at tissue level. J Eur Acad Dermatol Venereol. 2004;18:683–686. doi: 10.1111/j.1468-3083.2004.01080.x. [DOI] [PubMed] [Google Scholar]
- 50.Yildirim M, Baysal V, Inaloz HS, et al. The role of oxidants and antioxidants in generalized vitiligo. The Journal of dermatology. 2003;30:104–108. doi: 10.1111/j.1346-8138.2003.tb00356.x. [DOI] [PubMed] [Google Scholar]
- 51.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 52.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.