

Abstract
Some human populations interbred with Neanderthals and Denisovans, resulting in substantial contributions to modern-human genomes. Therefore, it is now possible to use genomic data to investigate mechanisms that shaped historical gene flow between humans and our closest hominin relatives. More generally, in eukaryotes, mitonuclear interactions have been argued to play a disproportionate role in generating reproductive isolation. There is no evidence of mtDNA introgression into modern human populations, which means that all introgressed nuclear alleles from archaic hominins must function on a modern-human mitochondrial background. Therefore, mitonuclear interactions are also potentially relevant to hominin evolution. We performed a detailed accounting of mtDNA divergence among hominin lineages and used population-genomic data to test the hypothesis that mitonuclear incompatibilities have preferentially restricted the introgression of nuclear genes with mitochondrial functions. We found a small but significant underrepresentation of introgressed Neanderthal alleles at such nuclear loci. Structural analyses of mitochondrial enzyme complexes revealed that these effects are unlikely to be mediated by physically interacting sites in mitochondrial and nuclear gene products. We did not detect any underrepresentation of introgressed Denisovan alleles at mitochondrial-targeted loci, but this may reflect reduced power because locus-specific estimates of Denisovan introgression are more conservative. Overall, we conclude that genes involved in mitochondrial function may have been subject to distinct selection pressures during the history of introgression from archaic hominins but that mitonuclear incompatibilities have had, at most, a small role in shaping genome-wide introgression patterns, perhaps because of limited functional divergence in mtDNA and interacting nuclear genes.
Keywords: coevolution, cytonuclear interactions, hybridization, human evolution, OXPHOS enzymes, speciation
Introduction
Understanding the mechanisms that lead to speciation is one of the defining challenges in the field of evolutionary biology, and numerous causes of reproductive isolation have been identified (Dobzhansky 1937; Coyne and Orr 2004). Natural curiosity about our own origins has spurred debate about how these mechanisms have shaped the evolution and diversification of hominins (Stringer 2002; Wood and Boyle 2016).
The advent of technologies to extract and sequence ancient DNA samples and the rapid increase in availability of population genomic data have revealed that modern-human populations that expanded out of Africa subsequently underwent hybridization and genetic admixture with related hominin lineages that were already present in Asia and Europe (Green et al. 2010; Reich et al. 2010; Meyer et al. 2012; Prüfer et al. 2014; Sankararaman et al. 2014, 2016; Vernot and Akey 2014; Vernot et al. 2016). Interbreeding with Neanderthals resulted in substantial introgression into modern human populations such that ∼2–4% of genome content in non-African individuals is now derived from Neanderthals (Green et al. 2010). Additional interbreeding appears to have occurred between modern humans and Denisovans, an enigmatic lineage of hominins known only from limited fossil remains found in Denisova Cave in the Altai Mountains of Siberia (Krause et al. 2010; Sawyer et al. 2015). Denisovan-derived alleles make up a substantial portion (up to ∼5%) of the genomes of modern humans from Melanesia and surrounding regions but appear to have made little or no contribution to other populations (Reich et al. 2010).
There is large variation across the human genome in the extent of introgression from Neanderthals and Denisovans (Sankararaman et al. 2014, 2016; Vernot and Akey 2014; Vernot et al. 2016), which is a common theme in hybridizing organisms (Baack and Rieseberg 2007). Substantial variation is expected even under neutral models of introgression, but differences can be magnified by selection for or against certain foreign alleles (Harrison and Larson 2014, 2016; Payseur and Rieseberg 2016; Schumer and Brandvain 2016). Much of the recent interest in introgression from archaic hominins has focused on loci with unexpectedly high frequencies of foreign alleles because they provide evidence that modern human populations acquired beneficial genetic variants from Neanderthals and Denisovans in the process of adapting to novel environments outside of Africa (Racimo et al. 2015).
Even so, regions of the genome that exhibit atypically low rates of introgression are also noteworthy, as they present the opportunity to infer causes of restricted gene flow and to draw parallels with more general mechanisms of reproductive isolation. For example, Neanderthal- and Denisovan-derived alleles are substantially underrepresented on the modern human X-chromosome, suggesting that X-linked loci might have contributed to early stages of reproductive isolation among hominin lineages (Sankararaman et al. 2016). This “large X-effect” on hybrid inviability and sterility is a common pattern from the field of speciation genetics (Presgraves 2008) and is often attributed to the exposure of recessive alleles because of hemizygosity of X-linked genes in males (Qvarnström and Bailey 2009; cf.Hu and Filatov 2016). In addition, there is evidence that levels of introgression from archaic hominins are lower for genes near structural rearrangements (Rogers 2015) and for genes that are preferentially expressed in the testes (Sankararaman et al. 2016). These patterns are consistent with findings from other organisms that structural rearrangements can act as a barrier to gene flow (Rieseberg et al. 1999; Rieseberg 2001; Feder et al. 2003; Lowry and Willis 2010) and that testis-specific genes are often involved in hybrid male infertility (Wu and Davis 1993; Presgraves 2008).
Another hypothesis that has emerged from the study of reproductive isolation in eukaryotes is that mitonuclear incompatibilities play a disproportionate role in the creation of species boundaries because hybridization can disrupt coadapted combinations of mitochondrial and nuclear alleles (reviewed in Gershoni et al. 2009; Burton and Barreto 2012; Hill 2016; Sloan et al. 2017). Mitonuclear coadaptations are thought to be driven by the rapid evolution of the mitochondrial genome and subsequent coevolutionary responses in nuclear genes that encode mitochondrially targeted proteins (N-mt genes) (Osada and Akashi 2012; Havird et al. 2017). Although mitochondria retain their own genome, the vast majority of the >1000 proteins that function in mitochondria are encoded in the nucleus (Gray 2015). Coevolutionary dynamics are expected to be the most intense for N-mt proteins that interact directly with mitochondrial gene products in multisubunit complexes such as oxidative phosphorylation (OXPHOS) enzymes and mitochondrial ribosomes (Rand et al. 2004). By incorporating models of molecular structures, various studies have investigated the possibility that physically interacting residues in mitochondrial- and nuclear-encoded subunits can undergo coevolutionary changes that lead to coadapted mitonuclear genotypes (Schmidt et al. 2001; Melvin et al. 2008; Osada and Akashi 2012; Aledo et al. 2014; Havird et al. 2015). However, researchers have only recently begun using genomic data sets to examine the effect of selection on maintaining associations between coadapted combinations of mitochondrial haplotypes and N-mt alleles during genetic admixture/hybridization (Trier et al. 2014; Bar-Yaacov et al. 2015; Beck et al. 2015; Sloan et al. 2015; McKenzie et al. 2016; Baris et al. 2017; Morales et al. 2017; Runemark et al. 2017). These studies have produced mixed results about the extent to which selection can preferentially act on N-mt loci relative to the rest of the nuclear genome. More generally, the hypothesis that mitonuclear interactions play a common or disproportionate role in speciation remains controversial (Eyre-Walker 2017) and seemingly at odds with evidence of widespread introgression of mtDNA in many hybrid zones (Sloan et al. 2017). Therefore, addressing this hypothesis is a pressing biological challenge.
The potential for mitonuclear interactions among hominins is of interest because, unlike in the nuclear genome, there has not been any detectable mtDNA introgression from Neanderthals or Denisovans into modern human populations (Krings et al. 1997; Serre et al. 2004). Regardless of what has caused this lack of mtDNA introgression, one consequence is that all introgressed Neanderthal and Denisovan nuclear alleles must function on a modern-human mitochondrial background, and it has been proposed that divergence in mitochondrial gene products between modern humans and archaic hominins may have altered physical interactions with N-mt proteins (Green et al. 2008). Accordingly, if coadapted combinations of mitochondrial and nuclear alleles have evolved within hominin lineages, incompatibilities between modern-human mtDNA and Neanderthal/Denisovan nuclear alleles may have preferentially restricted introgression at nuclear loci that encode N-mt proteins and are involved in mitonuclear interactions. Here, we investigate the hypothesis that mitonuclear incompatibilities have contributed to the earliest stages of reproductive isolation between modern humans and related hominins and shaped patterns of genetic admixture following hybridization.
Materials and Methods
Analysis of mtDNA Polymorphism and Divergence
To compare patterns of molecular evolution in mtDNA across hominin lineages, we obtained published mitochondrial genome sequences for 6 Neanderthal samples (NCBI accessions FM865407-FM865411 and AM948965), 3 Denisovan samples (KT780370, FN673705, FR695060), and 54 samples from globally diverse lineages of modern humans (AF346963-AF347015, NC_012920), which included the revised Cambridge Reference Sequence (Andrews et al. 1999) and the full sampling performed by Ingman et al. (2000). We also included the mitochondrial genome sequence (NC_023100) from an ancient hominin (∼400,000 years old) that was recovered from the Sima de los Huesos (SdlH) site in Spain (Meyer et al. 2014). Nuclear, morphological, and geographical data indicate that the SdlH sample is an early representative of the Neanderthal lineage (Meyer et al. 2016), but its mitochondrial haplotype is more closely related to Denisovan mtDNA (Meyer et al. 2014). The SdlH sample is not a primary focus of this study, but we included it in our mtDNA analysis to achieve a thorough sampling of available mitochondrial genomes from archaic hominins.
We aligned all sampled mitochondrial genome sequences with MAFFT v7.305b, using the FFT-NS-i algorithm with a maximum of 1000 iterations (supplementary file S1, Supplementary Material online) (Katoh and Standley 2013). After visual inspection and manual alignment editing, we used a custom Python script (available at https://github.com/jsharbrough/polymorphismsSubstitutions) to identify polymorphisms segregating within Neanderthals, Denisovans, and modern humans as well as fixed differences among the sampled hominin lineages (treating SdlH as a separate lineage; supplementary file S2, Supplementary Material online). To account for sites with multiple hits, we assumed that the number of changes was the minimum required to explain the data. In protein-coding sequences, we further assumed that any codons with multiple hits resulted from the minimum number of nonsynonymous changes needed to explain the data. We generated a pairwise mismatch distribution by quantifying the number of nucleotide substitutions between each modern human mitochondrial haplotype and each other modern human, Neanderthal, or Denisovan sample from the above alignment.
The primary goal of our mtDNA analysis was to distinguish ancestral divergence between modern humans and related hominin lineages from more recent changes segregating within these populations. For the purposes of this analysis, we considered variants to be fixed differences if they were not segregating in our sample of 54 modern human mitochondrial genomes even if they have been identified as low-frequency variants in larger samples of human mitochondrial diversity. To assess the effects of this sampling approach, we repeated the analysis of sequence polymorphism and divergence using two larger modern human mtDNA data sets: 114 mitochondrial genomes from Mishmar et al. (2003) and 9862 mitochondrial genomes from Levin et al. (2013). We also directly compared Neanderthal and Denisovan sequences to a reconstruction of the ancestral modern human mtDNA sequence from Behar et al. (2012).
Neanderthal Introgression Analysis
To test the hypothesis that mitonuclear incompatibilities have preferentially restricted the introgression of N-mt alleles from Neanderthals, we used the “introgression score” calculated by Sankararaman et al. (2014), with raw gene-specific values kindly provided by the authors of the original study (S. Sankararaman, pers. comm.). The introgression score was determined from a conditional random field analysis and based on the marginal probability of Neanderthal ancestry averaged across the entire length of a gene and across all sampled individuals, which were taken from the 1000 Genomes Project (1000 Genomes Project Consortium 2012). Distinct estimates were produced for European populations, Asian populations, and a combined European/Asian data set in the original analysis (Sankararaman et al. 2014). We used the set of high-confidence N-mt genes (“Tmito”) in the MitoCarta 2.0 classification (Pagliarini et al. 2008) to compare rates of introgression against all other nuclear protein-coding genes. We separated these high-confidence N-mt genes into two groups, distinguishing a subset of 150 genes that encode proteins that interact with mitochondrial gene products in multisubunit OXPHOS and ribosomal complexes from the rest of the N-mt gene set (following the classification scheme in Sloan et al. 2015). Differences in rates of introgression among gene classes were assessed with nonparametric Kruskal–Wallis rank sum tests.
To assess whether differences in levels of introgression may reflect a correlation between mitochondrial targeting and overall functional importance, we used two proxies for the functional importance of each gene. First, we used estimates of transcript abundance based on the premise that highly expressed genes are more functionally constrained (Zhang and Yang 2015). We obtained two separate measures of transcript abundance by averaging across 16 different human tissues from the Illumina Body Map data set (https://www.ebi.ac.uk/gxa/experiments/E-MTAB-513) and by averaging across median RPKM values from each of 53 different human tissues from the GTEx V6p release data set (The GTEx Consortium 2013). Second, we used gene-specific estimates for rates of nonsynonymous substitutions (dN) between humans and chimpanzees (The Chimpanzee Sequencing and Analysis Consortium 2005), with the expectation that slower rates of protein evolution are indicative of greater functional importance. Data sets were log10-transformed after adding 0.001 to introgression scores and dN values and adding 0.01 to mean expression values. After transformation, Pearson correlation coefficients were calculated between introgression score and each of these proxies for functional importance. We also extracted residuals from linear models and used them to repeat the above Kruskal–Wallis tests as a means for controlling for effects of expression or rate of sequence evolution. All statistical analyses were implemented in R v3.1.2 (R Core Team 2014), and all data plots were generated with the ggplot2 package (Wickham 2009). Introgression data are provided in (supplementary file S3, Supplementary Material online).
Denisovan Introgression Analysis
To perform a similar analysis of introgression from Denisovans, we used haplotype calls from a study of 35 Melanesian genomes (Vernot et al. 2016). The physical locations of individual Denisovan-derived haplotypes were obtained from supporting data sets posted on the personal website of one of the authors of the original study (http://akeylab.gs.washington.edu/vernot_et_al_2016_release_data/). We calculated frequencies of Denisovan-derived alleles for each nucleotide position in the hg19 human genome assembly (i.e., anywhere from 0 to 70 of the haplotypes from the 35 diploid Melanesian genomes). Gene-specific estimates were then calculated by averaging frequencies across the full length of each gene (supplementary file S3, Supplementary Material online). Using the same statistical approach described earlier for Neanderthal introgression, we compared rates of introgression among different gene classes. To assess the consequences of methodological differences between the Neanderthal (Sankararaman et al. 2014) and Denisovan (Vernot et al. 2016) data sets, we repeated our analyses using haplotype calls for Neanderthal-derived alleles in East Asian and European populations that were produced with the same methodology as the Denisovan haplotype predictions (supplementary file S3, Supplementary Material online; Vernot et al. 2016).
Identification of Contact Residues within the Structures of Mitonuclear Enzyme Complexes
To assess whether fixed differences between modern humans and ancient hominin lineages in mitochondrially encoded proteins and rRNAs may have affected mitonuclear interactions by altering interfaces with N-mt proteins, we mapped substitutions onto published mammalian structural models of OXPHOS complexes and the mitochondrial ribosome. The PDB accessions used for NADH dehydrogenase, cytochrome bc1, cytochrome c oxidase, ATP synthase, and the mitochondrial ribosome were 5LNK, 1BGY, 1V54, 5ARA, and 3J9M, respectively (Iwata et al. 1998; Tsukihara et al. 2003; Amunts et al. 2015; Zhou et al. 2015; Fiedorczuk et al. 2016). We used UCSF Chimera v1.11.2 (Pettersen et al. 2004) to visualize structures and identify mitonuclear contact residues, that is, those that occur at the interfacing surface between proteins or rRNAs encoded by the mitochondrial and nuclear genomes. To identify contact residues, we used the “find clashes/contacts” tool with default parameters, except that the minimum overlap was set to ≥ −1 Å to increase sensitivity.
Analysis of Sequence Divergence in N-mt Genes
To identify substitutions in N-mt genes encoding proteins that contact variable sites in mitochondrial subunits, we used genotype calls (relative to hg19) from a representative nuclear genome sequence from Neanderthals (Prüfer et al. 2014), Denisovans (Meyer et al. 2012), and modern humans (HuRef; Levy et al. 2007). We obtained RefSeq sequences for each N-mt gene from the UCSC genome browser (http://genome.ucsc.edu/) and extracted FASTA sequences from VCF files corresponding to the Neanderthal (http://cdna.eva.mpg.de/neandertal/altai/AltaiNeandertal/VCF) and Denisovan (http://cdna.eva.mpg.de/denisova/VCF/hg19_1000g/) data sets, using a custom tool called vcf2Fasta in the seqTools_v2.2 package (https://github.com/jsharbrough/seqTools). Variants were filtered based on basecall quality (≥Q30) and depth (≥10×). We mapped N-mt RefSeq genes to the HuRef alternate genome assembly (ftp://ftp.ncbi.nih.gov/genomes/Homo_sapiens/ARCHIVE/BUILD.37.3/Assembled_chromosomes/seq/) using BWA-MEM v0.7.15-r1140 (Li and Durbin 2010) with pairing and mate-pair rescuing turned off. We used these mapping data to determine N-mt gene positions within the HuRef genome and extracted the specified regions using the samtools faidx function implemented in SAMtools v1.3.1 (Li et al. 2009).
Protein-coding N-mt gene sequences were extracted from the mapped Neanderthal, Densiovan, and HuRef sequences based on annotated RefSeq exon coordinates. We then used MAFFT v7.305b to align these sequences with the FFT-NS-i algorithm and a maximum of 1000 iterations (supplementary file S1, Supplementary Material online). We applied the same custom Python package that was used for the mtDNA comparative analysis to identify all homozygous substitutions among the three hominin samples. Identified variants were cross-referenced against dbSNP (Sherry et al. 2001) to determine whether they were segregating within modern human populations, using dbSNP identifiers from the original VCF files. For variants that were unique to the Neanderthal sample, we assessed whether they were fixed within Neanderthals by comparing against additional Neanderthal genomic data sets (Green et al. 2010). UCSF Chimera was used to classify identified variants in N-mt proteins as either contact or noncontact sites as described earlier.
Results
mtDNA Polymorphism and Divergence in Modern Human, Neanderthals, and Denisovans
The original publication of Neanderthal and Denisovan mitochondrial genomes revealed that the level of sequence divergence between modern humans and these related hominins greatly exceeds the divergence between any pair of extant human mitochondrial haplotypes (fig. 1; Green et al. 2008; Krause et al. 2010). In addition, these sequencing efforts revealed that mitochondrial haplotypes of modern humans and Neanderthals share a more recent common ancestor with each other than with Denisovans (Krause et al. 2010), which is reflected in the much greater pairwise divergences with Denisovans (fig. 1 and table 1). Although early efforts were made to describe the functional changes in the mitochondrial genome that separate Neanderthals and modern humans (Green et al. 2008), it is worth revisiting these analyses in light of the subsequent increase in the availability of mitochondrial genomes from Neanderthals and Denisovans.
Fig. 1.
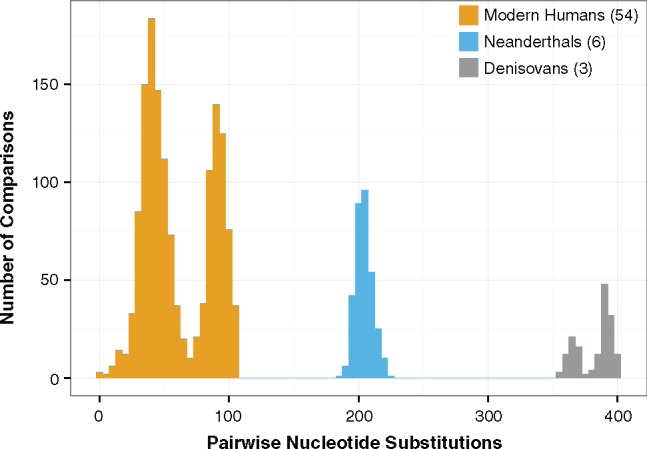
—Pairwise mismatch distribution for human–human (orange), human–Neanderthal (blue), and human–Denisovan (gray) comparisons of complete mtDNA sequences. This plot replicates previous findings that the sequence divergence between modern humans and archaic hominins is greater than the divergence between any pair of extant modern human mitochondrial haplotypes (Green et al. 2008) and that divergence between modern humans and Denisovans greatly exceeds the divergence between modern humans and Neanderthals (Krause et al. 2010). The number of genomes used from each group is indicated in parentheses. The bimodality in the modern-human distribution results from the recent expansion and diversification of human populations after leaving Africa. The first peak primarily consists of Eurasian versus Eurasian comparisons, whereas the second peak consists of African versus Eurasian and African versus African comparisons. The bimodality in the human–Denisovan distribution reflects the fact that one of recovered Denisovan mtDNA samples was from a fossil that was substantially older (and therefore less divergent in sequence from modern-human haplotypes) than the other two (Sawyer et al. 2015).
Table 1.
Fixed Nucleotide Substitutions between Modern Humans (MH), Neanderthals (Nean), and Denisovans (Den)
| Gene/region | Type | Length (aligned) | MH—Nean | MH—Den | Nean—Den |
|---|---|---|---|---|---|
| ATP6 | Protein | 681 | 4 (2) | 8 (3) | 12 (5) |
| ATP8 | Protein | 207 | 5 (1) | 7 (1) | 5 (0) |
| COI | Protein | 1,542 | 11 (0) | 18 (1) | 26 (2) |
| COII | Protein | 684 | 5 (3) | 18 (4) | 14 (2) |
| COIII | Protein | 784 | 5 (2) | 15 (2) | 17 (2) |
| CYTB | Protein | 1,141 | 13 (4) | 20 (3) | 26 (6) |
| ND1 | Protein | 956 | 6 (1) | 16 (1) | 21 (2) |
| ND2 | Protein | 1,044 | 7 (1) | 19 (2) | 18 (4) |
| ND3 | Protein | 346 | 6 (1) | 7 (1) | 9 (0) |
| ND4 | Protein | 1,378 | 6 (0) | 14 (0) | 22 (1) |
| ND4L | Protein | 297 | 2 (1) | 4 (1) | 5 (0) |
| ND5 | Protein | 1,812 | 15 (6) | 32 (5) | 41 (6) |
| ND6 | Protein | 525 | 1 (0) | 6 (0) | 9 (1) |
| RNR1 | rRNA | 954 | 1 | 5 | 11 |
| RNR2 | rRNA | 1,561 | 4 | 9 | 9 |
| TRNA | tRNA | 69 | 0 | 0 | 0 |
| TRNC | tRNA | 67 | 0 | 0 | 0 |
| TRND | tRNA | 68 | 0 | 0 | 0 |
| TRNE | tRNA | 69 | 0 | 1 | 1 |
| TRNF | tRNA | 71 | 0 | 1 | 1 |
| TRNG | tRNA | 68 | 0 | 0 | 0 |
| TRNH | tRNA | 69 | 1 | 2 | 1 |
| TRNI | tRNA | 69 | 0 | 1 | 1 |
| TRNK | tRNA | 70 | 0 | 0 | 0 |
| TRNL1 | tRNA | 75 | 0 | 1 | 2 |
| TRNL2 | tRNA | 71 | 0 | 1 | 1 |
| TRNM | tRNA | 68 | 0 | 1 | 1 |
| TRNN | tRNA | 73 | 1 | 0 | 1 |
| TRNP | tRNA | 68 | 0 | 0 | 0 |
| TRNQ | tRNA | 72 | 0 | 0 | 0 |
| TRNR | tRNA | 65 | 0 | 0 | 0 |
| TRNS1 | tRNA | 69 | 0 | 0 | 0 |
| TRNS2 | tRNA | 59 | 0 | 0 | 0 |
| TRNT | tRNA | 66 | 0 | 1 | 1 |
| TRNV | tRNA | 69 | 0 | 0 | 0 |
| TRNW | tRNA | 68 | 0 | 2 | 2 |
| TRNY | tRNA | 66 | 1 | 1 | 0 |
| D-Loop | Noncoding | 1,142 | 10 | 20 | 40 |
| Other intergenic | Noncoding | 95 | 1 | 0 | 1 |
| Whole genome | 16,599 | 105 (22) | 229 (24)a | 297 (31)a |
Note.—For coding regions, nonsynonymous changes are in parentheses. Corresponding divergences with the SdlH sample are reported in supplementary table S1, Supplementary Material online.
The sum of individual gene counts exceeds the whole genome count for comparisons involving Denisovans because of a substitution in a region of overlap between ATP6 and ATP8.
Despite the rapid rate of sequence evolution in mtDNA, the number of fixed nonsynonymous changes between modern humans and related hominins is small (table 1). Only 22 fixed amino acid substitutions distinguish modern humans and Neanderthals in the set of 13 protein-coding sequences found in the mitochondrial genome. Surprisingly, comparisons between modern humans and Denisovans revealed a nearly identical level of divergence in amino acid sequences (24 fixed differences; table 1) even though overall mtDNA sequence divergence is nearly twice as high (fig. 1). This pattern is consistent with previous observations that the ratio of nonsynonymous to synonymous changes (dN/dS) is higher in Neanderthals and modern humans than in other branches of the hominin tree (see supplementary fig. 5 in Krause et al. 2010). Comparisons involving modern humans show smaller numbers of fixed differences (table 1), in part because many variants are polymorphic within modern humans (supplementary table S2, Supplementary Material online).
Individual protein-coding genes have anywhere from zero to six fixed nonsynonymous changes between modern humans and archaic hominins (table 1). Total sequence divergence is even lower in structural RNA genes (tRNAs and rRNAs) than in protein-coding genes (table 1). There was also variation within these structural RNA genes, as the frequency of fixed substitutions was significantly different across the various structural regions within tRNAs (P = 3.8 × 10−5; Fisher’s Exact Test), with most changes found in the TΨC loop (fig. 2 and supplementary table S3, Supplementary Material online). Not surprisingly, changes were generally not observed at sites known to be crucial for tRNA identity and recognition by the nuclear-encoded aminoacyl-tRNA synthetases (aaRSs) that charge tRNAs with the proper amino acid. However, the identified variant in TRNL2 has been found to affect charging rates (Hao et al. 2005), and TRNW has experienced a change in the acceptor stem, which is typically involved in aaRS interactions (fig. 2). Although none of these tRNA substitutions were segregating in our sample of 54 modern human mitochondrial genomes, some of these variants have been identified at low frequencies in human populations or in heteroplasmic individuals and have been associated with disease phenotypes (Zhu et al. 2009; Lu et al. 2010).
Fig. 2.

—The secondary structure of all mitochondrial tRNAs that have fixed differences distinguishing modern humans from Neanderthals (red), Denisovans (blue), or both (purple). The primary sequences are from the revised Cambridge Reference Sequence (Andrews et al. 1999), and variants in archaic hominins are indicated next to each variable site. Secondary structures were generated with VARNA v3-93 based on published structural models (Darty et al. 2009).
For the purposes of this analysis, variants were classified as fixed differences if they were not segregating within our samples of 54 modern humans, 6 Neanderthals, and 3 Denisovans. We investigated the effects of sampling depth on our estimates by repeating analyses with additional human mtDNA data sets of varying sizes. As expected, performing comparisons directly against the reconstructed ancestral sequence for modern humans (Behar et al. 2012) led to in an increased number of fixed differences because some segregating polymorphisms are the result of derived, homoplasious changes within the modern-human genealogy (supplementary tables S4 and S5, Supplementary Material online). In accordance with this pattern, using larger samples of human mtDNA diversity (Mishmar et al. 2003; Levin et al. 2013) led to reclassification of some fixed differences as segregating polymorphisms (supplementary tables S4 and S5, Supplementary Material online). For example, we found 26 fixed nonsynonymous differences between the reconstructed ancestral modern-human sequence and Neanderthals, and this value declined to 22, 17, and 1, when using modern-human data sets consisting of 54, 114, and 9862, respectively.
Underrepresentation of N-mt Genes in Regions of Neanderthal Introgression
If mitonuclear incompatibilities lowered reproductive fitness in hybrids, then the nuclear loci involved in these incompatibilities should have experienced reduced introgression relative to background levels in the genome. We reasoned that such effects should preferentially involve N-mt genes, so we used gene-specific estimates of introgression from Neanderthals (Sankararaman et al. 2014) to compare N-mt genes against all other nuclear genes. We found that N-mt genes have undergone significantly less introgression into both Asian (P = 5 × 10−9) and European populations (P = 2 × 10−8; fig. 3). These results lend some support to the mitonuclear-incompatibility hypothesis and are consistent with findings that certain mitochondrial-related functional classes are significantly enriched among the set of genes with low levels of introgression (see supplementary table 6.1 in Sankararaman et al. 2014). Only a subset of N-mt genes encode proteins that combine with mitochondrial gene products to form multisubunit complexes such as OXPHOS enzymes and ribosomes. This subset of 150 “interacting” N-mt genes had even lower levels of introgression on average than other N-mt genes, but the difference was not statistically significant (fig. 3). Although the differences in rates of introgression between N-mt genes and other nuclear genes are highly statistically significant, the magnitude of these effects is modest. For example, in the combined analysis of Asian and European introgression data, the median introgression scores for N-mt genes and other nuclear genes are 0.009 and 0.014, respectively.
Fig. 3.
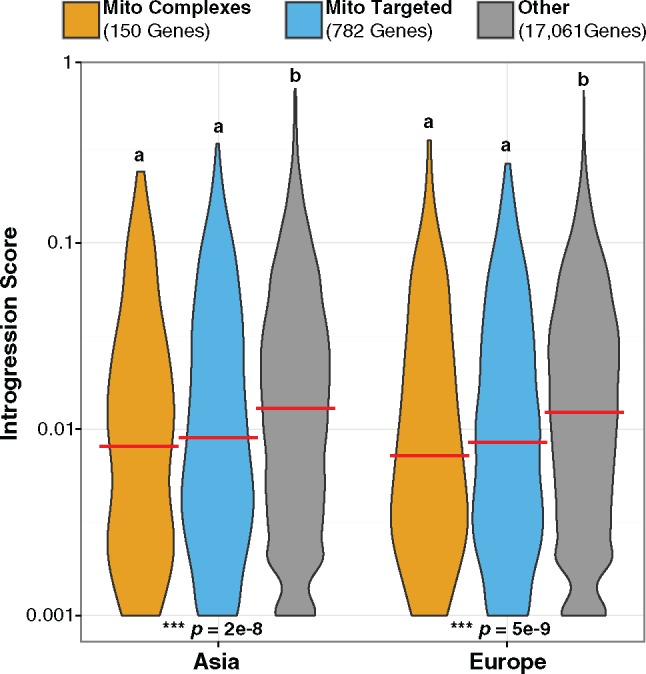
—A violin plot summarizing the distribution of gene-specific Neanderthal introgression scores in Asian (left) and European (right) populations of modern humans from Sankararaman et al. (2014). Separate distributions are shown for N-mt genes that contribute to mitonuclear OXPHOS and ribosomal complexes (orange), other N-mt genes (blue), and all other nuclear genes (gray). The median value for each distribution is indicated with a red bar. Overall statistical significance was assessed with a Kruskal–Wallis test (see reported P values), and lower case letters summarize significant differences that emerged from post hoc pairwise comparisons between each distribution. For the purposes of plotting on a log-scale, a minimum introgression score was set at 0.001.
Because regions of the genome that are subject to greater functional constraint exhibit lower rates of introgression (Sankararaman et al. 2014, 2016), one potential explanation for the lower rates of introgression for N-mt genes is that they are more functionally important on average than other nuclear genes. We used two proxies for functional importance to assess this possibility: 1) levels of transcript abundance and 2) level of amino acid divergence relative to chimpanzees. Using measures of transcript abundance from the Illumina Body Map data set, we found a significant, but very weak, negative association with introgression score in both Asian (P = 0.0073) and European populations (P = 0.0007; supplementary fig. S1, Supplementary Material online). Although this pattern is consistent with the expectation that more functionally important genes (i.e., those that are more highly expressed) will exhibit reduced rates of introgression, these effects explained a negligible proportion of the variance (R2 < 0.01 for both Asian and European data sets), and the relationship between mitochondrial targeting and introgression remained highly significant after controlling for this measure of transcript abundance (P < 1 × 10−6 in both Asian and European data sets). Furthermore, the relationship between expression level and introgression was even weaker (and nonsignificant) when the GTEx data set was used to measure transcript abundance (data not shown). We also found that genes with lower nonsynonymous substitution rates (i.e., more functional constraint) trended towards lower rates of introgression in both Asian (P = 1 × 10−6) and European populations (P = 9 × 10−7). However, as above, the magnitude of these effects was very small (supplementary fig. S1, Supplementary Material online), and the relationship between mitochondrial targeting and introgression remained highly significant after controlling for rate of nonsynonymous substitutions (P < 1 × 10−6 in both Asian and European data sets).
Analysis of Denisovan Introgression
If mitonuclear incompatibilities were responsible for the lower rate of introgression of N-mt genes from Neanderthals, we might expect a stronger historical effect on introgression from Denisovans because of the much higher level of mtDNA sequence divergence between modern humans and Denisovans (fig. 1). However, even in Melanesian populations, which have a sizeable amount of Denisovan-derived sequence, obtaining accurate gene-specific estimates of Denisovan introgression poses a technical challenge. It is difficult to confidently distinguish Denisovan alleles in a background that has already acquired alleles via admixture with at least one other archaic hominin lineage (Neanderthals). See Sankararaman et al. (2016) for discussion of these technical challenges.
Nevertheless, efforts have been made to identify Denisovan-derived haplotypes in a set of 35 published Melanesian genome sequences (Vernot et al. 2016). We used these haplotype calls to calculate an average proportion of Denisovan ancestry in the Melanesian population for each nuclear gene. We obtained a global average level of introgression of 0.0063, which is far lower than the estimates of Neanderthal introgression used in our previous analysis (mean introgression scores of 0.031 and 0.026 for Asian and European populations, respectively; Sankararaman et al. 2014). These gene-specific estimates of Denisovan introgression also produced an extremely high proportion of zero values (87.3% of genes). By contrast, zero values were relatively rare for Neanderthal introgression scores in Asian (5.1%) and European (5.6%) populations (Sankararaman et al. 2014). Given the evidence that Denisovan-derived alleles represent ∼5% of Melanesian genomes, which is more than the current contribution of Neanderthals to any modern human population (Green et al. 2010), it is likely that the values we used for Denisovan introgression are substantial underestimates, owing to the conservative nature of the published Denisovan haplotype calls (Vernot et al. 2016).
Using these estimates of Denisovan introgression into Melanesian genomes, we performed the same comparison between N-mt genes and other nuclear genes as described earlier for Neanderthal introgression. In this case, however, we did not find any significant differences in levels of introgression for N-mt genes relative to the rest of the genome (P = 0.26, Kruskal-Wallis test; supplementary fig. S2, Supplementary Material online). To assess whether this result represents a true biological difference between the histories of Denisovan versus Neanderthal introgression or a consequence of the more conservative calling of Denisovan-derived alleles, we analyzed a set of Neanderthal-derived haplotypes in Asians and Europeans from Vernot et al. (2016) that were obtained with the same methodology used to analyze Denisovan introgression in Melanesians. These gene-specific estimates of Neanderthal introgression exhibited a strongly significant positive correlation with estimates from Sankararaman et al. (2014) that were analyzed above, demonstrating a general consistency between the methods (P ≪ 0.001; r = 0.61 and 0.57 for Asian and European data sets, respectively; supplementary fig. S3, Supplementary Material online). However, the data from Vernot et al. (2016) yielded lower estimates and a much larger proportion of zero values even though they were derived from the same Asian and European populations as the estimates from Sankararaman et al. (2014). When we used these new estimates to compare levels of Neanderthal introgression for N-mt genes to background rates in the genome, the differences between N-mt genes and other nuclear genes were marginally significant at best (P = 0.03 and 0.07 for European and Asian data sets, respectively; supplementary fig. S3, Supplementary Material online). Therefore, the more conservative haplotype calling methods appear to result in a substantial reduction in statistical power to detect differences in rates of introgression, so we must be cautious in interpreting the biological significance of the difference in N-mt introgression results from Neanderthals (fig. 3) versus Denisovans (supplementary fig. S2, Supplementary Material online).
Molecular Interactions between Nuclear and Mitochondrial Gene Products
To test for sequence changes that potentially alter interactions between nuclear and mitochondrial gene products, we analyzed fixed differences in mitochondrially encoded proteins and structural RNAs to determine which sites physically contact nuclear-encoded proteins (fig. 4 and supplementary fig. S4, Supplementary Material online). Ten of the 22 amino acid substitutions in mitochondrially encoded proteins that distinguish humans and Neanderthals were identified as occurring at contact sites (supplementary table S6, Supplementary Material online). In total, these residues physically interact with nine different N-mt subunits within OXPHOS complexes (table 2). Similarly, 8 of the 24 amino acid substitutions that distinguish modern humans and Denisovans occurred at contact residues (supplementary table S6, Supplementary Material online), which interact with a total of eight different N-mt OXPHOS subunits (table 3). In mitochondrial rRNAs, four of the five nucleotide substitutions that distinguish modern humans and Neanderthals are at contact positions, which interact with a total of six N-mt ribosomal proteins (table 2 and supplementary table S6, Supplementary Material online). About 10 of the 14 rRNA substitutions that distinguish modern humans and Denisovans are at contact positions, which interact with a total of 13 N-mt ribosomal proteins (table 3 and supplementary table S6, Supplementary Material online). Overall, there was a slight trend towards enrichment or substitutions at contact sites relative to noncontact sites, but this was not significant in the Neanderthal data set (P = 0.10) or Denisovan data set (P = 0.17; Fisher’s exact tests). In addition, there are a total of 11 tRNAs with fixed nucleotide substitutions distinguishing modern humans from Neanderthals and/or Denisovans (table 1 and fig. 2). Although we have not specifically modeled the many protein–tRNA interactions that occur during mitochondrial tRNA maturation and charging (Pett and Lavrov 2015), we included the corresponding aaRS genes for these 11 tRNAs in our subsequent analysis of N-mt proteins.
Fig. 4.

—Fixed substitutions in mitochondrially encoded subunits between modern humans and Neanderthals mapped onto solved crystal structures for (A) NADH dehydrogenase, (B) cytochrome bc1, (C) cytochrome c oxidase, (D) ATP synthase, and (E) the mitochondrial ribosome. Mitochondrially encoded subunits are shown in green and N-mt subunits are shown in yellow. Substitutions at contact and noncontact residues are shown in red and black, respectively. Protein subunits are shown as spheres, while RNA subunits are shown as ribbons. For cytochrome bc1 and cytochrome c oxidase, substitutions are only highlighted in one of the two monomers for each subunit type. Note that only 25 of the 27 fixed differences from table 1 are shown because the ATP8 subunit is not included in the solved ATP synthase structure, and one of the ND5 substitutions is at the C-terminus, which is not included in the NADH dehydrogenase structure.
Table 2.
N-mt Genes That Directly Contact Sites with Fixed Substitutions in Mitochondrially Encoded Protein or Structural RNA Sequences between Modern Humans and Neanderthals
| N-mt Gene | Complex | Mito Contact(s) | Introgression |
|---|---|---|---|
| NDUFA13 | I | ND1 (186) | 2.1 |
| NDUFB5 | I | ND5 (24) | 65.2 |
| NDUFB9 | I | ND5 (515) | 41.2 |
| NDUFC2 | I | ND2 (346) | 34.2 |
| CYC1 | III | CYTB (233, 245) | 53.1 |
| COX5a | IV | COII (54) | 51.0 |
| COX6B1 | IV | COII (95) | 79.3 |
| COX6B2 | IV | COII (95) | 70.3 |
| COX6C | IV | COII (146) | 12.5 |
| ATP5F1 | V | ATP6 (176) | 7.3 |
| MRPL2 | Ribosome | RNR2 (855) | 69.1 |
| MRPL27 | Ribosome | RNR2 (1,163) | 19.5 |
| MRPL37 | Ribosome | RNR2 (39) | 46.1 |
| MRPL47 | Ribosome | RNR2 (39) | 62.3 |
| MRPL48 | Ribosome | RNR2 (1,163) | 41.1 |
| MRPL57 | Ribosome | RNR2 (386) | 78.5 |
| HARS2 | aaRS | TRNH (52) | 35.5 |
| NARS2 | aaRS | TRNN (57) | 5.6 |
| YARS2 | aaRS | TRNY (52) | 33.9 |
Note.—The mitochondrial gene products and contact residue positions based on the reference structures (in parentheses) are also indicated. An introgression ranking scaled between 0 and 100 is reported based on gene-specific introgression scores from Sankararaman et al. (2014). In cases where multiple genes shared the same introgression ranking, the reported value represents the midpoint of the range (e.g., 4.2% of genes had an introgression score of 0, so those genes have a ranking of 2.1). Note that both tissue-specific paralogs of COX6B are included.
Table 3.
N-mt Genes That Directly Contact Sites with Fixed Substitutions in Mitochondrially Encoded Protein or Structural RNA Sequences between Modern Humans and Denisovans
| N-mt Gene | Complex | Mito Contact(s) |
|---|---|---|
| NDUFB5 | I | ND5 (24) |
| NDUFB9 | I | ND5 (515) |
| CYC1 | III | CYTB (233) |
| UQCRC1 | III | CYTB (4) |
| COX4I1 | IV | COI (456) |
| COX4I2 | IV | COI (456) |
| COX6B1 | IV | COII (95) |
| COX6B2 | IV | COII (95) |
| COX6C | IV | COII (22, 146) |
| COX7B | IV | COI (456) |
| MRPL2 | Ribosome | RNR2 (734, 855) |
| MRPL15 | Ribosome | RNR2 (616) |
| MRPL16 | Ribosome | RNR2 (1,287) |
| MRPL27 | Ribosome | RNR2 (1,163) |
| MRPL37 | Ribosome | RNR2 (39, 736) |
| MRPL43 | Ribosome | RNR2 (616) |
| MRPL44 | Ribosome | RNR2 (616) |
| MRPL47 | Ribosome | RNR2 (39) |
| MRPL48 | Ribosome | RNR2 (1,163) |
| MRPL57 | Ribosome | RNR2 (386) |
| MRPS5 | Ribosome | RNR1 (281) |
| MRPS6 | Ribosome | RNR1 (362) |
| MRPS21 | Ribosome | RNR1 (362) |
| EARS2* | aaRS | TRNE (50) |
| FARS2* | aaRS | TRNF (20) |
| HARS2 | aaRS | TRNH (52, 56) |
| IARS2 | aaRS | TRNI (56) |
| LARS2* | aaRS | TRNL1 (48), TRNL2 (46) |
| MARS2 | aaRS | TRNM (55) |
| TARS2 | aaRS | TRNT (54) |
| WARS2 | aaRS | TRNW (5, 28) |
| YARS2* | aaRS | TRNY (52) |
Note.—The mitochondrial gene products and contact residue positions based on the reference structures (in parentheses) are also indicated. Asterisks indicate genes with identified Denisovan-derived alleles in Melanesian population based on analysis of Vernot et al. (2016). Note that both tissue-specific paralogs for COX6B and for COX4I are included.
To assess patterns of N-mt gene sequence divergence among hominins, we analyzed the set of 37 N-mt proteins that physically interact with the mitochondrial-encoded sites that have fixed differences distinguishing modern humans from Neanderthals and/or Denisovans. This gene set contains 14 OXPHOS subunits (including both tissue-specific paralogs for COX6B and for COX4I; Boczonadi et al. 2015), 13 ribosomal proteins, and 10 aaRSs (supplementary table S7, Supplementary Material online). There are only 10 aaRSs (despite there being 11 tRNAs with fixed differences) because the two leucine tRNAs are both charged by the same aaRS.
These 37 proteins totaled 11,604 amino acids in length (supplementary table S7, Supplementary Material online). Pairwise comparisons against the HuRef genome (Levy et al. 2007) identified only seven homozygous nonsynonymous differences relative to the Neanderthal nuclear genome (Prüfer et al. 2014) and only five relative to the Denisovan nuclear genome (Meyer et al. 2012). Because of the short divergence time between modern humans and Neanderthals/Denisovans, we are expected to share genetic variants with these related hominins, and all but two of these pairwise differences were amino acid polymorphisms that are segregating within modern human populations (supplementary table S8, Supplementary Material online). One of the two exceptions was a substitution (Y91C) in the complex III subunit UQCRC1 in Denisovans, which is a residue that does not contact any mitochondrially encoded subunits (fig. 5). The other exception is a premature stop codon in the ribosomal protein MRPL43 that appears to have been polymorphic within Neanderthals. This nonsense mutation truncates 86 amino acids from the longest predicted human isoform, but much of the truncated region is absent from some other reported human isoforms, and it is not found within the solved structure of the human mitochondrial ribosome (Amunts et al. 2015). Overall, we observed extremely little variation in the sequences of N-mt proteins. In particular, there were no cases of structurally correlated changes between directly interacting residues.
Fig. 5.
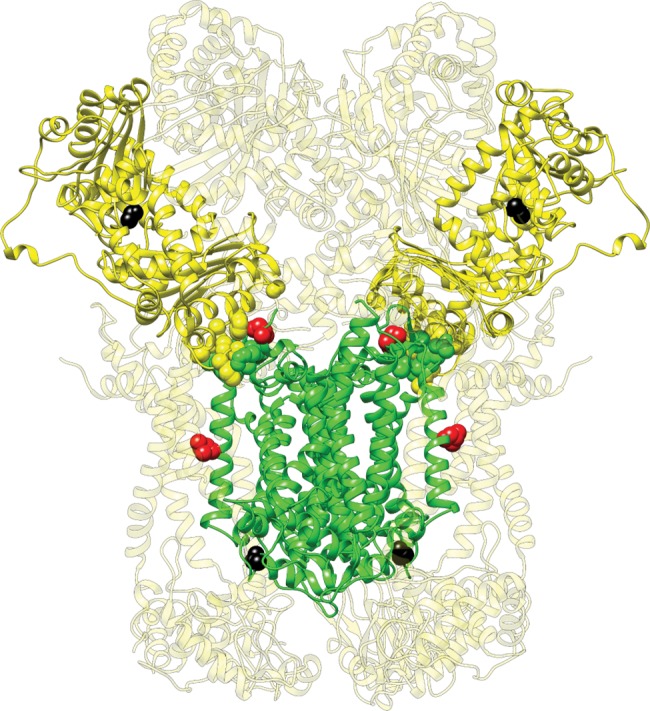
—The fixed amino acid substitution between modern humans and Denisovans in the N-mt protein UQCRC1 mapped onto the solved crystal structure for cytochrome bc1. The mitochondrially encoded CYTB subunits (two monomers) are shown in green and the UQCRC1 subunits (two monomers) are shown in bright yellow. The other N-mt subunits are shown in light yellow. Variable residues and those that form the interface between CYTB and UQCRC1 are shown as spheres. Substitution at contact residues are shown in red, while substitutions at noncontact residues (including the one in UQCRC1) are shown in black.
An additional predicted effect of structural interactions is that incompatibilities and restricted introgression would be most pronounced for N-mt genes encoding proteins that directly contact mitochondrially encoded sites with recent substitutions. However, we found little evidence to support this prediction. In the Neanderthal data set, only one such N-mt gene (NDUFA13) had an introgression score of 0 across both Asian and European population, and most of the other examples had introgression scores that are consistent with background levels in the genome (table 2). In the Denisovan data set, 28 of the 32 N-mt genes involved in interactions with altered mitochondrial sites had no detectable introgression (table 3), but this is consistent with the random expectation because most genes in the genome (87%) are in this category.
Discussion
The Role of Mitonuclear Incompatibilities during Hominin Introgression
The hypothesis that motivated our analysis was that incompatibilities (i.e., deleterious epistatic interactions) between Neanderthal/Denisovan nuclear alleles and modern-human mtDNA may have shaped patterns of introgression. In this light, the significant underrepresentation of N-mt loci in genomic regions of Neanderthal introgression suggests that mitonuclear interactions might have had an effect (fig. 3). However, it is possible that mitochondrial function per se has not affected introgression and is only correlated with some other important feature. For example, it has been shown that genomic regions with high levels of functional constraint tend to have lower frequencies of introgressed alleles in modern humans (Sankararaman et al. 2016), so it is possible that N-mt genes are simply more functionally important on average. However, the underrepresentation of introgressed alleles at N-mt loci remained significant even when we controlled for levels of expression and rates of sequence evolution (supplementary fig. S1, Supplementary Material online), which we used as proxies for functional importance.
It is also conceivable that N-mt genes are enriched in genomic regions that happen to be less prone to introgress for reasons such as low recombination rate (Baack and Rieseberg 2007). We consider this possibility unlikely, however, because the lone evidence for a nonrandom distribution of N-mt genes in the human genome is that they are underrepresented on the X-chromosome relative to autosomes (Drown et al. 2012). Because autosomes exhibit higher rates of introgression than the X-chromosome, the overrepresentation of N-mt genes on autosomes would tend to introduce a bias that is opposite the pattern that we observed in our data. We are also skeptical of any interpretation that N-mt alleles from archaic hominins suffered from a genotype × environment interaction. Mitochondrial biology has often been implicated in local adaptation and thermal tolerance, including in humans (Mishmar et al. 2003), but there is little reason to expect that the alleles in human populations expanding out of Africa would have been more locally adapted to Eurasian environments than those of Neanderthal and Denisovan populations that already resided there.
Therefore, although the cause of lower rates of introgression for N-mt genes is not entirely clear, we conclude that the most likely explanation involves deleterious epistatic interactions with modern-human nuclear and/or mitochondrial genetic backgrounds. However, it is very unlikely that such epistatic effects were mediated by substitutions at physically interacting sites even though direct structural interactions have been implicated in other studies of mitonuclear coevolution (Osada and Akashi 2012; Meiklejohn et al. 2013; Havird et al. 2015). A role for changes at directly interacting sites is inconsistent with the extremely low levels of primary sequence divergence in hominin N-mt proteins (table 3; supplementary tables S6–S8, Supplementary Material online). Moreover, rates of introgression appear to be lower for N-mt loci regardless of whether they contribute to direct molecular interactions in complexes with mitochondrially encoded gene products (fig. 3). It is possible that the rapid evolution of mtDNA has effects on diverse nuclear genes involved in mitochondrial function because epistasis does not necessarily require direct physical interaction between gene products (Moore and Williams 2005; Clark et al. 2012; Baris et al. 2017). Given the limited amounts of protein sequence divergence, it is likely that any functional and fitness effects that distinguish alleles from modern humans and related hominins largely reflect differences in gene expression and regulation.
We reasoned that, if changes in mtDNA were responsible for reduced introgression of Neanderthal N-mt alleles, such effects would have been even stronger during introgression from Denisovans because of their much more divergent mitochondrial haplotypes (fig. 1; Krause et al. 2010). Therefore, it was somewhat surprising that there was no detectable difference in rates of Denisovan introgression between N-mt genes and other nuclear loci (supplementary fig. S3, Supplementary Material online). However, there are both biological and methodological issues that potentially complicate the Denisovan analysis. First, despite the higher overall level of mtDNA divergence in Denisovans, it is not clear that this corresponds to a greater amount of functional divergence. Mitochondrial dN/dS values for modern humans and Neanderthals are substantially higher than those for Denisovans and internal branches in the hominin tree (table 1; Krause et al. 2010), suggesting a history of more efficient purifying selection in Denisovans against deleterious functional changes in mtDNA. Second, as we have already noted, the power to detect variation in rates of Denisovan introgression appears to be limited by the conservative methodology used to identify Denisovan-derived alleles (Vernot et al. 2016). Although methods based on conventional D-statistics can be used to estimate the total contribution of introgressed alleles from a given population, it is far more challenging to assign a probability of introgression for individual loci. Great progress has been made in addressing this challenge (Sankararaman et al. 2014; Vernot and Akey 2014), but it remains difficult to confidently identify Denisovan-derived alleles in Melanesian genomes because they already contain a large contribution of introgressed alleles from another source (Neanderthals).
Even if mitonuclear interactions were responsible for reduced introgression rates of Neanderthal-derived N-mt alleles, the magnitude of the effect appears to have been relatively small (fig. 3). Therefore, it is likely that these interactions have had limited consequences for the genome-wide landscape of introgression, at least in the European and Asian populations that were included in this analysis.
These limited effects could indicate that there is little functional divergence in mitochondrial haplotypes among hominin lineages, which would be consistent with recent inferences that mtDNA has introgressed between some hominin lineages. Even though there appears to have been no mtDNA introgression into modern human populations (Krings et al. 1997; Serre et al. 2004), the discordance between mitochondrial and nuclear genealogies in hominins has been interpreted as evidence for a mitochondrial replacement event late in the evolution of Neanderthals (Meyer et al. 2016). These findings are also relevant to recent debates about the extent of mitonuclear incompatibilities within modern humans, which have garnered attention with the advent of mitochondrial replacement therapy (Reinhardt et al. 2013; Chinnery et al. 2014; Morrow et al. 2015; Sloan et al. 2015; Rishishwar and Jordan 2017) and led to arguments that mitonuclear incompatibilities may be less widespread and slower to emerge than previously proposed (Eyre-Walker 2017). Of course, the lack of strong genome-wide signatures of mitonuclear incompatibilities does not preclude the possibility of strong effects on individual loci, and the existence of large introgression “deserts” within the modern human genome (Sankararaman et al. 2014) may be the result of linkage with loci involved in strongly deleterious epistatic interactions, including some N-mt genes (Runemark et al. 2017). Indeed, one of the pressing questions in the field of mitonuclear coevolution is whether incompatibilities emerge from individual changes of large effect or the more gradual and stepwise accumulation at many loci (McKenzie et al. 2003; Barnard‐Kubow et al. 2016).
Epistatic versus Additive Effects on Patterns of Gene Flow
Two general models have been put forth to explain the evidence for purifying selection against Neanderthal and Denisovan alleles in modern human populations. The first is based on incompatibilities between introgressing alleles from archaic hominins and the genetic background in modern humans (Currat and Excoffier 2011; Sankararaman et al. 2014, 2016; Kuhlwilm et al. 2016), which fits with a broader view that epistatic interactions have pervasive effects (Breen et al. 2012; Levin and Mishmar 2017). The second model is based on additive effects and does not require reproductive isolation or genetic incompatibilities among loci. Specifically, it has been argued that Neanderthals had lower effective population sizes than modern humans and, therefore, harbored larger numbers of weakly deleterious alleles because of inefficient selection (Harris and Nielsen 2016; Juric et al. 2016). Although this second model provides a cogent argument for why Neanderthal-derived alleles may have been generally disfavored in gene-rich regions (Sankararaman et al. 2014) and declined in frequency over time (Fu et al. 2016), it is less clear that it can explain the genome-wide underrepresentation of introgressed alleles in specific functional classes such as testis-expressed genes (Sankararaman et al. 2016) or the N-mt genes that were analyzed in our study. These functional gene classes would have to somehow be enriched for variants with deleterious effects that were weak enough to accumulate by drift in archaic hominin populations but strong enough to be effectively selected against in modern humans. While it is certainly possible that such variants are overrepresented in genes with mitochondrial- or testis-specific functions relative to the rest of the genome (Harris and Nielsen 2016), we are not aware of any a priori rationale for why this would be the case.
One prediction associated with an incompatibility model is that genes exhibiting limited introgression into modern humans would also have experienced lower levels of introgression moving in the opposite direction (i.e., from modern humans into Neanderthals and Denisovans). By contrast, if the historical accumulation of harmful mutations in Neanderthal and Denisovan populations is responsible for the underrepresentation of introgressed alleles at these loci in modern humans, we might expect that affected loci would have been subject to higher levels of introgression into archaic hominins as a form of adaptive replacement. We suggest that this offers a general framework for distinguishing between additive and epistatic effects in cases of bidirectional gene flow. Sign epistasis should contribute to a positive correlation between locus-specific rates of introgression in one direction versus the other because loci involved in incompatibilities would be restricted from moving in both directions. By contrast, differences in additive effects on fitness should lead to a negative correlation between locus-specific rates of introgression because the alleles that carry the heaviest deleterious mutation load will be least likely to introgress into a hybridizing population but most likely to be replaced themselves by introgression from that population. There is potential for future research to test this prediction in hominins because introgression appears to have occurred from modern humans into some eastern Neanderthal populations (Kuhlwilm et al. 2016). There is already evidence from this work that introgression has been restricted in both directions for genomics regions of greater functional importance, as might be expected in cases of epistatic incompatibilities.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank two anonymous reviewers and the lab groups of D.B.S. and Rachel Mueller for helpful comments on an earlier version of this manuscript and BZ 460 students at Colorado State University for stimulating discussion. We also thank Sriram Sankararaman for providing introgression score data and Joshua Akey for directing us to posted haplotype-call data. We are grateful to Thierry Chambert for helpful discussion and statistical advice. Our research on mitonuclear interactions is supported by a Division of Molecular and Cellular Biosciences grant at the National Science Foundation (MCB 1412260) and a National Institutes of Health Ruth L. Kirschstein National Research Service Award (F32GM116361).
Literature Cited
- Aledo JC, Valverde H, Ruíz-Camacho M, Morilla I, López FD.. 2014. Protein-protein interfaces from cytochrome c oxidase I evolve faster than nonbinding surfaces, yet negative selection is the driving force. Genome Biol Evol. 6:3064–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Toots J, Scheres SH, Ramakrishnan V.. 2015. The structure of the human mitochondrial ribosome. Science 348:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews RM, et al. 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 23:147.. [DOI] [PubMed] [Google Scholar]
- Baack EJ, Rieseberg LH.. 2007. A genomic view of introgression and hybrid speciation. Curr Opin Genet Dev. 17:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baris TZ, et al. 2017. Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genet. 13:e1006517.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard‐Kubow KB, So N, Galloway LF.. 2016. Cytonuclear incompatibility contributes to the early stages of speciation. Evolution 70:2752–2766. [DOI] [PubMed] [Google Scholar]
- Bar-Yaacov D, et al. 2015. Mitochondrial involvement in vertebrate speciation? The case of mito-nuclear genetic divergence in chameleons. Genome Biol Evol. 7:3322–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EA, Thompson AC, Sharbrough J, Brud E, Llopart A.. 2015. Gene flow between Drosophila yakuba and Drosophila santomea in subunit V of cytochrome c oxidase: a potential case of cytonuclear co‐introgression. Evolution 69:1973–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, et al. 2012. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet. 90:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczonadi V, et al. 2015. Investigating the role of the physiological isoform switch of cytochrome c oxidase subunits in reversible mitochondrial disease. Int J Biochem Cell Biol. 63:32–40. [DOI] [PubMed] [Google Scholar]
- Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA.. 2012. Epistasis as the primary factor in molecular evolution. Nature 490:535–538. [DOI] [PubMed] [Google Scholar]
- Burton RS, Barreto FS.. 2012. A disproportionate role for mtDNA in Dobzhansky-Muller incompatibilities? Mol Ecol. 21:4942–4957. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, et al. 2014. The challenges of mitochondrial replacement. PLoS Genet. 10:e1004315.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NL, Alani E, Aquadro CF.. 2012. Evolutionary rate covariation reveals shared functionality and coexpression of genes. Genome Res. 22:714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA.. 2004. Speciation. Sunderland (MA: ): Sinauer Associates, Inc. [Google Scholar]
- Currat M, Excoffier L.. 2011. Strong reproductive isolation between humans and Neanderthals inferred from observed patterns of introgression. Proc Natl Acad Sci. 108:15129–15134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y.. 2009. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. 1937. Genetics and the origin of species. Columbia University Press; New York. [Google Scholar]
- Drown DM, Preuss KM, Wade MJ.. 2012. Evidence of a paucity of genes that interact with the mitochondrion on the X in mammals. Genome Biol Evol. 4:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A. 2017. Mitochondrial replacement therapy: are mito-nuclear interactions likely to be a problem? Genetics 205:1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JL, Roethele JB, Filchak K, Niedbalski J, Romero-Severson J.. 2003. Evidence for inversion polymorphism related to sympatric host race formation in the apple maggot fly, Rhagoletis pomonella. Genetics 163:939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorczuk K, et al. 2016. Atomic structure of the entire mammalian mitochondrial complex I. Nature 538:406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, et al. 2016. The genetic history of Ice Age Europe. Nature 534:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, Templeton AR, Mishmar D.. 2009. Mitochondrial bioenergetics as a major motive force of speciation. BioEssays 31:642–650. [DOI] [PubMed] [Google Scholar]
- Gray MW. 2015. Mosaic nature of the mitochondrial proteome: implications for the origin and evolution of mitochondria. Proc Natl Acad Sci. 112:10133–10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, et al. 2008. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, et al. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao R, Zhao M-W, Hao Z-X, Yao Y-N, Wang E-D.. 2005. A T-stem slip in human mitochondrial tRNALeu (CUN) governs its charging capacity. Nucleic Acids Res. 33:3606–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, Nielsen R.. 2016. The genetic cost of Neanderthal introgression. Genetics 203:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RG, Larson EL.. 2014. Hybridization, introgression, and the nature of species boundaries. J Hered. 105:795–809. [DOI] [PubMed] [Google Scholar]
- Harrison RG, Larson EL.. 2016. Heterogeneous genome divergence, differential introgression, and the origin and structure of hybrid zones. Mol Ecol. 25:2454–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird JC, Trapp P, Miller C, Bazos I, Sloan DB.. 2017. Causes and consequences of rapidly evolving mtDNA in a plant lineage. Genome Biol Evol. 9:323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird JC, Whitehill NS, Snow CD, Sloan DB.. 2015. Conservative and compensatory evolution in oxidative phosphorylation complexes of angiosperms with highly divergent rates of mitochondrial genome evolution. Evolution 69:3069–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill GE. 2016. Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecol Evol. 6:5831–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X-S, Filatov DA.. 2016. The large-X effect in plants: increased species divergence and reduced gene flow on the Silene X-chromosome. Mol Ecol. 25:2609–2619. [DOI] [PubMed] [Google Scholar]
- Ingman M, Kaessmann H, Paabo S, Gyllensten U.. 2000. Mitochondrial genome variation and the origin of modern humans. Nature 408:708–713. [DOI] [PubMed] [Google Scholar]
- Iwata S, et al. 1998. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 281:64–71. [DOI] [PubMed] [Google Scholar]
- Juric I, Aeschbacher S, Coop G.. 2016. The strength of selection against Neanderthal introgression. PLoS Genet. 12:e1006340.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J, et al. 2010. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464:894–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings M, et al. 1997. Neandertal DNA sequences and the origin of modern humans. Cell 90:19–30. [DOI] [PubMed] [Google Scholar]
- Kuhlwilm M, et al. 2016. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature 530:429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin L, Zhidkov I, Gurman Y, Hawlena H, Mishmar D.. 2013. Functional recurrent mutations in the human mitochondrial phylogeny: dual roles in evolution and disease. Genome Biol. Evol. 5:876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin L, Mishmar D.. 2017. The genomic landscape of evolutionary convergence in mammals, birds and reptiles. Nat Ecol Evol. 1:0041.. [DOI] [PubMed] [Google Scholar]
- Levy S, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol. 5:e254.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Willis JH.. 2010. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 8:e1000500.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. 2010. Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A> G mutation. Mitochondrion 10:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie JL, Dhillon RS, Schulte PM.. 2016. Steep, coincident, and concordant clines in mitochondrial and nuclear‐encoded genes in a hybrid zone between subspecies of Atlantic killifish, Fundulus heteroclitus. Ecol Evol. 6:5771–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie M, Chiotis M, Pinkert CA, Trounce IA.. 2003. Functional respiratory chain analyses in murid xenomitochondrial cybrids expose coevolutionary constraints of cytochrome b and nuclear subunits of complex III. Mol Biol Evol. 20:1117–1124. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, et al. 2013. An Incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9:e1003238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin RG, Katewa SD, Ballard JW.. 2008. A candidate complex approach to study functional mitochondrial DNA changes: sequence variation and quaternary structure modeling of Drosophila simulans cytochrome c oxidase. J Mol Evol. 66:232–242. [DOI] [PubMed] [Google Scholar]
- Meyer M, et al. 2012. A high coverage genome sequence from an archaic Denisovan individual. Science 338:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, et al. 2014. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505:403–406. [DOI] [PubMed] [Google Scholar]
- Meyer M, et al. 2016. Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature 531:504–507. [DOI] [PubMed] [Google Scholar]
- Mishmar D, Ruiz-Pesini E, Golik P, et al. 2003. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci. 100:171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JH, Williams SM.. 2005. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays 27:637–646. [DOI] [PubMed] [Google Scholar]
- Morales HE, Sunnucks P, Joseph L, Pavlova A.. 2017. Perpendicular axes of differentiation generated by mitochondrial introgression. Mol Ecol. 26:3241–3255. [DOI] [PubMed] [Google Scholar]
- Morrow EH, Reinhardt K, Wolff JN, Dowling DK.. 2015. Risks inherent to mitochondrial replacement. EMBO Rep. 16:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Akashi H.. 2012. Mitochondrial-nuclear interactions and accelerated compensatory evolution: evidence from the primate cytochrome C oxidase complex. Mol Biol Evol. 29:337.. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134:112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payseur BA, Rieseberg LH.. 2016. A genomic perspective on hybridization and speciation. Mol Ecol. 25:2337–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pett W, Lavrov DV.. 2015. Cytonuclear interactions in the evolution of animal mitochondrial tRNA metabolism. Genome Biol Evol. 7:2089–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, et al. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Presgraves DC. 2008. Sex chromosomes and speciation in Drosophila. Trends Genet. 24:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, et al. 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvarnström A, Bailey RI.. 2009. Speciation through evolution of sex-linked genes. Heredity 102:4–15. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing.
- Racimo F, Sankararaman S, Nielsen R, Huerta-Sánchez E.. 2015. Evidence for archaic adaptive introgression in humans. Nat Rev Genet. 16:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Haney RA, Fry AJ.. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 19:645–653. [DOI] [PubMed] [Google Scholar]
- Reich D, et al. 2010. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468:1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt K, Dowling DK, Morrow EH.. 2013. Mitochondrial replacement, evolution, and the clinic. Science 341:1345–1346. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Whitton J, Gardner K.. 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152:713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol Evol. 16:351–358. [DOI] [PubMed] [Google Scholar]
- Rishishwar L, Jordan IK.. 2017. Implications of human evolution and admixture for mitochondrial replacement therapy. BMC Genomics 18:140.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL. 2015. Chromosomal rearrangements as barriers to genetic homogenization between archaic and modern humans. Mol Biol Evol. 32:3064–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runemark A, Trier CN, Eroukhmanoff F, Hermansen JS, Matschiner M, Ravinet M, Elgvin TO, Saetre G-P.. 2017. Variation and constraints in hybrid genome formation. bioRxiv. doi.org/10.1101/107508 [DOI] [PubMed] [Google Scholar]
- Sankararaman S, et al. 2014. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S, Mallick S, Patterson N, Reich D.. 2016. The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr Biol. 26:1241–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S, et al. 2015. Nuclear and mitochondrial DNA sequences from two Denisovan individuals. Proc Natl Acad Sci. 112:15696–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TR, Wu W, Goodman M, Grossman LI.. 2001. Evolution of nuclear-and mitochondrial-encoded subunit interaction in cytochrome c oxidase. Mol Biol Evol. 18:563–569. [DOI] [PubMed] [Google Scholar]
- Schumer M, Brandvain Y.. 2016. Determining epistatic selection in admixed populations. Mol Ecol. 25:2577–2591. [DOI] [PubMed] [Google Scholar]
- Serre D, et al. 2004. No evidence of Neandertal mtDNA contribution to early modern humans. PLoS Biol. 2:e57.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, et al. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Fields PD, Havird JC.. 2015. Mitonuclear linkage disequilibrium in human populations. Proc R Soc B Biol Sci. 282:20151704.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Havird JC, Sharbrough J.. 2017. The on-again, off-again relationship between mitochondrial genomes and species boundaries. Mol Ecol. 26:2212–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C. 2002. Modern human origins: progress and prospects. Philos Trans R Soc Lond B Biol Sci. 357:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87. [DOI] [PubMed] [Google Scholar]
- The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trier CN, Hermansen JS, Saetre GP, Bailey RI.. 2014. Evidence for mito-nuclear and sex-linked reproductive barriers between the hybrid Italian sparrow and its parent species. PLoS Genet. 10:e1004075.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, et al. 2003. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci. 100:15304–15309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernot B, Akey JM.. 2014. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343:1017–1021. [DOI] [PubMed] [Google Scholar]
- Vernot B, et al. 2016. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 352:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag. [Google Scholar]
- Wood B, Boyle EK.. 2016. Hominin taxic diversity: fact or fantasy? Am J Phys Anthropol. 159:S37–S78. [DOI] [PubMed] [Google Scholar]
- Wu C-I, Davis AW.. 1993. Evolution of postmating reproductive isolation: the composite nature of Haldane’s rule and its genetic bases. Am Nat. 142:187–212. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang J-R.. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16:409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, et al. 2015. Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. eLife 4:e10180.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H-Y, et al. 2009. Genetic variants in mitochondrial tRNA genes are associated with essential hypertension in a Chinese Han population. Clin Chim Acta 410:64–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.