

Supplemental Digital Content is available in the text.
Keywords: calcium, Cx43, cyclic nucleotides, endothelium, gap junction, pregnancy, vasodilation
Abstract
Cell–cell communication is dependent on GJ (gap junction) proteins such as Cx43 (connexin 43). We previously demonstrated the importance of Cx43 function in establishing the enhanced pregnancy vasodilatory phenotype during pregnancy in uterine artery endothelial cells from pregnant (P-UAEC) ewes. Cx43 is regulated by elevating cAMP and PKA (protein kinase A)–dependent Cx43 S365 phosphorylation–associated trafficking and GJ open gating, which is opposed by PKC (protein kinase C)–dependent S368 phosphorylation-mediated GJ turnover and closed gating. However, the role of cyclic nucleotide-mediated signaling mechanisms that control Cx43 and GJ function in P-UAECs is unknown. We hypothesize that cAMP will mediate increases in S365 phosphorylation, thereby, enhancing GJ trafficking and open gating, while cGMP will stimulate S368, but not S365, phosphorylation to enhance GJ turnover and closed gating in P-UAECs. Treatment with 8-Bromo (8-Br)-cAMP signal significantly (P<0.05) increased nonphosphorylated S365 signal and total Cx43 phosphorylation, but not S368 phosphorylation, while 8-Br-cGMP significantly (P<0.05) increased Cx43 C-terminus-S365 signal, S368, and total Cx43 phosphorylation. Inhibition of PKA, but not PKG (protein kinase G), abrogated the 8-Br-cAMP–stimulated increase in nonphosphorylated S365 and total Cx43 phosphorylation and inhibited S368 below basal levels, whereas inhibition of PKG blocked (P<0.05) the 8-bromo-cGMP-stimulated rises in nonphosphorylated S365, total Cx43, and S368 phosphorylation levels in P-UAECs. Functional studies showed that 8-Br-cAMP increased dye transfer and sustained calcium bursts, while 8-Br-cGMP decreased both. Thus, in P-UAECs, only 8-Br-cAMP and not 8-Br-cGMP effectively enhances nonphosphorylated S365 and total Cx43 expression that correspondingly reduces S368 phosphorylation, allowing increased GJ communication. This provides new insights into the regulatory mechanisms behind Cx43 function and GJ communication.
Cell to cell communication is vital for the development of cardiovascular adaptations during gestation and are reliant on GJ (gap junction) intercellular communication (GJIC).1–3 GJs are specialized membrane hemichannels composed of Cx (connexin) proteins that direct cell–cell diffusion of ions, small molecules, and second messengers (eg, IP3, Ca2+, cAMP, cGMP, ATP)4–6 that are important for synchronizing endothelial cell vasodilatory responses. Pregnancy is a physiological state of vasodilation that requires heightened cell coupling to maintain substantial increases in blood flow necessary for fetal growth and development.1,2,7,8 Endothelial-derived vasodilators, such as prostacyclin (PGI2) and nitric oxide (NO), are enhanced in the uterine circulation during pregnancy and serve as a pregnancy-adapted vasodilatory phenotype that sets the stage for the pronounced gestational elevations in uterine blood flow.3,8 We have shown that Cx43 is the major GJ protein present in uterine artery endothelial cells (UAECs) and controls pregnancy-specific Ca2+–mediated activation of endothelial NO synthase and NO production in vitro1 and ex vivo.2 In hypertensive states of pregnancy, reduced vasodilation is related to endothelial cell dysfunction characterized by reduced bioavailability of endothelium-derived vasodilators, in particular NO.1,2,7,8 Aberrant Cx43 expression, function,9 and lack of sufficient NO production are associated with hypertension.10 Because endothelial cell dysfunction is linked to impaired endothelium-dependent vasodilation mechanisms, hypertensive diseases of pregnancy, such as preeclampsia,9,11,12 may very well occur at least in part because of a failure to enhance the vasodilatory phenotype through Cx43 closure or internalization.3
Cx43 distribution at the plasma membrane seems to influence the conduction properties of cells.4–6 Cx43 distribution and conductivity are also dictated by phosphorylation by several site-specific signaling kinases.4,5,13–18 Phosphorylation of Cx43 at cAMP-dependent PKA (protein kinase A)–mediated serine (S)365 and PKC (protein kinase C)–dependent S368 sites differentially regulate GJIC via open16,19 and closed17,20 gating of GJ hemichannels, respectively. The S365 and S368 sites also differentially regulate GJ assembly and disassembly at the plasma membrane, respectively.16–20 The second messenger cAMP via PKA increases Cx43 phosphorylation at S365 and GJIC. The cAMP-enhanced GJ assembly directly results in the net movement of newly synthesized Cx43 to the plasma membrane,16,18,19 thereby, rapidly increasing the number and assembly of de novo GJs within minutes.19 In addition, cGMP-dependent Cx43 phosphorylation decreases GJ conductance,21 while PKC-dependent S368 phosphorylation decreases GJIC and dramatically inhibits new GJ assembly.22,23 The interaction between phosphorylation state at S365 and S368 inversely affects each other and is suggested to be mutually exclusive. This is known as the gatekeeper theory, where phosphorylated S365 plays a gatekeeper role by preventing Cx43 phosphorylation at S368. Thus, S365 phosphorylation may function to protect cells from downregulation of GJIC.5,24
Although cAMP and cGMP signaling is usually thought of as mutually redundant, it is possible that cAMP/PKA and cGMP/PKG (protein kinase G) pathways will have opposing functional effects on Cx43. To date, there is a lack of an understanding of their mutual roles in Cx43 phosphorylation and signaling events that affect vasodilatory processes in UAECs. In these studies, we, thus, contrasted cAMP and cGMP effects on (1) Cx43 phosphorylation state in nonpregnant (NP)-UAECs versus P-UAECs; (2) Cx43 phosphorylation state that occurs via PKA or PKG in P-UAECs; and (3) changes in Cx43 expression and phosphorylation that also correspondingly drive changes in GJIC communication and sustained calcium bursts in P-UAECs. We hypothesize that cAMP will mediate increases in Cx43 S365 phosphorylation that will enhance GJ trafficking and open gating, while cGMP will increase S368, but not S365, phosphorylation to enhance GJ turnover and closed gating in P-UAECs versus NP-UAECs.
Materials and Methods
Detailed methods are available in the online-only Data Supplement.
Cell Preparation and Culture
University of Wisconsin-Madison Animal Care Committee approved these protocols. Ovine UAECs were isolated from nonpregnant (luteal, n=4) and pregnant (120–130 days; term=147 days; n=4) ewes.3 Passage 4, UAECs (≈90% confluence) were cultured in endothelial basal medium for experimental treatments and either (1) lysed for Western analyses; (2) grown on chamber slides for Lucifer yellow dye transfer studies; or (3) grown on 5-mm glass-bottom micro-well dishes for Ca2+ imaging studies.
Experimental Treatments and Protein Kinases Inhibition
For exogenous cyclic nucleotide time course experiments, NP-UAECs and P-UAECs were treated with endothelial basal medium or endothelial basal medium containing 1 μmol/L or 1 mmol/L of 8-bromo (8-Br-)-cAMP or 8-Br-cGMP for 0 to 60 minutes or 12 hours. Based on these studies, P-UAECs were treated for 1 hour with cell-permeable 10 μmol/L of antagonists for PKA (PKI [protein kinase inhibitor] 14–22 amide, myristoylated) or PKG [2-Bromo-3,4-dihydro-3-[3,5-O-[(R)-mercaptophosphinylidene]-β-d-ribofuranosyl]-6-phenyl-9H-Imidazo[1,2-a]purin-9-one sodium salt] (Rp-8-Br-PET-cGMPs) followed by treatment with doses of either 1 μmol/L or 1 mmol/L of 8-Br-cAMP or 8-Br-cGMP for 30 minutes.
Protein Extraction and Western Immunoblotting
Western analyses were performed3 on whole cell lysates using antibodies for Cx43 C-terminus (CT1) S365 antibody (1:3000) specific for Cx43 when it is nonphosphorylated at S365 (online-only Data Supplement), S368 (1:1250) detects levels of Cx43 only when phosphorylated at serine 368, or total Cx43 (1:3000). Secondary antibodies, anti-rabbit or anti-mouse, were detected using enhanced chemiluminescence and HyperFilm. β-Actin was used as a loading control.2,3
Lucifer Yellow Dye Transfer
P-UAECs (<90% confluent) were grown on chamber slides3 and treated for 1 hour with 10 μmol/L PKI 14–22 amide, myristoylated, or Rp-8-Br-PET-cGMPs prior to treatment with vehicle or either 1 μmol/L or 1 mmol/L 8-Br-cAMP or 8-Br-cGMP for 30 minutes. UAECs were scraped with a sterile razor blade and incubated for 10 minutes with 0.05% Lucifer yellow and tetramethylrhodamine dextran (3 kDa),1,3,25 washed with Ca2+-free Krebs buffer containing an additional 50 μmol/L ethylene glycol tetraacetic acid,1,3 and imaged in triplicate using fluorescence microscope. Areas of dye spread (fluorescent areas) were quantified using ImageJ software.
Fura-2 [Ca2+]i Imaging Studies
P-UAECs were grown to >90% density, loaded with Fura-2AM (10 μmol/L) in Krebs buffer (125 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgSO4, 1 mmol/L KH2PO4, 6 mmol/L glucose, 2 mmol/L CaCl2, 25 mmol/L HEPES, pH 7.4), and individual endothelial cells (≈90 cells/dish) were preselected and imaged. Cells were recorded simultaneously for a 5-minute baseline and then stimulated by ATP (100 μmol/L; 30 minutes).1 Cells were washed and allowed to recover for 20 minutes and treated with 8-Br-cAMP or 8-Br-cGMP (1 μmol/L or 1 mmol/L; 30 minutes) before a subsequent treatment of ATP while recording continued (another 30 minutes). Cells that gave ≥2 Ca2+ bursts during the initial ATP treatment were counted before and after treatment as described previously.1
Statistical Analysis
Data presented as means±SEM fold of control (untreated). We performed statistical analyses between control and experimental groups using the Student’s t-test, 1-way analysis of variance, 2-way analysis of variance, or a Mann–Whitney rank-sum test as appropriate (SigmaPlot12). Statistical significance was established a priori at P<0.05.
Results
Effects of Cyclic Nucleotides on Cx43 Phosphorylation in UAECs
Using Western analysis, we examined Cx43 phosphorylation sites (S365 and S368) that may act inversely to control GJ assembly/disassembly and gating. Compared with NP-UAECs and untreated control, P-UAECs treated with 1 μmol/L and 1 mmol/L 8-Br-cAMP exhibited significant pregnancy-specific increases in CT1 S365 levels (nonphospho-specific antibody) of 3-fold and 5-fold (P<0.05) by 30 minutes, which maintained for ≤12 hours (≈2- to 3-fold; Figure 1A). In contrast, 8-Br-cAMP treatment only promoted a modest rise in Cx43 S368 phosphorylation signal (Figure 1B), which did not reach statistical significance, consistent with PKA regulating S365 and not the pS368. Interestingly, 1 μmol/L 8-Br-cAMP increased total Cx43 ≈2-fold by 60 minutes, while the 1 mmol/L dose significantly increased total Cx43 4-fold by 30 minutes, an increase maintained for ≤12 hours (P<0.05; Figure 1C). When P-UAEC S365 and pS368 data were normalized to total Cx43 (Figure 1D), P-UAECs incubated with 1 mmol/L 8-Br-cAMP still displayed an overall increase in CT1 S365 signal that is most likely caused by the substantial new synthesis of nonphosphorylated Cx43 protein. Because of this increase in total Cx43, the normalized pS368 Cx43 data displayed a relatively smaller, but still significant increase in CT1 S365 signal and an overall decrease (P<0.05) in Cx43 S368 phosphorylation.
Figure 1.
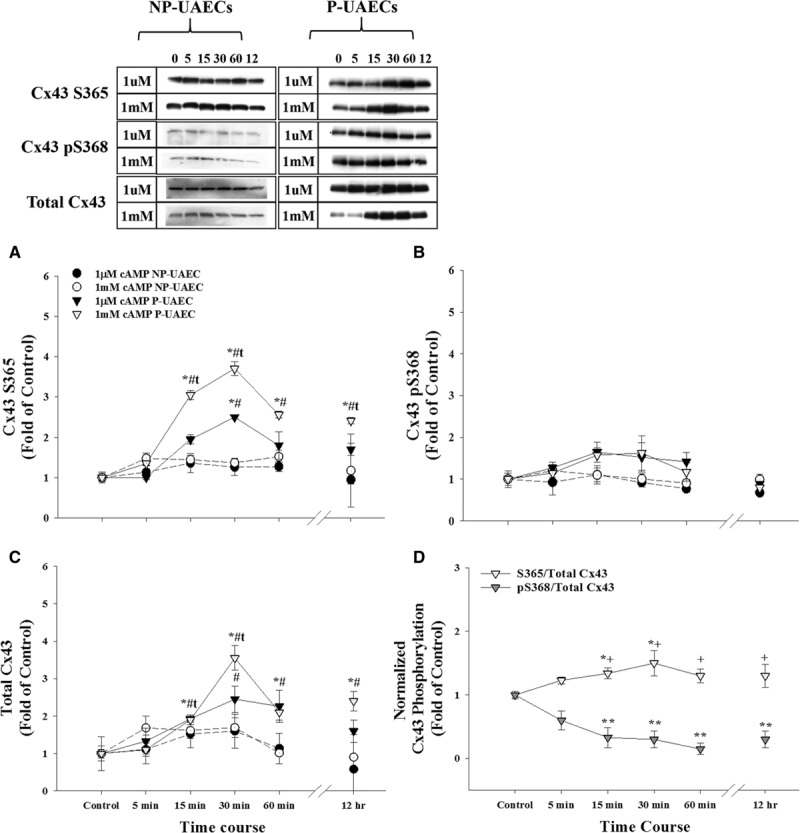
Effects of 8-Br (8-Bromo)-cAMP on Cx43 (connexin 43) phosphorylation in nonpregnant uterine artery endothelial cells (NP-UAECs) and pregnant uterine artery endothelial cells (P-UAECs). 8-Br-cAMP significantly increases nonphosphorylated Cx43 S365 levels, but not pS368. Western blot analyses showing the effects of 1 μmol/L and 1 mmol/L 8-Br-cAMP for 0 to 60 minutes or 12 hours in NP-UAECs vs P-UAECs on (A) nonphosphorylated Cx43 S365 levels detected by the Cx43 C-terminus (CT1) antibody, (B) Cx43 pS368 phosphorylation levels, (C) total Cx43 levels, and (D) Cx43 S365 and pS368 phosphorylation sites normalized to total Cx43 only for P-UAECs treated with 1 mmol/L 8-Br-cAMP. *, Increase (P<0.05, n=4) of cyclic nucleotide treatment vs untreated control. **, Decrease (P<0.05, n=4) of S365/total Cx43 vs pS368/total Cx43. t, Increase (P<0.05, n=4) of Cx43 phosphorylation in 1 mmol/L vs 1 μmol/L treatment. #, Increase (P<0.05, n=4) of Cx43 phosphorylation in NP-UAEC- vs P-UAEC-treated cells. +, Increase (P<0.05, n=4) of S365/total Cx43 vs pS368/total Cx43.
Treatment with 8-Br-cGMP also caused significant time- and dose-related increases of CT1 S365 signal but of smaller magnitude (≈2-fold; 30 minutes) than 8-Br-cAMP (Figure 2A). In contrast to 8-Br-cAMP, 8-Br-cGMP also significantly increased Cx43 S368 phosphorylation levels (3- to 4-fold; 30 minutes; P<0.05; Figure 2B). While 8-Br-cGMP also displayed both a time- and dose-related increase in total Cx43 (Figure 2C), when the S365 and pS368 signals were normalized to total Cx43 levels in P-UAECs, there was no significant increase (P>0.05) in S365 signal or significant decrease in pS368 signal (Figure 2D).
Figure 2.
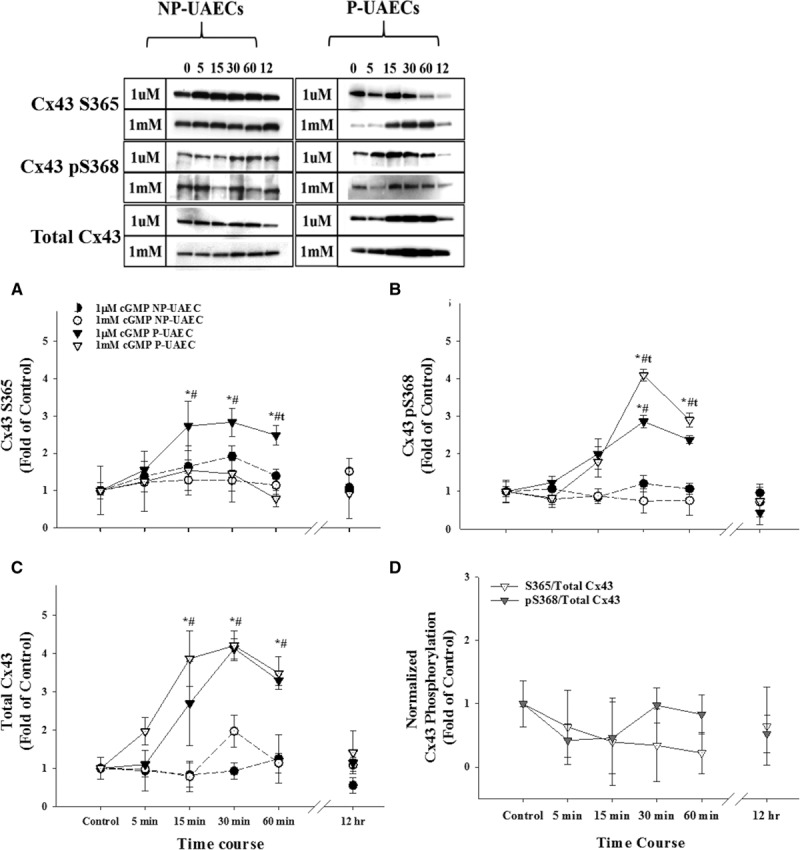
Effects of 8-Br (8-Bromo)-cAMP on Cx43 (connexin 43) phosphorylation in nonpregnant uterine artery endothelial cells (NP-UAECs) and pregnant uterine artery endothelial cells (P-UAECs). 8-Br-cGMP significantly increases both nonphosphorylated Cx43 S365 and pS368 levels. Western blot analysis showing the effects of 1 μmol/L and 1 mmol/L 8-Br-cGMP for 0 to 60 minutes or 12 hours in NP-UAECs vs P-UAECs on (A) nonphosphorylated Cx43 S365 levels detected by the Cx43 C-terminus (CT1) antibody, (B) Cx43 pS368 phosphorylation levels, (C) total Cx43 levels, and (D) Cx43 S365 and pS368 phosphorylation sites normalized to total Cx43 only for P-UAECs treated with 1 mmol/L 8-Br-cGMP. *, Increase (P<0.05, n=4) of cyclic nucleotide treatment vs untreated control. t, Increase (P<0.05, n=4) of Cx43 phosphorylation in 1 mmol/L vs 1 μmol/L treatment. #, Increase (P<0.05, n=4) of Cx43 phosphorylation in NP-UAEC- vs P-UAEC-treated cells.
Role of PKA or PKG in Mediating Cx43 Phosphorylation in P-UAECs
To test the role of PKA and PKG, we used kinase-specific inhibitors. We observed that P-UAECs treated with the PKA antagonist, PKI, alone significantly reduced CT1 S365 signal and total Cx43 levels and pS368 Cx43 phosphorylation. PKI treatment prevented 8-Br-cAMP-mediated increases and significantly reduced CT1 S365 (nonphosphorylated at S365), Cx43 pS368, and total Cx43 signals below basal (Figure 3A through 3C). PKI also inhibited the lesser 8-Br-cGMP-induced effects on the CT1 S365 signal and reduced total Cx43 levels below basal, consistent with a possible cross reaction or cross talk of 8-Br-cGMP with PKA. PKI treatments did not significantly alter the pS368 signal in 8-Br-cGMP-treated P-UAECs, in contrast to the significant (P<0.05) inhibition below basal when 8-Br-cAMP treatment was used (Figure 3B).
Figure 3.
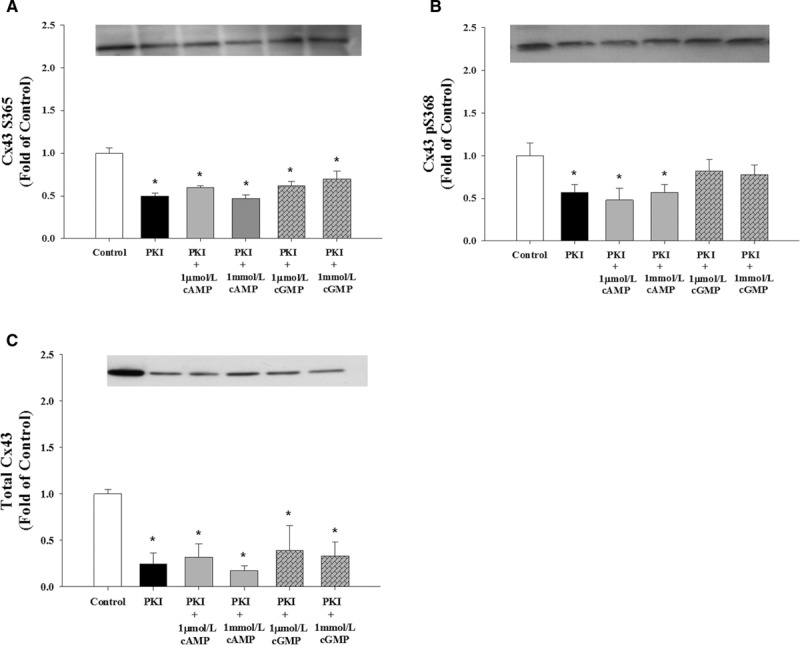
Role of PKA (protein kinase A) signaling pathways in Cx43 (connexin 43) phosphorylation in pregnant uterine artery endothelial cells (P-UAECs). Inhibition of PKA significantly reduced nonphosphorylated Cx43 S365 levels. Western blot analysis showing the effects of treatment with 10 μmol/L of PKA antagonist PKI (1 hour) on 1 μmol/L and 1 mmol/L 8-Br (8-Bromo)-cAMP-mediated and 8-Br-cGMP-mediated (30 minutes) changes of (A) nonphosphorylated Cx43 S365 levels detected by the Cx43 C-terminus (CT1) antibody, (B) Cx43 pS368 phosphorylation levels, and (C) total Cx43 levels. *, Decrease (P<0.05, n=4) in Cx43 phosphorylation state vs untreated control.
Use of the PKG inhibitor Rp-8-Br-PET-cGMPs alone appeared to not affect CT1 S365 signal (Figure 4A) or changes in pS368 Cx43 (Figure 4B) or total Cx43 (Figure 4C). Rp-8-Br-PET-cGMPs also failed to prevent 8-Br-cAMP-stimulated increases in CT1 S365 signal or total Cx43 in P-UAEC (Figure 4A and 4C). However, Rp-8-Br-PET-cGMPs blocked (P<0.05) 8-Br-cGMP-stimulated increases in CT1 S365 signal and total Cx43 (Figure 4A and 4C) and successfully blocked 8-Br-cGMP effects on S368 phosphorylation levels in P-UAECs (Figure 4B). Thus, these inhibitors successfully discriminated the roles of PKA versus PKG in each response at the dose used.
Figure 4.
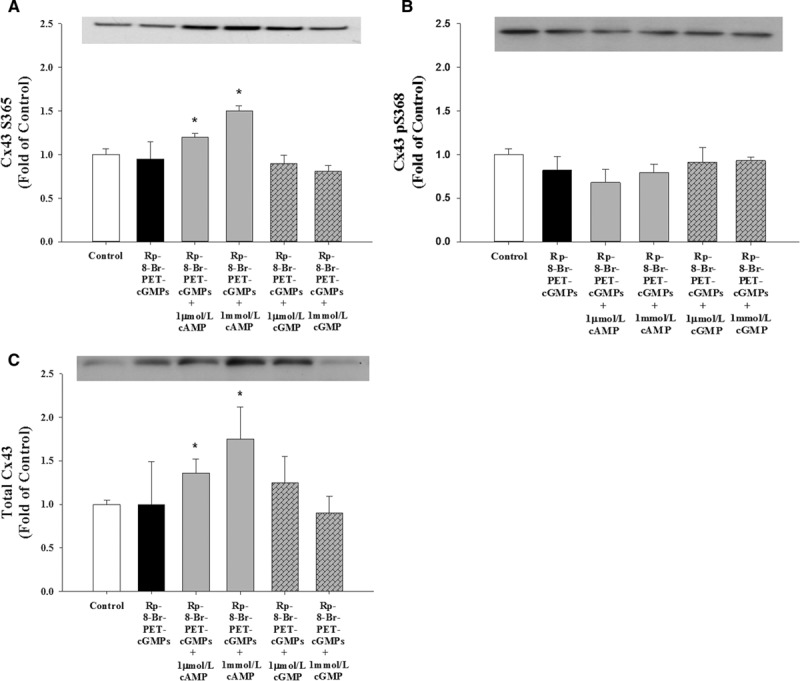
Role of PKG (protein kinase G) signaling pathways in Cx43 (connexin 43) phosphorylation in pregnant uterine artery endothelial cells (P-UAECs). Inhibition of PKG did not affect Cx43 phosphorylation. Western blot analysis of the effects of 10 μmol/L of PKG antagonist Rp-8-Br (8-Bromo)-PET-cGMPs (1 hour) on 1 μmol/L and 1 mmol/L 8-Br-cAMP-mediated and 8-Br-cGMP-mediated (30 minutes) changes of (A) nonphosphorylated Cx43 S365 levels detected by the Cx43 C-terminus (CT1) antibody, (B) Cx43 pS368 phosphorylation levels, and (C) total Cx43 levels. *, Increase (P<0.05, n=4) in Cx43 phosphorylation state vs untreated control.
Role of Cyclic Nucleotides Signaling Pathways in Regulating GJIC Function
Using Lucifer yellow dye transfer as a measure of GJIC and GJ functionality, we examined the role cyclic nucleotides play in GJIC. Under these conditions, there was no detectable difference in basal GJIC between NP-UAEC and P-UAEC (Figure 5A). P-UAECs incubated with 1 mmol/L 8-Br-cAMP (30 minutes) increased GJIC. However, selective inhibition of PKA using PKI, but not by PKG inhibitor Rp-8-Br-PET-cGMPs, completely abrogated GJIC stimulated by 8-Br-cAMP (Figure 5B). P-UAECs treated with 1 mmol/L 8-Br-cGMP decreased dye transfer (Figure 5C), but neither PKA nor PKG inhibitors (PKI and Rp-8-Br-PET-cGMPs) had an effect on 1 mmol/L 8-Br-cGMP-induced changes in GJIC (Figure 5C).
Figure 5.
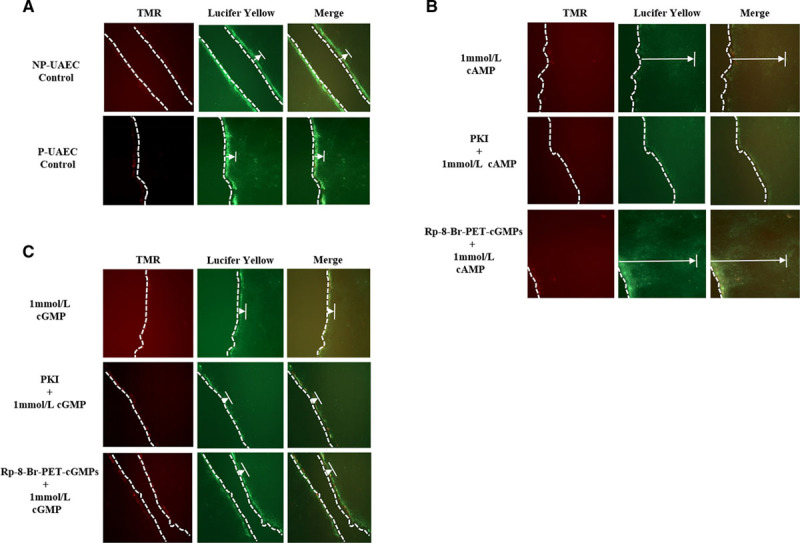
Effects of PKA and PKG (protein kinase A and G) inhibitors on cyclic nucleotide–induced gap junction intercellular communication (GJIC). PKA, but not PKG-mediated processes, increased Cx43 (connexin 43) Lucifer yellow dye transfer. Representative fluorescence dye microscopy images: (A) untreated control nonpregnant and pregnant uterine artery endothelial cells (NP-UAECs and P-UAECs), (B) P-UAECs pretreated with 10 μmol/L of either PKA inhibitor (PKI; 1 hour) or PKG inhibitor (Rp-8-Br [8-Bromo]-PET-cGMPs; 1 hour) then 1 mmol/L 8-Br-cAMP for 30 minutes, and (C) P-UAECs pretreated with 10 μmol/L of either PKA inhibitor (PKI; 1 hour) or PKG inhibitor (Rp-8-Br-PET-cGMPs; 1 hour) then 1 mmol/L of 8-Br-cGMP for 30 minutes. Left, Illustrates tetramethylrhodamine (TMR) dextran staining at the cell membrane. TMR dextran is too large to pass through gap junctions (GJs) and, thus, serves as a measure of cell damage at the wounding site and a marker for Lucifer yellow entry. Middle, Illustrates Lucifer yellow dye passing through GJs into adjacent cells. Right, An overlapping merged photo of the TMR dextran and Lucifer yellow dye images. Dotted lines (—) denote the wound site, whereas the extended arrows (|←) denote the distance Lucifer Yellow dye traveled from the wound site.
Cyclic Nucleotides Differentially Regulate Sustained-Phase [Ca2+]i Responses
In light of the Western analysis and direct GJIC studies above, we further investigated whether the cAMP- and cGMP-mediated control of Cx43 phosphorylation and GJIC is reflected by changes in ATP-induced sustained Ca2+ bursting that mediate the enhanced and sustained NO production in P-UAEC1,2 and subsequently the vasodilatory phenotype or P-UAECs that we recently defined.3 Although repeated doses of ATP itself show modest reductions in subsequent Ca2+ bursting by ≈10%, the pretreatment with cAMP completely abrogated this (Figure 6). Indeed, compared with control burst numbers, P-UAECs exposed to 1 μmol/L and 1 mmol/L 8-Br-cAMP increased the number of ATP-mediated sustained Ca2+ bursts by 5.4% and 27.9%, respectively (P<0.01). Conversely, treatment with 1 μmol/L 8-Br-cGMP led to a slight but not significant elevation (2.9%) in Ca2+ bursting, while 1 mmol/L 8-Br-cGMP significantly inhibited [Ca2+]i bursts by 11.5% less than control (P<0.05). This level is similar to ATP-mediated Ca2+ bursts previously observed in NP-UAECs,1 but greater than those seen when GAP27 was used to block Cx43-mediated activity and NO production.1,2
Figure 6.
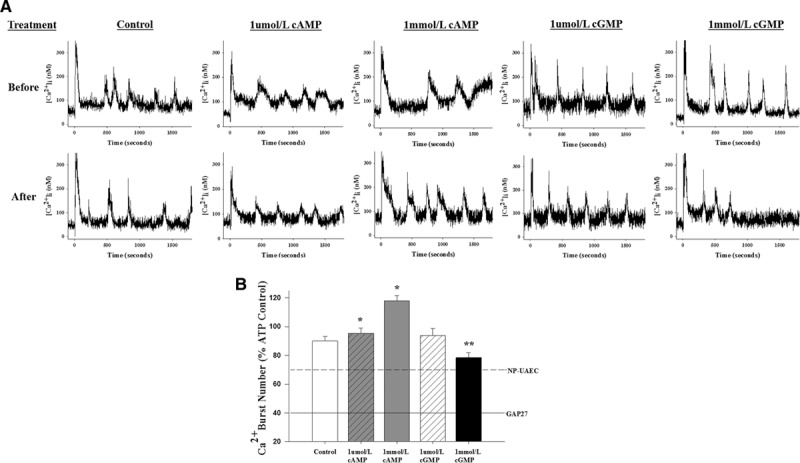
Effects of cyclic nucleotides on ATP-stimulated sustained [Ca2+]i bursts in pregnant uterine artery endothelial cells (P-UAECs). 8-Br (8-Bromo)-cAMP, but not 8-Br-cGMP, increases [Ca2+]i bursts in P-UAECs. A, Individual plots show representative [Ca2+]i single-cell tracings of P-UAECs in response to sequential stimulation with ATP (100 μM) in the absence or presence of 8-Br-cAMP or 8-Br-cGMP (1 μmol/L or 1 mmol/L). B, Quantitative analysis of [Ca2+]i bursts in P-UAECs responding to sequential stimulation with ATP (100 μmol/L; 30 minutes), washed, and restimulated with ATP again after a 30-minute treatment in the absence or presence of either 8-Br-cAMP or 8-Br-cGMP (1 μmol/L or 1 mmol/L). Data are restricted to cells showing ≥2 bursts on initial ATP stimulation and recorded as mean burst numbers±SEM. Data were gathered from 4 to 6 dishes with the individual cell count for control (n=196); 1 μmol/L cAMP (n=240); 1 mmol/L cAMP (n=246); 1 μmol/L cGMP (n=211); 1 mmol/L cGMP (n=231). Statistics were performed on raw data. *, Increases vs control (P<0.01). #, Decreases vs control (P<0.05). Comparisons of control vs different treatments are by rank-sum test. Dashed line indicates the equivalent level of Ca2+ bursts in nonpregnant uterine artery endothelial cells (NP-UAECs) on ATP stimulation,1 and solid line represents previously described Cx43 peptide inhibitor (43,37)Gap27 pre-exposure effects on ATP stimulation.1,2
Discussion
Previously, we demonstrated the importance of Cx43 function in establishing the enhanced pregnancy vasodilatory phenotype of UAECs during pregnancy that includes increases in cyclic nucleotides.1–3,7,13 This process is highlighted by the requirement of a functional Cx43 GJs for normal pregnancy-enhanced Ca2+ responses to increase NO production. Accumulating evidence has shown that cAMP plays an important regulatory role in Cx43 expression and functionality in many cell types,15 yet there are few reports investigating a role for cAMP versus cGMP in regulating Cx43 in endothelial cell function during the pregnancy state. In this report, we investigated the involvement of the classical cAMP/PKA and the lesser-studied cGMP/PKG signaling pathway on Cx43 phosphorylation states of sites S365 and S368 that are associated with GJ assembly/disassembly and gating5,14,16,24 and function in UAECs during pregnancy. The key findings of the present study are: (1) pregnancy-specific P-UAEC Cx43 phosphorylation state is differentially regulated by both cyclic nucleotides; (2) cAMP/PKA activation mediates a rise in CT1 nonphosphorylated Cx43 S365 signal; (3) cGMP/PKG pathway stimulates a greater rise in S368 phosphorylation; and (4) overall, PKA specifically regulates in vitro increases in ATP-stimulated GJIC function and sustained phase [Ca2+]i responses that are necessary for enhanced NO production in normal pregnancy, while PKG can oppose these actions through Cx43 S368 phosphorylation. Therefore, this study provides the first direct evidence for the mechanistic basis of the cAMP-mediated and cGMP-mediated signaling pathways opposing each other and that cAMP increases the rapid appearance of new Cx43 protein, for reasons that have yet to be determined, to regulate gap junctional communication in UAECs from the pregnant state.
In the current study, we were surprised to observe that in exposing UAECs to 8-Br-cAMP and 8-Br-cGMP, both cyclic nucleotides decreased S365 signaling in P-UAECs. However, only 8-Br-cAMP stimulated a net decrease in S365 signaling, especially in a pregnancy-specific fashion because this was not observed in NP-UAECs. Most reports studying 8-Br-cAMP actions on S365 signaling found an increase in S365 phosphorylation15,16,24 and not the net decrease as we observed. Others have reported that the CT1 Cx43 antibody preferentially recognizes predominantly nonphosphorylated S365 that is newly synthesized in the Golgi complex and not present in Cx43 GJ plaques in the plasma membrane.16,26 In this regard, our data show that 8-Br-cAMP caused a rapid appearance of newly synthesized nonphosphorylated Cx43 protein. We also observed that 8-Br-cAMP did not increase the S368 phosphorylation that previous reports have shown correlates with reduced GJIC, channel conductance, and half-life.13,14,20 This observation is consistent with other studies that demonstrated that 8-Br-cAMP did not have any effects on the PKC-sensitive phosphorylation site S368.27 Moreover, reports have described S365 as the gatekeeper that prevents S368 phosphorylation until S365 is dephosphorylated,24 thus, showing that phosphorylation and dephosphorylation events contribute to GJIC. Remarkably, because of the 8-Br-cAMP-stimulated decreases in S365 signal, we expected a corresponding increase in S368 phosphorylation; however, this was not the case. Of note, these present data in P-UAECs, however, is not fully consistent with this hypothesis. Using the CT1 Cx43 antibody, we provide direct evidence that 8-Br-cAMP promotes a substantial increase in total Cx43 and net decrease in S365 phosphorylation, but also shows little increase in absolute S368 phosphorylation and a concurrent decrease in overall S368 phosphorylation in P-UAECs.
This is the first report showing that 8-Br-cGMP can also increase the availability of newly released Cx43 in P-UAECs or in any cell type. However, we caution that we only saw this increase by 8-Br-cGMP with the higher 1 mmol/L dose. Selective kinase antagonist studies suggest that 8-Br-cGMP stimulation of PKA at these higher doses or cross talk through cGMP-mediated inhibition of phosphodiesterase PDE3, subsequently increasing cAMP levels, may have occurred.28 Cross talk between the different kinases via phosphodiesterases has been reported in consideration of the NO/cGMP signaling cascade.15,18,28Yao et al29 previously reported that activation of the NO/cGMP pathway increases GJIC and Cx43 expression acting through PKA activity. We also show that 8-Br-cGMP has less effect on new protein synthesis or overall S365 phosphorylation and yet increases Cx43 S368 phosphorylation beyond any effect of 8-Br-cAMP. Indeed, this increase in S368 phosphorylation parallels most closely the cGMP-stimulated rises in total Cx43. Thus, while an increase in newly synthesized proteins, presumably devoid of Cx43 S365 phosphorylation, may be available for S368 phosphorylation, this scenario would fit the gatekeeper hypothesis. The fact that both 8-Br-cAMP and 8-Br-cGMP can stimulate substantial increases in new Cx43 protein, but have different outcomes at pS368, suggests that the hypothesis that phosphorylation of sites S365 and S368 are mutually exclusive may not be the only determinant of Cx43 function in P-UAECs. Other studies have suggested that an initial PKA activation may even accelerate subsequent PKC phosphorylation of Cx43 in other cell types.30 However, considering our own data, this was not the case as it relates to our results in response to 8-Br-cAMP. It would seem that, even in the face of increased total Cx43 with both 8-Br-cAMP and 8-Br-cGMP, the maintenance of S368 phosphorylation by 8-Br-cGMP otherwise seen to decline with 8-Br-cAMP is enough to prevent any immediate functional advantage.
In addition to the role of cyclic nucleotides in Cx43 regulation, we also present evidence that specific kinases play an important role in Cx43 phosphorylation. Several reports have shown that increased cAMP increases Cx43 expression, functionality, and phosphorylation,15 and specifically PKA seems to mediate the increase in Cx43 expression.31 The finding that the PKA antagonist inhibits the effects of 8-Br-cAMP on S365, pS368, and total Cx43 confirms that changes in total Cx43 are PKA-mediated and possibly that mass action of such new protein synthesis is the single greatest force that underlies observed changes in phosphorylation at S365 and at S368. The present finding that 8-Br-cAMP also drives parallel increases in nonphosphorylated S365 and total Cx43 is in agreement with several reports that cAMP events are associated with an increased Cx43 export to the plasma membrane, incorporation into functional GJs, and open gating via PKA-sensitive mechanisms.16,18,19,31 The inability of 8-Br-cAMP to overcome PKA inhibition and return responses to levels higher than the control is consistent with PKI being an effective and specific inhibitor of PKA and further confirms that PKA also primarily regulates the basal state of Cx43 in P-UAECs. Interestingly, we also show that cGMP is not protective against PKA inhibition. As described earlier, cGMP may be able to compensate for cAMP inhibition in other cell types, but in P-UAECs, it would seem that normally PKA alone positively regulates Cx43. We also show that PKG inhibition had little or no effect on 8-Br-cAMP-induced changes in total Cx43 or Cx43 phosphorylation levels, but did block the effects of 8-Br-cGMP, including on S368 phosphorylation. This observation is consistent with reports that PKG activation has similar effects to PKC-dependent signaling on Cx43 expression and GJIC,20,23,27 suggesting cGMP negatively regulates Cx43 trafficking and GJ plaques in the plasma membrane in a manner not seen for cAMP.
Although 8-Br-cAMP and 8-Br-cGMP both independently increased Cx43 phosphorylation alone, it was only with 8-Br-cAMP that significantly decreased net S365 signal and increased ATP-mediated GJIC and Ca2+ bursts. Functionally, we previously reported that P-UAECs and NP-UAECs exhibit the same expression levels of Cx43, but that P-UAECs show greater ATP-stimulated Cx43 GJ function,1 which we confirm here in this study. Moreover, we have previously shown that inhibition of Cx43 completely abrogates ATP-induced Ca2+-mediated rises in NO production,1 a hallmark part of the pregnancy-enhanced vasodilatory phenotype.3 Several reports have shown that increased cAMP increases Cx43 functionality, as well as expression and phosphorylation,15 indicating that GJIC and Cx43 phosphorylation are tightly regulated. Specifically, PKA31 mediates increases in Cx43 expression, and other studies have reported that cAMP/PKA pathway regulates GJIC.15,18 We now demonstrate herein for the first time that cAMP/PKA signaling pathway regulates dye transfer and relative ATP-induced Ca2+ bursting in P-UAECs, which is consistent with studies where cAMP-stimulated Cx43 phosphorylation promotes greater GJIC and the number and size of GJ plaques.19 Consistent with other reports,13,15,16,19 treatments with 8-Br-cAMP increased GJIC; however, we show in the present study that PKA (not PKG) inhibition specifically abrogated cAMP-induced rises in GJIC. These data further confirm in P-UAECs that the classical cAMP/PKA signaling pathway primarily increases GJIC, perhaps, by way of both increased GJ open gating and new Cx43 protein availability and translocation through the Trans Golgi Network, without a substantial absolute or relative increase in Cx43 S365. Indeed, the overall slight reduction in S365 is paralleled by a greater decrease in inhibitory S368 phosphorylation, demonstrating that GJIC control by cAMP and cGMP is dependent on different site-specific Cx43 phosphorylation profiles. Thus, physiologically, PKA may be a proximal stimulus of endothelial vasodilatory phenotype and function because it may accelerate the relative delivery of newly available Cx43 to the plasma membrane and facilitate open gating, while free of Cx43 S368 inhibitory phosphorylation. Notably, PKG inhibition failed to exert noticeable effects on GJIC, while exogenous treatment with 8-Br-cGMP decreased GJIC below the pregnant control levels. Although 8-Br-cGMP has been observed to increase GJIC in other tissues,29 similar effects on GJIC were not observed in the current or others studies.21,27 It is noteworthy that the differences observed in 8-Br-cGMP-mediated GJIC may be because of treatment times. In our study, we used 8-Br-cGMP acutely for 30 minutes, whereas others reported27 observed increases in GJIC after a 24-hour period. The purpose of using a shorter time point was to avoid potential changes in Cx43 expression levels and to investigate if rapid phosphorylation of Cx43 coincided with changes in GJIC. In addition, although the 1 μmol/L dose of 8-Br-cAMP increased Cx43 phosphorylation, only the 1 mmol/L dose changed GJIC. This may be because of increased cross talk because of the higher concentration of cyclic nucleotides or possibly by a much smaller mass action effect in our cell type.
While the uterine artery endothelium expresses an abundance of Cx43 protein, Cx43 function is far more related to Cx43 phosphorylation state and location within GJs. Changes in Cx43 function controls communication between cells, and states of high Cx43 connectivity are a part of pregnancy adaptations of endothelial cell function.2,3 The biggest adaptations occur within the uterine artery endothelium where increases in ATP-stimulated sustained Ca2+ bursting and NO output occur. However, endothelium adaptations in the systemic and fetal arteries and veins may also occur to different degrees.13 Furthermore, hypertensive disorders of pregnancy such as preeclampsia are associated with a loss of sustained Ca2+ signaling in both the uterine and systemic vasculature13 and possibly a decrease in cyclic nucleotide production. Because the uterine artery endothelium exhibits pregnancy-specific endogenous rises in prostacyclin, NO, cAMP, and cGMP production compared with the systemic vasculature,3 this reinforces the importance of enhanced GJ function via cAMP, as described herein. Overall, our data suggest that 8-Br-cAMP causes a decrease in S365 signal because of a rapid appearance in newly synthesized Cx43 for reasons that have yet to be determined, which was exceeding PKA-mediated phosphorylation of existing Cx43. This study further implicates cAMP/PKA and cGMP/PKG pathways in the differential modification of Cx43 phosphorylation sites associated with the assembly/disassembly and gating of GJs, as well as linked to GJ communication and function.
Perspectives
The tightly regulated GJ cell–cell communication process by the actions of cAMP/PKA and cGMP/PKG pathways that inversely modulate both Cx43 phosphorylation and GJ functionality during pregnancy were specifically defined. Previously, studies demonstrated the importance of Cx43 function in regulating vasodilatory pathways, including ATP-induced Ca2+-mediated endothelial NO synthase activation and NO production. We report herein that the specific actions of cAMP, but not cGMP, mediate the vasodilatory phenotype in P-UAECs. These current data suggest that cAMP stimulates both the rapid increase in de novo Cx43 trafficking to the membrane and the open gating associated with a rise in GJIC, while cGMP serves to increase the capacity of P-UAECs to respond to stimuli, but not to an immediate increase in function. Such processes increase P-UAECs capacity to enhance vasodilatory phenotypes during pregnancy to maintain rises in uterine blood flow. Further direct studies of Cx43 trafficking, assembly, and function would be of value to identify how pregnancy-specific vasodilatory mechanisms fail in hypertensive diseases of pregnancy, such as preeclampsia.
Acknowledgments
We thank Mayra B. Pastore, Rosalina Villalon-Landeros, Vladimir E. Vargas, Chi Zhou, S. Omar Jobe, Jason L. Austin, Terrance M. Phernetton, Gladys E. Lopez, Cindy Goss, and Jessica H. Youngblood. We thank Dr Paul Lampe at the Fred Hutchinson Cancer Research Center for the Cx43 CT1-antibody.
Sources of Funding
This work was supported by National Institutes of Health (NIH) P01HD38843, HL49210, HL87144, HL117341 (R.R. Magness), R25-GM083252 (M.L. Carnes), and T32-HD041921 (I.M. Bird).
Disclosures
None.
Supplementary Material
Footnotes
Current Address for R.R. Magness: 12901 Bruce B Down Blvd MDC48, Tampa, FL 33612.
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.117.09113/-/DC1.
Novelty and Significance
What Is New?
cAMP/PKA (protein kinase A), but not cGMP/PKG (protein kinase G), regulate enhanced connexin 43 trafficking and function at the plasma membrane to increase the capacity for pregnancy in uterine artery endothelial cell cell–cell communication during pregnancy.
cAMP/PKA signaling pathway regulate the pregnancy-specific vasodilatory phenotype associated with elevations in ATP-induced Ca2+-mediated endothelial nitric oxide synthase phosphorylation and nitric oxide production.
What Is Relevant?
cAMP/PKA-mediated enhancement of gap junction intercellular communication is a uterine artery endothelial cells pregnancy–specific process to maintain nitric oxide–mediated elevations in uterine blood flow and other vasodilatory phenotypes. Understanding gap junction regulation gives more insight of the mechanisms controlling normal uterine blood flow during gestation, which may be dysfunctional in preeclampsia.
Summary
These findings demonstrate that in the uterine vasculature during pregnancy, cAMP and not cGMP signaling processes promote connexin 43 trafficking to the endothelial plasma membrane and gap junction open gating to increase gap junction intercellular communication associated with the enhanced pregnancy-specific vasodilatory phenotype to maintain sufficient uterine blood flow.
References
- 1.Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, Bird IM. Pregnancy enhances sustained Ca2+ bursts and endothelial nitric oxide synthase activation in ovine uterine artery endothelial cells through increased connexin 43 function. Biol Reprod. 2010;82:66–75. doi: 10.1095/biolreprod.109.078253. doi: 10.1095/biolreprod.109.078253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morschauser TJ, Ramadoss J, Koch JM, Yi FX, Lopez GE, Bird IM, Magness RR. Local effects of pregnancy on connexin proteins that mediate Ca2+-associated uterine endothelial NO synthesis. Hypertension. 2014;63:589–594. doi: 10.1161/HYPERTENSIONAHA.113.01171. doi: 10.1161/HYPERTENSIONAHA.113.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ampey BC, Morschauser TJ, Ramadoss J, Magness RR. Domain-specific partitioning of uterine artery endothelial connexin43 and caveolin-1. Hypertension. 2016;68:982–988. doi: 10.1161/HYPERTENSIONAHA.116.08000. doi: 10.1161/HYPERTENSIONAHA.116.08000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodenough DA, Paul DL. Gap junctions. Cold Spring Harb Perspect Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 7.Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J, Magness RR. Pregnancy-dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology. 2000;141:1107–1117. doi: 10.1210/endo.141.3.7367. doi: 10.1210/endo.141.3.7367. [DOI] [PubMed] [Google Scholar]
- 8.Magness RR, Zheng J. Maternal cardiovascular alterations during pregnancy. In: Gluckman PD, Heymann MA, editors. In: Pediatrics and Perinatology: The Scientific Basis. 2nd ed. London: Arnold Publishing; 1996. pp. 762–772. [Google Scholar]
- 9.Rummery NM, Hill CE. Vascular gap junctions and implications for hypertension. Clin Exp Pharmacol Physiol. 2004;31:659–667. doi: 10.1111/j.1440-1681.2004.04071.x. doi: 10.1111/j.1440-1681.2004.04071.x. [DOI] [PubMed] [Google Scholar]
- 10.Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelium-dependent contractions and endothelial dysfunction in human hypertension. Br J Pharmacol. 2009;157:527–536. doi: 10.1111/j.1476-5381.2009.00240.x. doi: 10.1111/j.1476-5381.2009.00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumwell S, Karumanchi SA. Pre-eclampsia: clinical manifestations and molecular mechanisms. Nephron Clin Pract. 2007;106:c72–c81. doi: 10.1159/000101801. doi: 10.1159/000101801. [DOI] [PubMed] [Google Scholar]
- 12.Roberts JM, Taylor RN, Goldfien A. Clinical and biochemical evidence of endothelial cell dysfunction in the pregnancy syndrome preeclampsia. Am J Hypertens. 1991;4:700–708. doi: 10.1093/ajh/4.8.700. [DOI] [PubMed] [Google Scholar]
- 13.Ampey BC, Morschauser TJ, Lampe PD, Magness RR. Gap junction regulation of vascular tone: implications of modulatory intercellular communication during gestation. Adv Exp Med Biol. 2014;814:117–132. doi: 10.1007/978-1-4939-1031-1_11. doi: 10.1007/978-1-4939-1031-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384:205–215. doi: 10.1006/abbi.2000.2131. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 15.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–1429. doi: 10.1016/j.febslet.2014.01.049. doi: 10.1016/j.febslet.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yogo K, Ogawa T, Akiyama M, Ishida-Kitagawa N, Sasada H, Sato E, Takeya T. PKA implicated in the phosphorylation of Cx43 induced by stimulation with FSH in rat granulosa cells. J Reprod Dev. 2006;52:321–328. doi: 10.1262/jrd.17107. [DOI] [PubMed] [Google Scholar]
- 19.TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwak BR, Sáez JC, Wilders R, Chanson M, Fishman GI, Hertzberg EL, Spray DC, Jongsma HJ. Effects of cGMP-dependent phosphorylation on rat and human connexin43 gap junction channels. Pflugers Arch. 1995;430:770–778. doi: 10.1007/BF00386175. [DOI] [PubMed] [Google Scholar]
- 22.Rörig B, Sutor B. Nitric oxide-stimulated increase in intracellular cGMP modulates gap junction coupling in rat neocortex. Neuroreport. 1996;7:569–572. doi: 10.1097/00001756-199601310-00046. [DOI] [PubMed] [Google Scholar]
- 23.Kwak BR, Jongsma HJ. Regulation of cardiac gap junction channel permeability and conductance by several phosphorylating conditions. Mol Cell Biochem. 1996;157:93–99. doi: 10.1007/BF00227885. [DOI] [PubMed] [Google Scholar]
- 24.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.el-Fouly MH, Trosko JE, Chang CC. Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res. 1987;168:422–430. doi: 10.1016/0014-4827(87)90014-0. [DOI] [PubMed] [Google Scholar]
- 26.Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem J. 2007;408:375–385. doi: 10.1042/BJ20070550. doi: 10.1042/BJ20070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi CN, Martin DN, Shaver P, Madamanchi C, Muller-Borer BJ, Tulis DA. Control of vascular smooth muscle cell growth by connexin 43. Front Physiol. 2012;3:220. doi: 10.3389/fphys.2012.00220. doi: 10.3389/fphys.2012.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37:671–681. [PubMed] [Google Scholar]
- 29.Yao J, Hiramatsu N, Zhu Y, Morioka T, Takeda M, Oite T, Kitamura M. Nitric oxide-mediated regulation of connexin43 expression and gap junctional intercellular communication in mesangial cells. J Am Soc Nephrol. 2005;16:58–67. doi: 10.1681/ASN.2004060453. doi: 10.1681/ASN.2004060453. [DOI] [PubMed] [Google Scholar]
- 30.Shah MM, Martinez AM, Fletcher WH. The connexin43 gap junction protein is phosphorylated by protein kinase A and protein kinase C: in vivo and in vitro studies. Mol Cell Biochem. 2002;238:57–68. doi: 10.1023/a:1019902920693. [DOI] [PubMed] [Google Scholar]
- 31.Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF, Johnson RG. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113(pt 17):3037–3049. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]